

Abstract
Protein kinase D is a novel family of serine/threonine kinases and diacylglycerol receptors that belongs to the calcium/calmodulin-dependent kinase superfamily. Evidence has established that specific PKD isoforms are dysregulated in several cancer types, and PKD involvement has been documented in a variety of cellular processes important to cancer development, including cell growth, apoptosis, motility, and angiogenesis. In light of this, there has been a recent surge in the development of novel chemical inhibitors of PKD. This review focuses on the potential of PKD as a chemotherapeutic target in cancer treatment and highlights important recent advances in the development of PKD inhibitors.
Keywords: Protein kinase D, cancer, small molecule inhibitors
1. Introduction
PKD constitutes a novel family of serine/threonine kinases and diacylglycerol (DAG) receptors that signal downstream of G protein-coupled receptors (GPCRs) and tyrosine kinase receptors. Now classified as a subfamily of the calcium/calmodulin-dependent kinase (CaMK) superfamily, PKD has emerged as a key regulator of many important cellular processes. Major advances in our understanding of PKD signaling have been made on multiple fronts: (1) basic signaling mechanisms [1, 2], where PKD has been identified as a protein kinase C (PKC) effector [3]; (2) protein trafficking [4–6], in which PKD, a Golgi-resident kinase, has been shown to regulate protein transport from the Golgi to the plasma membrane [7–11], a significant function of PKD that underlies its critical role in multiple secretory processes including the secretion of insulin from pancreatic beta cells [12] and cell motility [13, 14]; (3) immune system function [15], where PKD was found to play an important role in mediating antigen-receptor signaling [15, 16] and in regulating lymphocyte adhesion and motility [17]; (4) oxidative stress response [18, 19], where PKD has been shown to be activated by oxidative stress and to promote cell survival through activation of nuclear factor-kappaB (NFκB) signaling [19–21]; (5) angiogenesis [22, 23] and cardiovascular biology [24, 25], where PKD can mediate vascular endothelial growth factor (VEGF) signaling [26] and promote angiogenesis in part through regulation of class IIa histone deacetylase (HDAC) activity [27–29], which plays a critical role in pathological cardiac remodeling in the heart [30–32]; and (6) drug discovery, where the field culminates with the recent surge of several novel chemical probes that target PKD with remarkable potency (single digit nanomolar), high selectivity, and in vivo activity [33–35] (reviews and major papers in these areas are cited). The basic functions of PKD revealed in these studies are intimately coupled to its role in tumor development, particularly in the regulation of protein trafficking, oxidative stress, angiogenesis, and immune system function. PKD is truly a unique and important family of signaling proteins that has recently drawn increasing attention through efforts to understand its role in cancer.
Three isoforms of PKD have been identified (PKD1/PKCμ [36, 37], PKD2 [38], and PKD3/PKCν [39]), which share a distinct structure that includes a catalytic domain, a pleckstrin homology (PH) domain, and an N-terminal cysteine-rich DAG/phorbol ester binding domain (the C1 domain) [36–39]. High homology between the three isoforms exists, particularly in the catalytic domain and C1 domain, though there are differences in the N-terminal region and in regions flanked by the C1 and PH domains which may confer isoform-specific functions [40]. The PH domain has a negative regulatory effect on PKD catalytic activity, as deletion of this domain leads to constitutive activation of the kinase [41].
The regulatory mechanisms that control PKD activity have been well-documented. Studies have shown that PKD is activated through direct phosphorylation of two conserved serine residues in the activation loop by DAG-responsive PKC isoforms [2, 3, 42]. Subsequent autophosphorylation then confers full, sustained activation [43, 44]. This canonical PKC/PKD activation pathway can be further “tuned” by other factors and pathways. For example, the Src-Abl pathway has been shown to prime PKD for activation by PKCδ through tyrosine phosphorylation in the PH domain [19, 45]. As a DAG target, PKD is also subjected to spatial regulation by DAG or phorbol esters. Binding of DAG to the C1 domain allows PKD to localize to the plasma membrane and trans Golgi network (TGN), mediating site-specific functions [40]. PKD also shuttles between the cytoplasm and the nucleus and can demonstrate transient nuclear accumulation upon activation or through specific signals [46–48]. PKD can be activated in response to a variety of stimuli, including DAG, phorbol esters, growth factors, GPCR agonists, and hormones [26, 49–52].
2. Signaling mechanisms of PKD: relevance to tumor cell biology
Emerging evidence links PKD to a diverse set of signal transduction pathways involved in tumor development and cancer progression (Figure 1). Here, we will discuss the potential roles and signaling mechanisms of PKD in cancer-associated biological responses.
Figure 1.

PKD has been implicated in the regulation of multiple cancer-promoting pathways. PKC-mediated activation of PKD has been shown to regulate such cellular functions as proliferation, apoptosis, angiogenesis, migration, and invasion. Dysregulation of these fundamental pathways can lead to the development, progression, and metastasis of cancer.
2.1. Proliferation, Survival, and Apoptosis
Uncontrolled cell growth and resistance to apoptosis are among the hallmarks of cancer development. Functional studies have described PKD as a potent promoter of cell growth and proliferation in multiple cellular systems, suggesting that PKD may possibly contribute to the cancer phenotype. For example, in Swiss 3T3 cells, overexpression of PKD has been shown to potentiate DNA synthesis in response to bombesin, vasopressin, and phorbol esters [53], and expression of PKD1 has been shown to correlate with levels of proliferating-cell nuclear antigen (PCNA) and proliferation state in mouse keratinocytes [54]. Furthermore, treatment with the PKC/PKD inhibitor Gö6976 inhibits 3H-thymidine incorporation in keratinocytes [55], supporting a role for a PKC/PKD axis in the regulation of keratinocyte proliferation. A similar role for PKD was also demonstrated in prostate and pancreatic cancer [56, 57].
