

Abstract
Interleukin-22 (IL-22), a recently identified member of the IL-10 family of cytokines that is produced by Th17 and NK cells, plays an important role in controlling bacterial infection, homeostasis, and tissue repair. In this paper, we tested the effect of IL-22 on alcohol-induced liver injury in a murine model of chronic-binge ethanol feeding. Feeding male C57BL/6 mice with a Lieber-DeCarli diet containing 5% ethanol for 10 days, followed by a single dose of 5g/kg ethanol gavage, induces significant fatty liver and liver injury with peak serum levels of about 250 IU/L ALT and 420 IU/L AST 9 hours post gavage. Moreover, chronic-binge ethanol administration increases expression of hepatic and serum inflammatory cytokines and hepatic oxidative stress. Using this model, we demonstrate that treatment with IL-22 recombinant protein activates hepatic STAT3 and ameliorates alcoholic fatty liver, liver injury, and hepatic oxidative stress. Administration with IL-22 adenovirus also prevents alcohol-induced steatosis and liver injury. Deletion of STAT3 in hepatocytes abolishes the hepatoprotection provided by IL-22 in alcoholic liver injury. In addition, IL-22 treatment downregulates the hepatic expression of fatty acid transport protein, but upregulates several antioxidant, antiapoptotic genes, and antimicrobial genes. Finally, expression of IL-22R1 is upregulated while IL-22 is undetectable in the livers from mice with chronic-binge ethanol feeding or patients with alcoholic hepatitis. In conclusion, chronic-binge ethanol feeding may be a useful model to study the early stage of alcoholic liver injury. IL-22 treatment could be a potential therapeutic option to ameliorate alcoholic liver disease due to its antioxidant, antiapoptotic, antisteatotic, proliferative, and antimicrobial effects with the added benefit of potentially few side effects.
Keywords: alcohol, liver, IL-22, STAT3
Introduction
Excessive alcohol drinking is a leading cause of chronic liver disease worldwide, which results in a spectrum of liver disorders, ranging from simple fatty liver to more severe forms of liver injury such as alcoholic hepatitis, cirrhosis, and hepatocellular carcinoma.1, 2 Fatty liver is the earliest response to alcohol drinking and occurs in almost everyone who drinks heavily,3 while more severe forms of alcoholic liver injury typically develop in up to 35% of heavy drinkers.1, 2 No specific medical treatment is needed for patients with simple alcoholic fatty liver, which usually resolves within several weeks of alcohol withdrawal. More severe forms of alcoholic liver disease such as alcoholic hepatitis require treatment. Current therapeutic options for alcoholic hepatitis include corticosteroids or TNF-α inhibitor therapy; however, these treatments have generated controversial results and are associated with increased rates of infection. 2, 4–7
Interleukin-22 (IL-22), a recently identified cytokine that is produced by Th17 cells and natural killer cells, has been shown to play an important role in controlling bacterial infection, homeostasis, and tissue repair. 8, 9 The biological effect of IL-22 was believed to be mediated mainly via activation of the signal transducer and activator of transcription 3 (STAT3) signaling pathway through binding of IL-22R1 and IL-10R2, although high concentrations of IL-22 can also activate many other signaling pathways including STAT1, STAT5, MAPKs, AKT, NF-κB, AP-1 etc. 8 Recently, we and others have demonstrated that IL-22 treatment prevents T cell hepatitis,10 stimulates liver regeneration,10, 11 and improves fatty liver.12 Thus, we hypothesize that IL-22 treatment could be a potential therapeutic option to ameliorate alcoholic liver disease. In this paper, we test this hypothesis in a murine model of alcoholic liver injury induced by chronic-binge ethanol feeding.
During the last 5 decades, many animal models of alcoholic liver injury have been developed,13–17 which have significantly helped us understand the molecular mechanisms of alcoholic liver disease. Presently, the most commonly used model for alcoholic liver injury in rodents is voluntary feeding with the Lieber-DeCarli liquid ethanol-containing diet.17–22 However, this model only induces minor hepatic lesions such as fatty liver and slight elevation of serum ALT, especially in male mice.17–22 In contrast, the Tsukamoto-French model, which administers a higher dose of ethanol through continuous intragastric feeding, causes more severe forms of liver injury such as steatosis and mild liver inflammation and fibrosis.13, 23 However, this model has not been widely used in many laboratories because of its technical difficulty, requirement for animal husbandry, and the expense of equipment.23 Lastly, another popular animal model of liver injury features ad libitum feeding of a liquid diet containing ethanol followed by a challenge with endotoxin or hepatotoxins, etc. Although this model is used by many investigators, it is sometimes difficult to rule out whether the enhanced hepatocellular damage is due to the ethanol or toxin challenge.16 Therefore, the present study was designed todevelop a murine model of chronic-binge ethanol administration to induce significant liver injury and to test the hypothesis that treatment of IL-22 ameliorates alcoholic liver injury by using this model. Our findings demonstrated that chronic feeding plus a single dose of ethanol delivered by oral gavage induced more severe forms of liver injury and fatty liver than chronic feeding or single ethanol gavage alone. By using this model, we also demonstrated that IL-22 treatment ameliorated alcoholic liver injury, suggesting therapeutic potential for IL-22 in treating alcoholic liver disease.
Materials and Methods
Mice
C57BL/6N mice were purchased from the NCI (Frederick, MD). Hepatocyte-specific STAT3 knockout (STAT3Hep−/−) mice and wild-type mice were described previously.24 All male mice were used unless specified. All animal experiments were approved by the NIAAA Animal Care and Use Committee.