Mechanistically, PKD has been linked to several pathways known to control cell proliferation, most notably the extracellular signal-regulated kinase (ERK) signaling pathway. Inhibition of PKD expression and activity has been shown to attenuate ERK signaling, while overexpression of PKD has been shown to potentiate ERK activity in response to growth factors in multiple cell types including endothelial cells, Swiss 3T3 cells, and prostate cancer cells [26, 56, 58, 59]. It has been proposed that PKD modulates the Ras-Raf-MEK-ERK pathway and promotes proliferation, possibly through direct phosphorylation of the Ras effector protein Ras and Rab interactor 1 (RIN1) [60, 61]. Separate studies have suggested PKD may mediate endothelial cell proliferation and growth through regulation of class IIa HDACs [29].
PKD is also implicated in the regulation of cell survival and apoptosis. Substantial work has shown that PKD promotes survival and inhibits apoptosis through modulation of the NF-κB and c-Jun N-terminal kinase (JNK) pathways [18, 21, 62]. Interestingly, in an opposing role, PKD can promote anoikis, cell death resulting from detachment from the extracellular matrix, by modulating the phosphorylation and subsequent localization of Bcl-2 inhibitor of transcription 1 (Bit1) [63]. Additionally, PKD was found to be cleaved by caspase-3 in response to certain genotoxic agents, a process that generates catalytically active PKD fragments that may play a role in sensitizing cells to the cytotoxic effects of these agents [64, 65]. Furthermore, several PKD substrates including sphingosine kinase 2 (SphK2) and heat-shock protein 27 (Hsp27) have also been connected to the apoptotic response, but the functional relevance of their regulation by PKD in apoptosis remains to be determined [66–68].
2.2. Cell Migration and Invasion
PKD has been implicated in several pathways mediating cell migration, invasion, and motility – processes that are critical to cancer progression and metastasis. Consistent with its important role as a modulator of protein trafficking, PKD1 regulates fibroblast motility and Rac-1-dependent leading edge activity through modulation of anterograde membrane traffic from the TGN to the plasma membrane [14]. Furthermore, PKD directs the transport of αvβ3 integrin to focal adhesions, affecting cell migration [13]. PKD-mediated phosphorylation of several proteins important to cell migration and invasion, including cortactin [69], Par-1 [70], and slingshot 1 like (SSH1L) [71], has also been documented. SSH1L is a phosphatase whose activity causes reactivation of the actin-remodeling protein cofilin at the leading edge of migrating cells [72]. Pioneering work by the Storz group showed that phosphorylation of SSH1L by PKD1 leads to its dissociation from the actin cytoskeleton and subsequent inhibition of cell motility through sustained cofilin inactivation [71]. Additional studies have shown that PKD and its substrates are involved in the regulation of matrix metalloproteinase (MMP) expression [28, 73, 74]. MMPs are matrix-degrading enzymes that control cell migration and invasion and have been linked to the progression of many types of cancer [75].
2.3. Angiogenesis
Angiogenesis is the process through which new blood vessels are formed. In tumor development, angiogenesis is required to provide growing primary and secondary tumors with oxygen and nutrients [76]. A study in 2005 by Wong and Jin showed that PKD was activated by VEGF downstream of PKCα and regulated endothelial cell proliferation [26]. Further studies showed that VEGF-induced endothelial cell proliferation, migration, and in vivo angiogenesis all required PKD activity [77]. Subsequently, researchers from two separate groups reported that VEGF-stimulated activation of PKD leads to PKD-dependent phosphorylation of HDAC7 and increased migration in endothelial cells [28, 29]. This pathway was also shown to promote several additional processes required for angiogenesis including proliferation [29], tube formation, and microvessel sprouting [28]. These studies stemmed from the seminal work by Vega et al. that first identified class IIa HDACs as direct substrates of PKD [30]. Class IIa HDACs, including HDAC4, -5, -7, and -9, catalyze the deacetylation of lysine residues in histone amino-terminal tails and, as part of large, multiprotein transcriptional co-repressor complexes, regulate the activity of transcription factors such as myocyte enhancer factor-2 (MEF2) [78]. Direct phosphorylation of class IIa HDACs by PKD leads to nuclear exclusion of these proteins and subsequent expression of MEF2 target genes, including the angiogenesis-promoting MMPs [28, 29].
3. PKD in Cancer: Expression, Activity and Localization
Emerging as a major player in cell proliferation, survival, motility and angiogenesis pathways, it is no surprise that PKD has recently received considerable attention as a potential target in the treatment of cancer. Studies assessing the expression levels of all three isoforms have shown that indeed, PKD is dysregulated in several cancer types (Table 1).
Table 1.