A Mouse model of chronic plus single binge ethanol consumption (referred as chronic-binge model)
Eight to 10-week-old male C57BL/6N mice were fed a nutritionally adequate liquid control diet (Bioserv, Frenchtown, NJ) for 5 days, then divided into two groups (supplemental Fig. S1a): ethanol groups were fed a liquid diet containing 5% ethanol for 10 days; control groups were pair-fed control diet for 10 days; At day 11, mice in ethanol groups were gavaged a single doses of ethanol (5g/kg body weight, 20% ethanol), while mice in control groups were gavaged isocaloric dextrin maltose. The gavage was always performed in the early morning. After gavage, mice were kept on control or ethanol diet and kept in the cages on the warm blanket with circulating water. All mice survived after chronic-binge ethanol feeding. After gavage, mice were slow-moving, but conscious and regained normal behavior within 4–6 h. The mice were always euthanized 9 h post gavage when the serum levels of ALT and AST reached to the peak. In some experiments, mice were also euthanized at different time points post gavage as specified.
Treatment of mice with recombinant IL-22 protein
Mice were fed ethanol diet for 10 days and then treated (i.p) with a single dose of recombinant murine IL-22 protein (1 μg/g) (GenScript, Piscataway, NJ) or saline. After treatment with IL-22 for 3 h, mice were gavaged with a single dose of ethanol (5g/kg) or maltose and sacrificed 9 h post gavage.
Administration of mice with IL-22 adenovirus
IL-22 adenovirus was made by cloning mouse IL-22 cDNA (544 bp) into the pENTR/D-TOPO system (Invitrogen), followed by using Invitrogen Gateway system to perform a LR reaction with pAd/CMV/V5-DEST to make the expression vector pAd/CMV/mIL-22. Mice were fed ethanol diet or control diet for 10 days, then injected (i.v) with adenovirus-IL-22 (2×108pfu) or adenovirus-empty vector (2×108pfu) and kept on ethanol or control diet for 48 h, followed by gavage with a single dose of ethanol. Mice were sacrificed 9 h post gavage.
Other methods
The following methods are described in supplemental materials, including blood chemistry (ALT, AST, TG, and Cholesterol), hepatic lipid contents, blood ethanol concentration, histology, serum cytokine levels, real time PCR, lipid peroxidation, and GSH assay.
Patients with alcoholic hepatitis and selection of control livers
Thirty two consecutive patients were admitted at the Liver Unit (Hospital Clínic, Barcelona, Spain) with clinical and analytical features of alcoholic hepatitis as described previously25 and in the supplemental materials and table S2. In all cases, liver biopsy tissues were immediately submerged in an RNA stabilization solution and stored at −80°C until RNA extraction. The protocol was approved by the Ethics Committee of the Hospital Clínic and all patients gave informed consent. Normal livers were obtained from optimal cadaveric liver donors (n=3) or resection of liver metastases (n=3) before vascular clamp. All controls had normal serum aminotranferases and normal liver histology.
Statistical Analysis
Data are expressed as means ± SD. To compare values obtained from two groups, the Student’s t-test was performed. To comparevalues obtained from three or more groups, one-way ANOVA was performed followed by Tukey’s post hoc test. A value of P < 0.05 was considered significant.
Results
A model of alcoholic liver injury induced by chronic feeding plus single gavage of ethanol (chronic-binge model)
The most commonly used voluntary feeding model with Liber-DeCali diet containing ethanol only induced mild liver injury in male C57BL/6 mice, with the peak serum levels of 60–100 IU/L ALT.17–21 In order to induce more severe form of liver injury, mice were fed chronically ethanol diet for 10 days followed by single gavage of ethanol. Control group mice were pair-fed control diets without ethanol for 10 days followed by single gavage of maltose. Similar body weight was gained in both groups and liver/body weight ratios were higher in ethanol-treated mice compared to control diet-fed mice (Supplemental Fig. S1b). The peak levels of blood ethanol concentration reached 140 mM 1 and 2 h post ethanol gavage in chronic-binge ethanol-fed mice (supplemental Fig. S1c).
As illustrated in Figs. 1A–C, the basal levels (0 h gavage) of serum ALT, liver and serum triglyceride were significantly higher after 10-day ethanol feeding than those after 10-day control diet feeding. Compared to control groups, chronic-binge ethanol induced significantly higher levels of serum ALT and AST, with peak effects 9 h post gavage reaching about 250 IU/L ALT and 420 IU/L AST. In addition, chronic-binge ethanol induced higher levels of serum ALT and AST in female mice compared to male mice (supplemental Fig. S1d). Hepatic and serum levels of triglyceride were much higher in chronic-binge group than control group, while liver and serum levels of cholesterol were comparable between these 2 groups. Liver histology and Oil Red O staining analyses showed significant microsteatosis in the livers from all ethanol-fed mice. Macrosteatosis and necrosis were also often observed in some ethanol-fed mice (Figs. 1E–D). Sirius red and α-SMA staining revealed no obvious liver fibrosis in this model (data not shown).
Fig. 1. A mouse model of alcoholic liver injury induced by chronic-binge ethanol feeding.

(A–D) C57BL/6N mice were fed control or ethanol diet for 10 days, followed by single gavage of maltose or ethanol, respectively. Mice were euthanized at various time points post gavage. (A) Serum ALT, AST; (B, C) Liver and serum triglyceride (TG) and cholesterol (Chol) levels; (D, E) Representative H&E staining and Oil Red O staining of liver tissues 9 h post gavage. a: microsteatosis; b: macrosteatosis; c: macrosteatosis plus necrosis. (F) Serum ALT, AST from mice treated with different ethanol feeding protocols. Values represent means ± SD. In panes A–C: *P<0.05; ** P<0.01 vs corresponding control groups (n=5–15). In panel F, **P<0.01 in comparison with other 3 groups (n=4–8).
Fig. 1F shows that chronic-binge induced the highest levels of serum ALT and AST compared to 4-week, 10-day ethanol feeding, or single ethanol gavage alone. The detailed comparison of liver injury between chronic-binge and single ethanol gavage alone was further examined (supplemental Fig. S2), which clearly showed that chronic-binge induced much higher peak levels of liver injury (ALT and AST) than single ethanol gavage 6 and 9 hours post gavage. Single ethanol gavage alone also elevated hepatic triglyceride levels, which was similar to chronic-binge group (Fig. S2b) and consistent with previous reports.26
Fig. 2A shows that chronic-binge but not control group induced liver inflammation as indicated by elevation of inflammatory markers (CCR2 for monocytes; F4/80 for macrophages) and pro-inflammatory cytokines. Expression of neutrophils marker MPO was undetectable in both groups (data not shown). Serum levels of proinflammatory cytokines were higher in chronic-binge-treated mice than those with control diet (Fig. 2B). Finally, Fig. 2C shows that compared to the control group, the levels of oxidative stress marker 4-NHE in the liver were elevated while hepatic GSH levels were decreased in the chronic-binge group.
Fig. 2. Liver inflammation and oxidative stress are elevated after chronic-binge ethanol feeding.

Mice were fed control or ethanol diet as described in Fig. 1. (A) Real-time PCR analyses; (B) Serum levels of cytokines; (C) Hepatic levels of oxidative stress. Values represent means ± SD (n=5–8). *P<0.05; ** P<0.01.
The expression of hepatic fat metabolism-associated genes was also examined. As shown in Fig. 3, hepatic expression of several lipogenesis genes (SREBP-1, FAS, LXRα, ACC, and SCD1) was upregulated while fat oxidation gene (PPARα) was decreased in chronic-binge group compared to control groups.
Fig. 3. Regulation of fat metabolism genes after chronic-binge ethanol feeding.
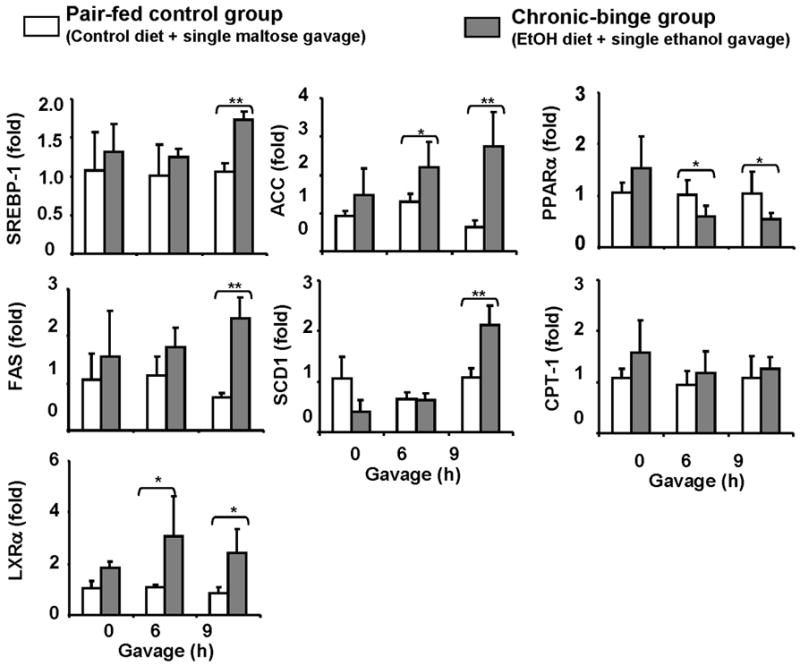
Mice were fed control or ethanol diet as described in Fig. 1. Real-time PCR analyses of fat metabolism-associated genes. Values represent means ± SD (n=4–8). *P<0.05, ** P<0.01.
Treatment with recombinant IL-22 protein ameliorates chronic-binge ethanol-induced liver injury
To explore the therapeutic potential of IL-22 in treating alcoholic liver disease, pair-fed or chronic-binge-fed mice were treated with IL-22. Liver injury, oxidative stress, and inflammation were then assessed. As illustrated in Figs. 4A–B, treatment with recombinant IL-22 reduced serum ALT and AST as well as liver triglyceride levels but did not affect cholesterol levels. The protective effect of IL-22 on alcoholic fatty liver was further confirmed by liver histology (Fig. 4C). Interestingly, serum levels of triglyceride were elevated after IL-22 treatment while cholesterol levels remained unchanged (Fig. 4D). In addition, IL-22 treatment prevented ethanol-mediated induction of 4-NHE and depletion of GSH levels in the liver (Fig. 4E). Finally, IL-22 treatment did not affect the hepatic and serum levels of proinflammatory cytokines from both groups but upreglated slightly the hepatic expression of F4/80 and CCR2 (supplemental Fig. S3).
Fig. 4. IL-22 treatment prevents chronic-binge ethanol-induced liver injury.

Pair-or ethanol-fed mice were treated with recombinant IL-22 as described in Materials and Methods. (A) Serum ALT, AST; (B) Hepatic lipid contents; (C) Representative H&E and Oil red O staining of the liver; (D) Serum lipid contents; (E) Liver oxidative stress markers. Values represent means ± SD (n=4–7). *P<0.05,**P<0.01.