Dysregulation of PKD isoforms in human tumor samples determined by immunohistochemistry analysis
| PKD Isoform | Tumor Type | Expression | Correlation/Notes | Refs. |
|---|---|---|---|---|
| PKD1 | Breast | ↓ | Reduced PKD1 in invasive tumors | [73] |
| Pancreatic | ↑ | Increased PKD1 and activated PKD1/2 in tumors | [57, 97] | |
| Prostate | ↑ ↓ | Increased PKD1 in tumors Reduced PKD1 in androgen-independent tumors |
[56, 105] | |
| Basal Cell Carcinoma | ↑ | Elevated and misdistributed PKD1 in the hyperproliferative human skin disorders, BCC and psoriasis | [114] | |
| Gastric | ↓ | Decreased PKD1 in tumors, correlated with increased PKD1 promoter methylation | [115] | |
| PKD2 | Breast | no change | - | [73] |
| Lymphoma | ↑ ↓ | PKD2 expression pattern in different lymphomas correspond to its expression in normal counterparts | [118] | |
| PKD3 | Breast | no change | - | [73] |
| Prostate | ↑ | Increased PKD3 in tumors Increased PKD3 nuclear accumulation, correlated with pathological grade |
[56] |
3.1. Breast Cancer
PKD enzymes were first studied in the context of breast cancer over a decade ago [79]. Despite this early start, the mechanisms through which PKD may contribute to breast cancer progression are not yet clear. Analysis of invasive human breast tumors has revealed that PKD1 expression is downregulated in infiltrating ductal carcinoma compared to normal breast tissue [73]. In this study, no differences were observed in PKD2 or PKD3 expression when comparing normal versus malignant tissue, suggesting that decreased expression or activity of PKD1, but not that of PKD2 or PKD3, may be important for the progression of breast cancer. Supporting these results, several transcriptional microarray studies of gene expression in normal breast tissue and of early and advanced stage breast tumors have shown reduction of the PRKD1 (PKD1) gene correlating with increased invasiveness and cancer progression [80–84]. In cellular models, similar expression patterns have been described. For example, the highly invasive cell lines SKBR3, T47D, and MDA-MB-231 have been shown to express little or no PKD1, while normal breast cell lines and very low-invasive breast cancer cell lines such as MCF-7 and BT-474 have been shown to express PKD1 at moderate levels, though considerable variation exists in the reported literature when different antibodies for PKD1 are utilized [73, 79]. Interestingly, the low level of PKD1 expression in highly invasive lines was due to epigenetic silencing by DNA methylation [73].
Functionally, studies investigating the role of PKD in breast cancer progression have focused on the processes of invasion and adhesion. As early as 1999, Mueller and colleagues described an interaction between PKD1, paxillin, and cortactin at sites of invadopodia in MDA-MB-231 breast cancer cells [79]. Invadopodia are actin-containing protrusions that extend outward into the extracellular matrix (ECM) and participate in degradation of the ECM [85]. This interaction, present in invasive breast cancer cells but not in non-invasive lines, suggested that PKD1 may regulate the function or formation of the paxillin/cortactin complex to promote invasion. Furthermore, cortactin was recently determined to be a PKD1 substrate [69], though it remains to be determined whether this phosphorylation event and the PKD1/cortactin/paxillin association does indeed promote invasion. Additionally, PKD1 also phosphorylates EVL-1, a splice variant of Ena/VASP like protein [86]. Ena/VASP proteins negatively regulate cell mobility through rearrangement of the actin cytoskeleton and are upregulated in estrogen-responsive breast cancer [87, 88]. While the exact function is not yet completely clear, the data suggests that impairment of PKD1-mediated EVL-1 phosphorylation may promote lamellipodia ruffling. Though membrane ruffling is often associated with invasiveness, it has been shown that increased local activity of Ena/VASP proteins at the leading edge leads to the growth of elongated, less-branched actin filaments which promote the formation of ruffles and results in an overall reduction in cell mobility [86, 89–91]. Future studies are needed to fully elucidate the role of PKD in these events.
Multiple separate studies have strongly supported an opposing role for PKD in breast cancer cell invasion and adhesion. Innovative studies by Storz and colleagues have shown that invasion in 2D and 3D environments was reduced with expression of a constitutively active PKD1 mutant in MDA-MB-231 cells [73]. Moreover, researchers have shown that PKD1 translocates to the plasma membrane and promotes adhesion in MDA-MB-435 cells in response to fatty acid treatment [92, 93]. Based on mechanistic argumentation, it has been suggested that the regulation of adhesion and invasion by PKD1 may be related to MMP expression. Activated PKD1 caused reduced expression of MMP-2, MMP-7, MMP-9, MMP-10, MMP-11, MMP-13, MMP-14, and MMP-15, while upregulating expression of MMP-3 [73]. As described earlier, MMPs are endopeptidases involved in the degradation of the ECM, a process that is required for the development of malignant tumors. In breast cancer, MMP-2 in particular has been identified as an indicator of potential malignancy [94]. Thus, it is possible that PKD1 may inhibit breast cancer invasion and metastasis through regulation of MMP expression. Another critical target through which PKD1 exerts its inhibitory effect on cell migration is SSH1L, which was first shown in MTLn3 rat breast carcinoma cells [71]. Later, a separate study by Hausser and colleagues showed that both PKD1 and PKD2 negatively regulate cell migration through direct phosphorylation of SSH1L in MCF-7 and MDA-MB-231 breast cancer cells [95]. Collectively, these studies suggest a potential significant role for PKD in the suppression of breast cancer metastasis.