Treatment with IL-22 adenovirus ameliorates chronic-binge ethanol-induced liver injury
The hepatoprotective effect of IL-22 on alcoholic liver injury was further confirmed by adenovirus IL-22 treatment. As illustrated in Fig. 5, administration of mice with IL-22 adenovirus resulted in elevation of serum IL-22 levels reaching about 4500 pg/ml, and reduced serum ALT levels and liver triglyceride levels without affecting liver cholesterol levels (Figs. 5B–D). Serum levels of triglyceride and cholesterol were not affected after treatment with IL-22 adenovirus. Liver histology also confirmed less steatosis in the mice treated with IL-22 adenovirus compared to those treated with adenovirus with empty vector (data not shown).
Fig. 5. Injection of IL-22 adenovirus prevents chronic-binge ethanol-induced liver injury.
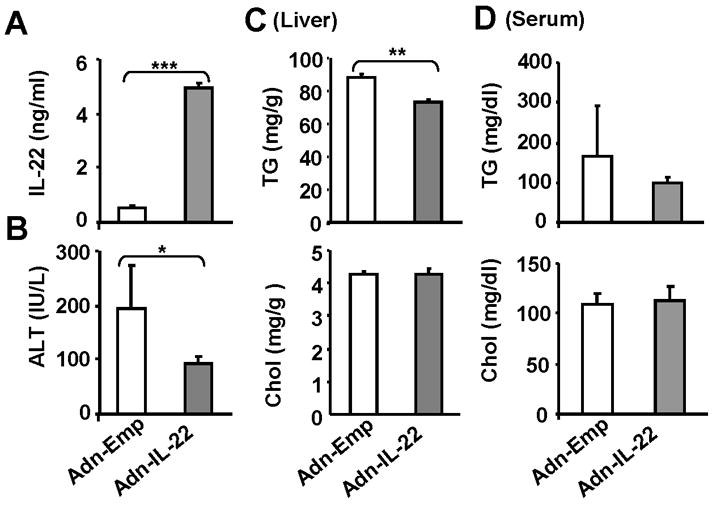
Ethanol-fed mice were treated with adenovirus-IL-22 or adenovirus-empty vector for 48 h as described in Materials and Methods. (A) Serum IL-22 levels; (B) Serum ALT; (C) Hepatic lipid contents; (D) Serum lipid levels. Values represent means ± SD (n=5). *P<0.05, **P<0.01, and ***P<0.001.
Hepatoprotective effect of IL-22 on alcoholic liver injury is dependent on STAT3 in hepatocytes
To determine the mechanisms underlying IL-22 protection against alcoholic liver injury, we examined the effect of recombinant IL-22 protein treatment on STAT3 activation in the liver. As illustrated in Fig. 6A, injection of IL-22 protein induced STAT3 activation in the liver with peak effect occurring at 1 hour post injection. Next we tested the role of STAT3 in IL-22 protection against ethanol-induced liver injury using hepatocyte-specific STAT3 knockout (STAT3Hep−/−) mice. As shown in Figs. 6B–C, IL-22 treatment reduced serum ALT and AST and hepatic triglyceride in wild-type mice but not in STAT3Hep−/−mice fed with chronic-binge ethanol. Liver histology also showed that the protective effect of IL-22 on steatosis was observed in wild-type mice but diminished in STAT3Hep−/−mice (Fig. 6D).
Fig. 6. Hepatoprotective effect of IL-22 on chronic-binge ethanol-induced liver injury is mediated via activation of hepatic STAT3.

(A) Western blot analyses of liver tissues from C57BL/6N mice treated with IL-22 protein. (B–D) STAT3Hep−/− and wild-type mice were treated with chronic-binge ethanol feeding and injected (i.p) with IL-22 protein as described in Materials and Methods. (B) Serum ALT, AST; (C) Liver triglyceride; (D) Representative H&E staining of liver tissues. Values represent means ± SD (n=4–6). *P<0.05 and **P<0.01.
Regulation of hepatic fat-metabolism, antioxidant, antiapoptotic, and antimicrobial genes by IL-22 treatment in vivo
To further understand the mechanisms underlying IL-22 protection against alcoholic liver injury, we examined the effects of IL-22 on expression of fat metabolism, antioxidant, and antiapoptotic genes. As illustrated in Fig. 7A, treatment with recombinant IL-22 protein markedly downregulated the expression of fatty acid transport protein (FATP) in the livers from chronic-binge-treated mice but not from pair-fed mice. Interestingly, IL-22 treatment had no effect on the expression of many other fat metabolism-related genes (supplemental Fig. S4). Furthermore, IL-22 downergulation of FATP was not observed in C57BL/6N mice without ethanol feeding or in chronic-binge-treated STAT3Hep−/− mice (Fig. 7C).
Fig. 7. Regulation of fat metabolism, antioxidant, antiapopotic, and antimicrobial genes by IL-22 treatment in vivo.
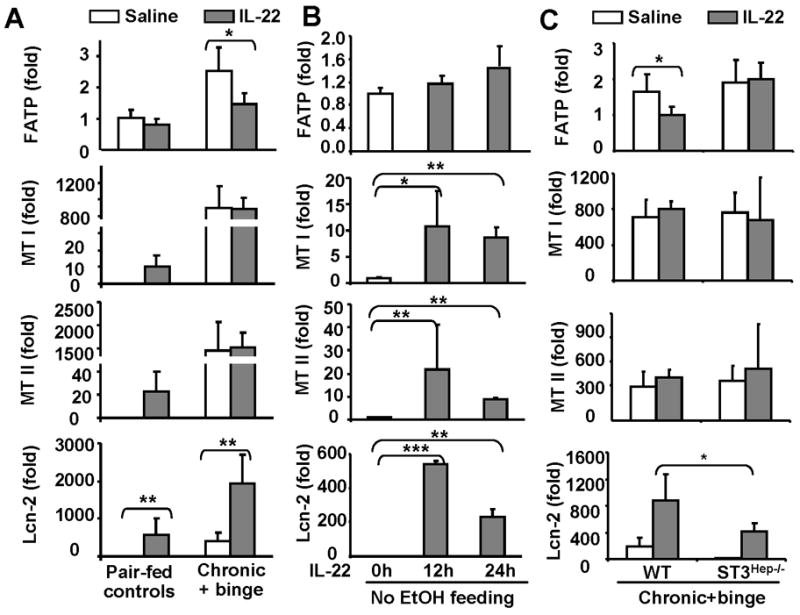
(A) Mice were fed ethanol diet or pair-fed, followed by treating with IL-22 as described in Fig. 4. (B) Normal C57BL/6 mice were treated with IL-22 for 12 and 24 hours. (C) STAT3Hep−/− and wild-type mice were treated with chronic-binge ethanol feeding and treated with IL-22 as described in Fig. 6. Liver tissues from panels A, B, and C were then collected for real-time PCR analyses. Values represent means ± SD (n=5–7). *P<0.05, **P<0.01,***P<0.001.
Fig. 7A shows that alcohol feeding significantly increased the expression of the antioxidant gene metallothionein (MTI/II) in the liver, which is consistent with previous reports showing that short ethanol exposure elevated hepatic MT levels.27 IL-22 treatment increased MTI/II expression in the livers from pair-fed mice (Fig. 7A) and C57BL/6N mice (Fig. 7B), but did not further increase expression of MTI/II in the chronic-binge-treated mice (Fig. 7A). The lack of further induction of MT1/II by IL-22 in ethanol-fed mice may be due to high basal levels of MTI/II in these mice.
The antimicrobial effect of IL-22 has been well documented, which is mediated via induction of several antimicrobial genes.8 Here we demonstrated that IL-22 treatment also elevated the hepatic expression of antimicrobial genes such lipocalin 2 in pair-fed and chronic-binge-fed mice (Fig. 7A) as well as in C57BL/6N mice (Fig. 7B). IL-22 induction of lipocalin-2 was partially diminished in STAT3Hep−/− mice compared to wild-type mice (Fig. 7C).
Alcohol consumption upregulates IL-22R1 expression without affecting IL-22 expression in the liver
Previous studies have reported that the number of IL-17+ cells (Th17) is increased in alcoholic liver disease.28 Since IL-17 is mainly produced by Th17 cells that may also secrete IL-22,8 we examined whether IL-22 was also upregulated in alcoholic liver disease. Surprisingly, the levels of IL-22 mRNA expression were undetectable in the liver from chronic-binge-treated mice or from patients with alcoholic hepatitis (Figs. 8A–B). On the contrary, hepatic expression of IL-22R1 was upregulated from chronic-binge-fed mice or from patients with alcoholic hepatitis (Figs. 8A–B) (Fig. 8B)
Fig. 8. Hepatic expression of IL-22 receptors and IL-22 from mice with chronic-binge ethanol (A) or patients with alcoholic hepatitis (B).
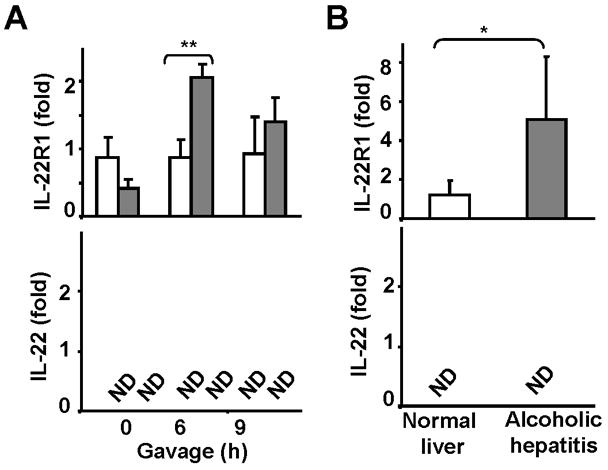
(A) Mice were fed ethanol diet or pair-fed as described in Fig. 1. Liver tissues were collected 6 and 9 h post gavage for real-time PCR analyses. (B) Real-time PCR analyses of liver tissues from normal liver and patients with alcoholic hepatitis. Values represent means ± SD. In panel A (n-5–7), *P<0.05, **P<0.01; In panel B (normal liver n=6; alcoholic hepatitis n=32), *P<0.05, **P<0.01.
Discussion
Two important findings are presented in the current study. First, we developed a murine model of alcoholic liver injury induced by chronic-binge ethanol feeding, which induces significant steatosis and liver injury. Second, using this model, we have demonstrated that treatment with IL-22 ameliorates ethanol-induced liver injury, suggesting therapeutic potential of IL-22 in treating alcoholic liver disease.
Currently the most commonly used model for alcoholic liver injury in rodents is voluntary feeding with the liquid diet containing ethanol; however, this model only induces minor liver injury with slight elevation of serum ALT.17–22 In particular, male mice seem to be more resistant to liver injury induced by voluntary ethanol feeding than female mice. Thus many laboratories used female mice instead of male mice to investigate ethanol-induced hepatocellular damage; however, serum ALT levels were still only slightly elevated in female mice, ranging from 50–116 IU/L.18, 22 In the present study, we demonstrated that chronic-binge ethanol feeding significantly elevated serum ALT and AST levels with the peak levels of about 250 IU/L ALT and 420 IU/L AST in male C57BL/6 mice, which are equivalent to the levels obtained from mice fed intragastrically ethanol diet,29 and are much higher than that from mice fed a voluntary ethanol diet.17–21 Because chronic alcohol feeding increases CYP2E1 protein expression, whose role in alcoholic liver injury has been well documented,30, 31 we speculate that chronic ethanol feeding elevates CYP2E1 protein, which subsequently accelerates binge ethanol-induced hepatocellular damage in this chronic-binge model.