3.2. Pancreatic Cancer
Ductal adenocarcinoma of the pancreas is a devastating disease with a very low 5-year survival rate (5%) [96]. Studies in pancreatic cancer cells are some of the earliest works indicating a potential role for PKD in cancer. In human ductal adenocarcinoma of the pancreas, studies showed PKD1 to be overexpressed compared to normal pancreatic tissues [57]. Furthermore, recent studies have shown an increase in expression of activated PKD1/2 in pancreatic cancer tissue microarrays [97]. No studies of PKD2 or PKD3 expression in human tumors have been published, though PKD2 expression has been demonstrated in PANC-1 cells [98].
In pancreatic cancer cell lines, studies on the function of PKD have revealed a role in cell proliferation and apoptosis. Trauzold et al. demonstrated that PKD1 overexpression in Colo357 cells led to: 1) increased viability following treatment with the anti-CD95 antibody CH11, 2) increased cell proliferation rate, and 3) upregulation of survivin and FLICE-inhibitory protein (c-FLIPL) [57]. In PANC-1 cells, PKD stimulated DNA synthesis and mitogenic signaling and was activated by neurotensin (NT), a mitogenic neuropeptide that has been implicated in the autocrine/paracrine growth stimulation of human pancreatic cancer [52]. A separate study showed that PKD1 and PKD2 phosphorylated Hsp27 in response to NT in PANC-1 cells [98]. Hsp27 is an important protein and molecular chaperone linked to a variety of cellular processes including protein folding, cytoskeleton rearrangement, cell mobility, cell survival, and proliferation, and its expression is increased in many types of cancer [99–102]. Further validation of the role of PKD in pancreatic cancer was recently revealed in significant xenograft studies. Researchers showed that treatment with the novel PKD inhibitor CRT0066101 reduced tumor growth in both subcutaneous PANC-1 tumors and in orthotopically implanted PANC-1 cells [97]. Taken together, these studies reveal the pro-survival and pro-proliferation functions of PKD1 in pancreatic cancer cells, and implicate PKD as a potential therapeutic target in pancreatic cancer treatment.
3.3. Prostate Cancer
Prostate cancer was the second leading cause of death from cancer among men in the U.S. in 2009 [103]. Despite recent advances in early diagnosis and screening procedures, there are currently no effective therapeutic treatments once tumors have metastasized [104]. We still have an incomplete understanding of the molecular mechanisms that cause prostate cancer to become invasive.
Recently, evidence for the role of PKD in the progression of prostate cancer has been revealed. Studies have shown that PKD1 and PKD3 expression levels are elevated in human prostate carcinoma tissues compared to normal prostate epithelial tissue, and advanced-stage tumors were found to have increased PKD3 nuclear accumulation [56]. In contrast, androgen-independent tumors showed reduced PKD1 expression [105]. Common prostate cancer cell lines also display differential expression of the PKD isoforms. The LNCaP cell line, an androgen-sensitive and less metastatic cell line, expresses PKD1 and PKD2 only, while DU145 and PC3 cells, both androgen-insensitive and highly metastatic prostate tumor cells, express primarily PKD3, with moderate expression of PKD2 and no detectable PKD1 [56]. The differential expression and distribution of PKD in these cell lines and in tumor samples suggests PKD might be important for the progression of prostate cancer.
Functional analysis of PKD has revealed distinct roles for the PKD isoforms in prostate cancer. Balaji and colleagues [106] demonstrated a tumor suppressor-like function for PKD1 in prostate cancer. PKD1 was found to negatively regulate androgen receptor (AR) function and prostate cancer cell migration and proliferation. Specifically, expression of PKD1 in DU145 cells reduced cell proliferation, while co-expression of kinase-dead PKD1 with AR attenuated the AR-mediated increase in cell-proliferation [106]. Furthermore, expression of either PKD1 or the kinase-dead PKD1 mutant reduced AR-mediated transcription. Additional studies have suggested that repression of AR-dependent transcription by PKD1 may be mediated through Hsp27 [68]. They have also demonstrated that overexpression of PKD1 may promote cell aggregation, inhibit migration, and reduce cell proliferation through phosphorylation of E-cadherin and regulation of β-catenin activity, linking PKD1 to the Wnt signaling pathway [107–110]. These studies are suggestive of a possible role for PKD1 in suppressing the development of the androgen-independent state, common to more advanced, aggressive prostate cancer.
In contrast, separate studies conducted in our laboratory have demonstrated a positive role for the PKD3 isoform in prostate cancer cell proliferation and survival [33, 56]. In PC3 cells, reduction of PKD3 levels using siRNA caused potent inhibition of cell proliferation. Analysis of a potential mechanism revealed that PKD3 activity stimulated prolonged activation of Akt and ERK1/2. This regulation of Akt and ERK1/2 may account for the effects on proliferation, and also may affect other steps in prostate cancer progression. Akt, commonly found to be hyperactive in prostate cancer due to a phosphatase and tensin homolog (PTEN)-null phenotype, has been implicated in angiogenesis and metastasis in addition to its fundamental roles in survival and proliferation [111, 112]. We also found that inhibition of PKD activity using a novel chemical inhibitor of PKD not only reduced proliferation in LNCaP, DU145, and PC3 prostate cancer cells, but also significantly slowed migration and invasion of PC3 and DU145 cells [33].
These studies not only highlight the significance of PKD signaling in prostate cancer progression, but also strongly suggest isoform-specific functions and contrasting roles for PKD1 and PKD3 in prostate cancer cells. Further studies are needed to determine whether indeed there are distinct and opposing roles for the PKD isoforms in prostate cancer and what implications this might have in the development of PKD inhibitors for potential use as chemotherapeutic agents in the treatment of prostate cancer.