Using this model, we demonstrated that IL-22 treatment reduces ethanol-induced liver injury (Figs. 4–6). IL-22 treatment induces activation of STAT3, whose role in protecting against liver injury has been well documented.32 This leads us to speculate that the hepatoprotection of IL-22 is likely mediated via activation of STAT3 in hepatocytes. In deed, deletion of STAT3 in hepatocytes abolished the hepatoprotective effects of IL-22 (Fig. 6), confirming the critical role of STAT3 in IL-22 protection against alcoholic liver injury. In addition, high concentrations of IL-22 can also activate many other signaling pathways in addition to STAT3, such as STAT1, STAT5, MAPKs, AKT, NF-κB, AP-1 etc.8 Some of these pathways may also attribute to the hepatoprotection of IL-22 in alcoholic liver injury. Moreover, Yang et al recently reported that IL-22 treatment ameliorates obesity-associated fatty liver by downregulating several lipogenesis- and triglyceride synthesis-related genes.12 However, we found that IL-22 treatment significantly downregulates expression of FATP, but not other fat metabolism-associated genes (Fig. 7 and Fig. S3). The discrepancy between these studies may be due to the different models employed. Yang et al. used mice fed a high fat diet for 6 months that had severe hepatic steatosis,12 while we used mice treated with chronic-binge feeding only for 10 days that had mild steatosis. In our model, downregulation of FATP likely contributes to the protective effect of IL-22 on ethanol-induced fatty liver, as inactivation of FATP has been shown to ameliorate high fat diet-induced fatty liver.33 In addition, we have demonstrated that IL-22 treatment elevates expression of MT I/II (Fig. 7), two antioxidant genes that play an important role in protecting against alcoholic liver injury,27 suggesting that induction of MT I/II may contribute to IL-22 hepatoprotection against ethanol-induced hepatocellular damage.
Similar to IL-22, IL-6 also activates STAT3 in hepatocytes and protects against ethanol-induced liver injury.34 However, treatment with IL-6 may generate many side effects such as fever and inflammation etc,35 which is due to the ubiquitous expression of IL-6 receptors and its gp130 signal chain in a wide variety of cell types, and thereby limits its clinical application for treating patients. In contrast, IL-22 may have better therapeutic potential in combination with current therapy of corticosteroids or TNF-α inhibitors in treating alcoholic hepatitis (see discussion below).
Corticosteroids are widely used and TNF-α inhibitors have been tested in treating alcoholic hepatitis, but the results have been controversial.2, 4–7 This is likely because treatments with these 2 drugs have anti-inflammatory effects, which are beneficial for alcoholic hepatitis, but can also inhibit liver regeneration 36, 37 and increase the rate of bacterial infection.4–6 The latter two events are potentially fatal to patients with severe alcoholic hepatitis and are probably responsible for the poor outcomes associated with these treatments.4–6 Findings from this study and previous studies suggest that treatment with IL-22 in combination with corticosteroids or TNF-α inhibitors may have many beneficial effects in treating alcoholic hepatitis. First, IL-22 treatment not only ameliorates steatosis,12 but also protects against alcoholic liver injury (this study) as well as other forms of liver injury;10 Second, since the antimicrobial effect of IL-22 has been well documented,9 application of IL-22 may prevent bacterial infection associated with corticosteroid or TNF-α inhibitor therapy.2, 4–7 Third, IL-22 treatment may overturn corticosteroid- or TNF-α inhibitor-mediated inhibition of liver regeneration, as IL-22 can stimulate hepatocyte proliferation and promote liver regeneration.10, 11 Fourth, IL-22 is not elevated, while expression of IL-22R1 is upregulated in the liver from mice treated with chronic-binge ethanol (Fig. 8). Just like in our animal model, IL-22 is not detected, while expression of IL-22R1 is elevated (5 fold) in the liver of patients with alcoholic hepatitis (Fig. 8). A potential limitation of these clinical samples is that we only include patients with severe alcoholic hepatitis. Further studies should evaluate if the results obtained in these patients are also found in the livers with mild to moderate liver injury. Collectively, these findings suggest that patients with alcoholic hepatitis may be sensitive to IL-22 treatment due to low levels of endogenous IL-22 and elevated levels of IL-22R1 in the liver. Finally, side effects from IL-22 treatment may be minimal as IL-22 receptor expression is restrictedly to epithelial cells such as hepatocytes.8
In summary, IL-22 treatment appears to have multiple beneficial effects on alcoholic hepatitis, such as preventing hepatocelluar damage, promoting hepatocyte proliferation, and inhibiting bacterial infection. Clinical trials examining combination therapy with IL-22 plus corticosteroids or plus TNF-α inhibitor for patients with severe alcoholic hepatitis is warranted.
Supplementary Material
Acknowledgments
This work was supported by the intramural program of NIAAA, NIH.
Abbreviations
- IL-22
interleukin-22
- STAT3
signal transducer and activator of transcription 3
- STAT3Hep−/− mice
Hepatocyte-specific STAT3 knockout mice
- TNF-α
Tumor necrosis factor α
Footnotes
No conflicts of interest exist for all authors.
References
- 1.O’Shea RS, Dasarathy S, McCullough AJ. Alcoholic liver disease. Hepatology. 2010;51:307–328. doi: 10.1002/hep.23258. [DOI] [PubMed] [Google Scholar]
- 2.Lucey MR, Mathurin P, Morgan TR. Alcoholic hepatitis. N Engl J Med. 2009;360:2758–2769. doi: 10.1056/NEJMra0805786. [DOI] [PubMed] [Google Scholar]
- 3.You M, Crabb DW. Recent advances in alcoholic liver disease II. Minireview: molecular mechanisms of alcoholic fatty liver. Am J Physiol Gastrointest Liver Physiol. 2004;287:G1–6. doi: 10.1152/ajpgi.00056.2004. [DOI] [PubMed] [Google Scholar]
- 4.Boetticher NC, Peine CJ, Kwo P, Abrams GA, Patel T, Aqel B, et al. A randomized, double-blinded, placebo-controlled multicenter trial of etanercept in the treatment of alcoholic hepatitis. Gastroenterology. 2008;135:1953–1960. doi: 10.1053/j.gastro.2008.08.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Louvet A, Wartel F, Castel H, Dharancy S, Hollebecque A, Canva-Delcambre V, et al. Infection in patients with severe alcoholic hepatitis treated with steroids: early response to therapy is the key factor. Gastroenterology. 2009;137:541–548. doi: 10.1053/j.gastro.2009.04.062. [DOI] [PubMed] [Google Scholar]
- 6.Naveau S, Chollet-Martin S, Dharancy S, Mathurin P, Jouet P, Piquet MA, et al. A double-blind randomized controlled trial of infliximab associated with prednisolone in acute alcoholic hepatitis. Hepatology. 2004;39:1390–1397. doi: 10.1002/hep.20206. [DOI] [PubMed] [Google Scholar]
- 7.Tilg H, Day CP. Management strategies in alcoholic liver disease. Nat Clin Pract Gastroenterol Hepatol. 2007;4:24–34. doi: 10.1038/ncpgasthep0683. [DOI] [PubMed] [Google Scholar]
- 8.Wolk KWE, Witte K, Warszawska K, Sabat R. Biology of interleukin-22. Semin Immunopathol. 2010 doi: 10.1007/s00281-009-0188-x. in press. [DOI] [PubMed] [Google Scholar]
- 9.Aujla SJ, Kolls JK. IL-22: a critical mediator in mucosal host defense. J Mol Med. 2009;87:451–454. doi: 10.1007/s00109-009-0448-1. [DOI] [PubMed] [Google Scholar]
- 10.Radaeva S, Sun R, Pan HN, Hong F, Gao B. Interleukin 22 (IL-22) plays a protective role in T cell-mediated murine hepatitis: IL-22 is a survival factor for hepatocytes via STAT3 activation. Hepatology. 2004;39:1332–1342. doi: 10.1002/hep.20184. [DOI] [PubMed] [Google Scholar]
- 11.Ren XHB, Colletti LM. IL-22 is involved in liver regeneration after hepatectomy. Am J Physiol Gastrointest Liver Physiol. 2010;298:G74–G80. doi: 10.1152/ajpgi.00075.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yang L, Zhang Y, Wang L, Fan F, Zhu L, Li Z, Ruan X, Huang H, Wang Z, Huang Y, Yan Z, Chen Y. Amelioration of high fat diet induced lipogenesis and hepatic steatosis by interleukin-22. J Hepat. 2010 doi: 10.1016/j.jhep.2010.03.004. in press. [DOI] [PubMed] [Google Scholar]
- 13.de la MHP, Lieber CS, DeCarli LM, French SW, Lindros KO, Jarvelainen H, et al. Models of alcoholic liver disease in rodents: a critical evaluation. Alcohol Clin Exp Res. 2001;25:254S–261S. doi: 10.1097/00000374-200105051-00041. [DOI] [PubMed] [Google Scholar]
- 14.Tsukamoto H, French SW, Benson N, Delgado G, Rao GA, Larkin EC, et al. Severe and progressive steatosis and focal necrosis in rat liver induced by continuous intragastric infusion of ethanol and low fat diet. Hepatology. 1985;5:224–232. doi: 10.1002/hep.1840050212. [DOI] [PubMed] [Google Scholar]
- 15.Nanji AA, French SW. Animal models of alcoholic liver disease--focus on the intragastric feeding model. Alcohol Res Health. 2003;27:325–330. [PMC free article] [PubMed] [Google Scholar]
- 16.Tsukamoto H, Machida K, Dynnyk A, Mkrtchyan H. “Second hit” models of alcoholic liver disease. Semin Liver Dis. 2009;29:178–187. doi: 10.1055/s-0029-1214373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ramaiah S, Rivera C, Arteel G. Early-phase alcoholic liver disease: an update on animal models, pathology, and pathogenesis. Int J Toxicol. 2004;23:217–231. doi: 10.1080/10915810490502069. [DOI] [PubMed] [Google Scholar]
- 18.Roychowdhury S, McMullen MR, Pritchard MT, Hise AG, van Rooijen N, Medof ME, et al. An early complement-dependent and TLR-4-independent phase in the pathogenesis of ethanol-induced liver injury in mice. Hepatology. 2009;49:1326–1334. doi: 10.1002/hep.22776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hritz I, Mandrekar P, Velayudham A, Catalano D, Dolganiuc A, Kodys K, et al. The critical role of toll-like receptor (TLR) 4 in alcoholic liver disease is independent of the common TLR adapter MyD88. Hepatology. 2008;48:1224–1231. doi: 10.1002/hep.22470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kang X, Zhong W, Liu J, Song Z, McClain CJ, Kang YJ, et al. Zinc supplementation reverses alcohol-induced steatosis in mice through reactivating hepatocyte nuclear factor-4alpha and peroxisome proliferator-activated receptor-alpha. Hepatology. 