3.4. Basal Cell Carcinoma and Skin Cancer
Basal cell carcinoma (BCC) is an extremely common form of malignant skin cancer [113]. Analysis of BCC lesions showed increased expression of PKD1 when compared with normal epidermis [114]. Furthermore, PKD1 expression in normal epidermis was primarily restricted to the stratum basalis, the proliferative compartment of the epidermis, supporting the concept that PKD promotes hyperproliferative disorders of the skin [54, 55]. Additional studies are necessary to confirm whether PKD plays a significant role in the development of BCC or other hyperproliferative skin conditions.
3.5. Gastric Cancer
Similar to the studies in breast cancer, analysis of PKD1 levels in primary gastric tumors and gastric cancer cell lines has revealed decreased protein expression due to hypermethylation of the PKD1 promoter region [115]. Interestingly, the authors demonstrated a gradual increase in methylation of the PKD1 promoter in aging, but normal, gastric mucosal tissues. Since the incidence of gastric cancers is positively correlated with age [116], it is possible that PKD1 silencing may occur early on in the development of gastric cancer or, at the very least, predispose individuals to gastric cancer. Moreover, the lack of PKD1 may promote the development of metastases, since knockdown of PKD1 using siRNA led to increased migration and invasion of SNU-484 and SNU-668 gastric cancer cells.
The epigenetic silencing of PKD1 in invasive gastric and breast cancer cell lines and tumors suggests it could potentially act as a tumor suppressor at certain stages of tumor development. Considering the large body of evidence suggesting that PKD1 positively regulates cell survival and proliferation, it is possible that the expression and silencing of PKD1 are concerted events occurring at different stages of tumor development. In this scenario, the presence of PKD1 may be required for early tumor promoting events such as enhanced survival and proliferation, while silencing of PKD1 may be necessary to allow tumor progression into invasive stages. Nonetheless, this concept might not apply to gastric cancer, since PKD1 silencing is most likely an early event in gastric carcinogenesis. Analysis of PKD1 expression and activity in different stages of tumors will provide better support for this concept.
3.6. Lung Cancer and Lymphoma
Very little is known about the role of PKD in small cell lung cancer (SCLC). One study has shown that the PKC-PKD signaling pathway exists in the SCLC cell lines H69, H345, and H510, and that PKD can be activated by phorbol esters and bombesin in these cells [117]. To date, there have been no reports on PKD expression and distribution in actual human tumor samples.
Immunohistochemistry analysis of multiple types of human malignant lymphoma have revealed varying expression of PKD2 protein and activity [118]. No correlation between aggressiveness of the lymphoma and PKD2 expression was observed, and the authors suggest that PKD2 expression often was very similar to the normal lymph tissue from which the particular tumors were derived. This study suggests that while PKD2 is expressed in many types of lymphoma, it may not be involved in progression of the disease; however, more studies are required to support these conclusions.
4. Chemical Inhibitors of PKD: Old and New
Novel biotechnologies such as RNAi-mediated knockdown of endogenous proteins now allow the study protein function through direct targeting of specific proteins. The limitations of these approaches, however, are that they deplete the entire protein and they are extremely difficult to apply in vivo. On the other hand, the development of chemical probes with high potency and selectivity provides the opportunity to directly and reversibly target the enzyme activity of PKD in cells and animals. Moreover, one can titrate the level of inhibition. In the past, PKD inhibition was achieved by various general kinase inhibitors or through compounds that were primarily developed as PKC inhibitors. Recently however, novel chemical inhibitors targeting PKD in various disease states including cancer have emerged, with several new compounds being reported in the past year (Table 2).
Table 2.
In vitro IC50 values for chemical inhibitors of PKD1
In earlier studies, several compounds capable of also inhibiting PKD among a range of other kinases were reported. The pan-kinase inhibitor staurosporine and staurosporine-related compounds such as K252a have been reported to inhibit PKD in the low nanomolar range (Figure 2). However, these compounds lack the specificity necessary to interrogate PKD in cells [119]. The indolocarbazole Gö6976 inhibits PKD with an IC50 around 20 nM and is commonly used to inhibit PKD in various cellular contexts. However, this inhibitor is foremost a classical PKC inhibitor with single digit nanomolar IC50s [8, 119]. Other compounds reported to inhibit PKD include the antioxidant and chemopreventive agent resveratrol. Resveratrol inhibits many proteins in addition to PKD, and high micromolar concentrations are required for inhibition of PKD [25, 120, 121]. Despite these limitations, resveratrol has been extensively studied as a potential chemotherapeutic and chemopreventive agent, showing potent antitumor activity in multiple animal models, and it is possible that a small part of its effectiveness is indeed due to inhibition of PKD activity [121]. In contrast to the abovementioned agents, suramin, a hexasulfonated naphthylurea, has been shown to be a novel and effective activator of PKD. Although suramin is a valuable tool for discriminating the activities of PKC isozymes, it too is rather unselective, thereby limiting its effectiveness as a chemical probe for the study of PKD in a relevant biological environment [122].
Figure 2.

Staurosporin, Gö6976, K252a, and resveratrol inhibit PKD, but are rather unselective. Suramin, a PKD activator, is a useful tool for differentiating PKC isoforms, but its application is similarly hampered by a lack of selectivity.