2009;50:1241–1250. doi: 10.1002/hep.23090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shen Z, Liang X, Rogers CQ, Rideout D, You M. Involvement of adiponectin-SIRT1-AMPK signaling in the protective action of rosiglitazone against alcoholic fatty liver in mice. Am J Physiol Gastrointest Liver Physiol. 2010;298:G364–374. doi: 10.1152/ajpgi.00456.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gustot T, Lemmers A, Moreno C, Nagy N, Quertinmont E, Nicaise C, et al. Differential liver sensitization to toll-like receptor pathways in mice with alcoholic fatty liver. Hepatology. 2006;43:989–1000. doi: 10.1002/hep.21138. [DOI] [PubMed] [Google Scholar]
- 23.Tsukamoto H, Mkrtchyan H, Dynnyk A. Intragastric ethanol infusion model in rodents. Methods Mol Biol. 2008;447:33–48. doi: 10.1007/978-1-59745-242-7_3. [DOI] [PubMed] [Google Scholar]
- 24.Horiguchi N, Wang L, Mukhopadhyay P, Park O, Jeong WI, Lafdil F, et al. Cell type-dependent pro- and anti-inflammatory role of signal transducer and activator of transcription 3 in alcoholic liver injury. Gastroenterology. 2008;134:1148–1158. doi: 10.1053/j.gastro.2008.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Colmenero J, Bataller R, Sancho-Bru P, Bellot P, Miquel R, Moreno M, et al. Hepatic expression of candidate genes in patients with alcoholic hepatitis: correlation with disease severity. Gastroenterology. 2007;132:687–697. doi: 10.1053/j.gastro.2006.12.036. [DOI] [PubMed] [Google Scholar]
- 26.Beier JI, Luyendyk JP, Guo L, von Montfort C, Staunton DE, Arteel GE. Fibrin accumulation plays a critical role in the sensitization to lipopolysaccharide-induced liver injury caused by ethanol in mice. Hepatology. 2009;49:1545–1553. doi: 10.1002/hep.22847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhou Z, Sun X, James Kang Y. Metallothionein protection against alcoholic liver injury through inhibition of oxidative stress. Exp Biol Med (Maywood) 2002;227:214–222. doi: 10.1177/153537020222700310. [DOI] [PubMed] [Google Scholar]
- 28.Lemmers A, Moreno C, Gustot T, Marechal R, Degre D, Demetter P, et al. The interleukin-17 pathway is involved in human alcoholic liver disease. Hepatology. 2009;49:646–657. doi: 10.1002/hep.22680. [DOI] [PubMed] [Google Scholar]
- 29.Esfandiari F, Medici V, Wong DH, Jose S, Dolatshahi M, Quinlivan E, et al. Epigenetic regulation of hepatic endoplasmic reticulum stress pathways in the ethanol-fed cystathionine beta synthase-deficient mouse. Hepatology. 2019;51:932–941. doi: 10.1002/hep.23382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Butura A, Nilsson K, Morgan K, Morgan TR, French SW, Johansson I, et al. The impact of CYP2E1 on the development of alcoholic liver disease as studied in a transgenic mouse model. J Hepatol. 2009;50:572–583. doi: 10.1016/j.jhep.2008.10.020. [DOI] [PubMed] [Google Scholar]
- 31.Lu Y, Zhuge J, Wang X, Bai J, Cederbaum AI. Cytochrome P450 2E1 contributes to ethanol-induced fatty liver in mice. Hepatology. 2008;47:1483–1494. doi: 10.1002/hep.22222. [DOI] [PubMed] [Google Scholar]
- 32.Gao B. Cytokines, STATs and liver disease. Cell Mol Immunol. 2005;2:92–100. [PubMed] [Google Scholar]
- 33.Doege H, Grimm D, Falcon A, Tsang B, Storm TA, Xu H, et al. Silencing of hepatic fatty acid transporter protein 5 in vivo reverses diet-induced non-alcoholic fatty liver disease and improves hyperglycemia. J Biol Chem. 2008;283:22186–22192. doi: 10.1074/jbc.M803510200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hong F, Radaeva S, Pan HN, Tian Z, Veech R, Gao B. Interleukin 6 alleviates hepatic steatosis and ischemia/reperfusion injury in mice with fatty liver disease. Hepatology. 2004;40:933–941. doi: 10.1002/hep.20400. [DOI] [PubMed] [Google Scholar]
- 35.Kammuller ME. Recombinant human interleukin-6: safety issues of a pleiotropic growth factor. Toxicology. 1995;105:91–107. doi: 10.1016/0300-483x(95)03128-3. [DOI] [PubMed] [Google Scholar]
- 36.Castellano TJ, Schiffman RL, Jacob MC, Loeb JN. Suppression of liver cell proliferation by glucocorticoid hormone: a comparison of normally growing and regenerating tissue in the immature rat. Endocrinology. 1978;102:1107–1112. doi: 10.1210/endo-102-4-1107. [DOI] [PubMed] [Google Scholar]
- 37.Akerman P, Cote P, Yang SQ, McClain C, Nelson S, Bagby GJ, et al. Antibodies to tumor necrosis factor-alpha inhibit liver regeneration after partial hepatectomy. Am J Physiol. 1992;263:G579–585. doi: 10.1152/ajpgi.1992.263.4.G579. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.