In 2008, we reported the identification and characterization of CID755673, a benzoxoloazepinolone that inhibited all three isoforms of PKD with an IC50 of around 200 nM (Figure 3) [33]. CID755673 showed some specificity toward PKD over several related kinases, and it did not inhibit the in vitro activity of PKCα, PKCβ, PKCδ, or CaMKIIα. Importantly, CID755673 retained cellular activity, albeit with a lower potency, inhibiting PKD1 autophosphorylation with an apparent IC50 of around 10 μM. Cellular studies also revealed that CID755673 reduced cell proliferation, migration, and invasion in prostate cancer cells, highlighting the potential significance of the novel chemical structure in future drug development. A recent study suggested that this compound might have some unintended PKD-independent effects [123]. Specifically, CID755673 was found to enhance PDBu- and growth factor-stimulated DNA synthesis and G1/S cell cycle transition independent of PKD1 in Swiss 3T3 cells, which correlated to a concentration-dependent increase in cyclin D1 and cyclin D3 levels in prostate cancer cells, as shown in our latest report [123, 124]. However, this unintended effect did not alter the ultimate cell fate, i.e. cell growth arrest and subsequent cell death. These new findings suggest that this class of compounds may target additional proteins, resulting in complex effects on cell cycle progression. More encouragingly, however, is the activity profile of new analogs of CID755673 which show reduced off-target effects and markedly increased potency in tumor cells (Figure 4) [124]. A novel lead compound, kb-NB142-70, inhibits PKD1 with an IC50 of 28 nM in vitro and shows significantly increased cellular activity, inhibiting PKD1 autophosphorylation at Ser916 with an IC50 of around 2 μM in LNCaP cells. All of these new analogs show increased potency with respect to inhibition of prostate cancer cell proliferation, migration, and invasion when compared to the parental compound, CID755673, demonstrating the potential value for future development of this series of novel PKD inhibitors in anticancer therapy.
Figure 3.
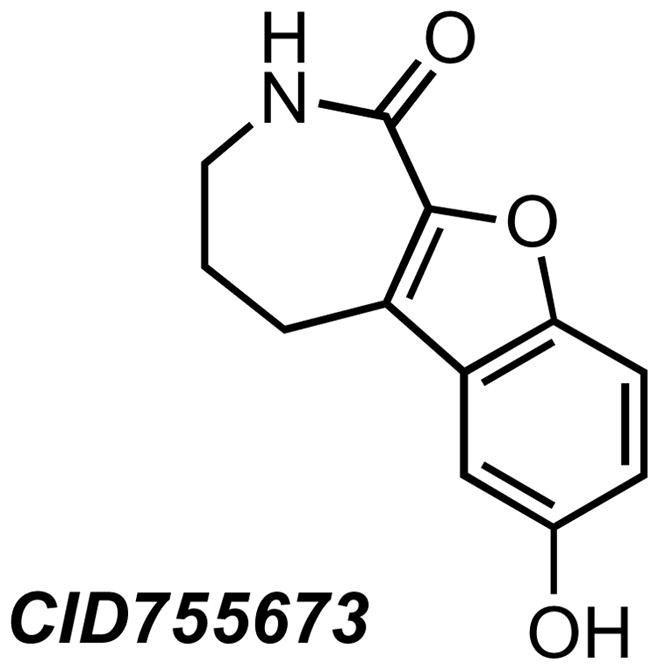
The selective PKD inhibitor CID755673. The inhibitory effect of this compound is most likely mediated through an allosteric effect.
Figure 4.

Analogs of CID755673 show increased potency toward PKD inhibition and some increased selectivity.
In addition to CID755673 and its analogs, an interesting bipyridyl, BPKDi (2′-(cyclohexylamino)-6-(piperazin-1-yl)-[2,4′-bipyridine]-4-carboxamide), was recently identified as a very potent inhibitor of PKD1, PKD2, and PKD3 (IC50 of 1–9 nM) in a >650,000 compound high-throughput screen (Figure 5) [34]. BPKDi had very little or no activity toward CaMKIδ, CaMKIIα, CaMKIIβ, CaMKIIδ, CaMKIV, MARK1, MARK2, SIK1, GRK5, PKCδ, or PKCε. The compound caused substantial inhibition of PKD1 autophosphorylation at 1 μM concentration. Furthermore, treatment of cardiac myocytes with BPKDi resulted in reduced phosphorylation and increased nuclear retention of HDAC4 and HDAC5, validating these proteins as PKD substrates.
Figure 5.
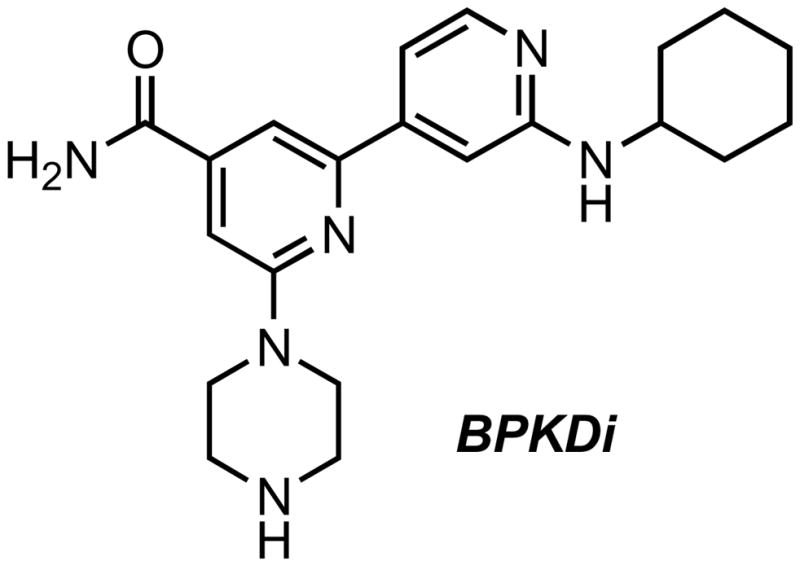
BPKDi, a potent and selective active site inhibitor of PKD.
It is important to note that both of the parental PKD inhibitory chemotypes (CID755673 and BPKDi) were identified using PKD1 biochemical high throughput screens [33, 34] and display inhibitory activity against all three PKD isoforms. The individual screening strategies (i.e., PKD isoform and substrate pairing) used to identify these small molecule inhibitors may have directly impacted the specificity (or lack thereof) of the identified chemotypes. A more advantageous strategy may focus on a particular PKD-isoform paired with its isoform-specific substrate. As our understanding of the roles of each PKD isoform expands, and with the emergence of isoform-specific substrates, this strategy may prove to be more advantageous.
Additional inhibitors continue to emerge in the literature. One class of inhibitors includes the amino-ethyl-amino-aryl compounds 1, 2, and 3 (Figure 6) [125]. Compound 1 was shown to inhibit PDBu-stimulated PKD1 Ser916 phosphorylation in PANC-1 cells with an IC50 of 4 μM, while compounds 2 and 3 showed inhibition of PKD1 in the low and sub-nanomolar range, respectively. Additionally, compound 3 was reported to inhibit PKD2 with an IC50 of 4.1 nM. Furthermore, the pyrazine and pyridine benzamides 5 and 6 were reported to be inhibitors of PKD1 with low nanomolar IC50 values. Benzamides 4 and 6 were similarly potent, exhibiting IC50 values of 420 nM and 32 nM, respectively, against PKD1 [126]. Compounds containing the pyrimidinediamine scaffold, such as compound 7, also demonstrated potent inhibition of PKD in the low nanomolar range [127].
Figure 6.

Recent active site PKD inhibitors reported in the research and patent literature.
During the review of this manuscript, another pan-PKD inhibitor, CRT0066101 (Figure 6, compound 8), was reported [97]. CRT0066101 inhibits PKD1, PKD2, and PKD3 with in vitro IC50s of 1, 2.5, and 2 nM, respectively, and has a cellular IC50 of 0.5 μM for PKD1 in intact PANC-1 cells. The structure of CRT0066101 appears to be derived from the core structure of compounds 1, 2, and 3 (Figure 6). The interesting scaffold 8 was reported to inhibit cell proliferation, induce apoptosis, and reduce viability of pancreatic cancer cells. Furthermore, treatment with CRT0066101 was shown to affect levels of PKD biomarkers including phosphorylated Hsp27, activated NFκB, and expression of NFκB-regulated genes. The most attractive features of this inhibitor are that it is orally available and efficacious in vivo. CRT0066101 given orally at 80 mg/kg/d significantly suppressed the growth of s.c. (qd for 24 days) and orthotopic (qd for 21 days) pancreatic tumor xenografts in mice. This study provides strong support for targeting PKD in pancreatic cancer therapy [97], and the efficacy of this compound in other tumor types awaits further evaluation. Taken together, efforts at developing PKD-selective inhibitors have so far consistently demonstrated PKD as an effective target for cancer therapy.
A significant obstacle in the development of novel PKD inhibitors with high potency and selectivity is the current lack of information regarding the three-dimensional (3D) structure of PKD, preventing the use of structure-based drug design as a strategy for future development of PKD chemical probes. Structure-based drug design is a powerful approach that has been widely used to develop targeted therapies against many cancer targets, including BCR-ABL, phosphoinositide 3-kinase (PI3K), Hsp90, p53, and epidermal growth factor receptor (EGFR) [128]. The availability of structural information on PKD would greatly accelerate the optimization of current lead compounds and would potentially facilitate the targeting of specific interactions between PKD and its substrates or binding partners. Furthermore, it might provide significant insight into structural differences between the PKD isoforms that could be exploited in the development of isoform-selective inhibitors.
5. PKD as a Chemotherapeutic Target?
Extensive evidence indicates that PKD expression is deregulated in multiple cancer types and plays an active role in a variety of cancer-associated biological processes including proliferation, survival, apoptosis, migration, invasion, and angiogenesis, making PKD an attractive target for drug development. However, the prominent lack of animal models to support the potential role of PKD in cancer remains a significant gap in knowledge in this field. To date, there have been no reports of genetic PKD animal models related to cancer. However, despite this, pharmacological inhibition of PKD has now shown to be effective at suppressing growth of pancreatic tumor xenografts [35], which greatly enhances the validity of PKD as a chemotherapeutic target.
A wide range of studies have characterized PKD as being a promoter of cell proliferation and survival. It is important to note that PKD may have cell-specific and isoform-specific functions, especially with regards to motility, and data suggests that the PKD1 isoform may even act as a tumor suppressor in certain stages of tumor development. To date, no PKD inhibitors reported exhibit isoform-selectivity. In prostate cancer, where reports have indicated potential opposing roles for PKD1 and PKD3 [33, 56, 105, 110], isoform selectivity may prove to be a limiting factor in treatment efficacy.
6. Concluding Remarks
A rapidly growing body of evidence demonstrates that PKD is significant to cancer progression. The apparent opposing roles of PKD in several cancer types and potential isoform-specific functions of members of this kinase family leave many questions as of yet unanswered: Are there significant isoform-specific differences in function that may limit the use of pan-PKD inhibitors to treat certain types of cancer? Is it possible to develop active site drugs that exhibit exquisite isoform selectivity? Will allosteric inhibitors provide greater isoform selectivity? What would be the optimal drug discovery strategy to identify potential PKD isoform specific chemotypes? However, despite these unanswered questions, exciting new discoveries have validated PKD as a potential chemotherapeutic target in pancreatic cancer, and the recent outstanding developments in the chemical inhibition of PKD may prove to provide a new therapeutic paradigm in the treatment of cancers.
Acknowledgments
This study was supported in part by the National Institutes of Health [Grants R03 MH082038-01, R01 DK066168, R01CA129127-01 and R01CA142580-01], the NIH Roadmap Program [Grants 1U54MH074411, GM067082], and the Elsa Pardee Foundation.
Abbreviations
- PKD
protein kinase D
- DAG
diacylglycerol
- CaMK
calcium/calmodulin-dependent kinase
- PH
pleckstrin homology
- PKC
protein kinase C
- GPCR
G protein-coupled receptor
- ERK
extracellular signal-regulated kinase
- HDAC
histone deacetylase
- SphK2
sphingosine kinase 2
- Hsp
heat-shock protein
- SSH1L
slingshot 1 like
- MMP
matrix-metalloproteinase
- VEGF
vascular endothelial growth factor
- MEF2
myocyte enhancer factor-2
- ECM
extracellular matrix
- AR
androgen receptor
- BCC
basal cell carcinoma
- SCLC
small cell lung cancer
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Rozengurt E, Rey O, Waldron RT. Protein kinase D signaling. J Biol Chem. 2005;280:13205–13208. doi: 10.1074/jbc.R500002200. [DOI] [PubMed] [Google Scholar]
- 2.Waldron RT, Iglesias T, Rozengurt E. Phosphorylation-dependent protein kinase D activation. Electrophoresis. 1999;20:382–390. doi: 10.1002/(SICI)1522-2683(19990201)20:2<382::AID-ELPS382>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 3.Zugaza JL, Sinnett-Smith J, Van Lint J, Rozengurt E. Protein kinase D (PKD) activation in intact cells through a protein kinase C-dependent signal transduction pathway. Embo J. 1996;15:6220–6230. [PMC free article] [PubMed] [Google Scholar]
- 4.Bankaitis VA. Cell biology. Slick recruitment to the Golgi. Science. 2002;295:290–291. doi: 10.1126/science.1068446. [DOI] [PubMed] [Google Scholar]
- 5.Cuenda A, Nebreda AR. p38delta and PKD1: kinase switches for insulin secretion. Cell. 2009;136:209–210. doi: 10.1016/j.cell.2009.01.005. [DOI] [PubMed] [Google Scholar]
- 6.Ghanekar Y, Lowe M. Protein kinase D: activation for Golgi carrier formation. Trends Cell Biol. 2005;15:511–514. doi: 10.1016/j.tcb.2005.08.001. [DOI] [PubMed] [Google Scholar]
- 7.Jamora C, Yamanouye N, Van Lint J, Laudenslager J, Vandenheede JR, Faulkner DJ, Malhotra V. Gbetagamma-mediated regulation of Golgi organization is through the direct activation of protein kinase D. Cell. 1999;98:59–68. doi: 10.1016/S0092-8674(00)80606-6. [DOI] [PubMed] [Google Scholar]
- 8.Martiny-Baron G, Kazanietz MG, Mischak H, Blumberg PM, Kochs G, Hug H, Marme D, Schachtele C. Selective inhibition of protein kinase C isozymes by the indolocarbazole Go 6976. J Biol Chem. 1993;268:9194–9197. [PubMed] [Google Scholar]
- 9.Liljedahl M, Maeda Y, Colanzi A, Ayala I, Van Lint J, Malhotra V. Protein kinase D regulates the fission of cell surface destined transport carriers from the trans-Golgi network. Cell. 2001;104:409–420. doi: 10.1016/s0092-8674(01)00228-8. [DOI] [PubMed] [Google Scholar]
- 10.Yeaman C, Ayala MI, Wright JR, Bard F, Bossard C, Ang A, Maeda Y, Seufferlein T, Mellman I, Nelson WJ, Malhotra V. Protein kinase D regulates basolateral membrane protein exit from trans-Golgi network. Nat Cell Biol. 2004;6:106–112. doi: 10.1038/ncb1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bossard C, Bresson D, Polishchuk RS, Malhotra V. Dimeric PKD regulates membrane fission to form transport carriers at the TGN. J Cell Biol. 2007;179:1123–1131. doi: 10.1083/jcb.200703166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sumara G, Formentini I, Collins S, Sumara I, Windak R, Bodenmiller B, Ramracheya R, Caille D, Jiang H, Platt KA, Meda P, Aebersold R, Rorsman P, Ricci R. Regulation of PKD by the MAPK p38delta in insulin secretion and glucose homeostasis. Cell. 2009;136:235–248. doi: 10.1016/j.cell.2008.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Woods AJ, White DP, Caswell PT, Norman JC. PKD1/PKCmu promotes alphavbeta3 integrin recycling and delivery to nascent focal adhesions. Embo J. 2004;23:2531–2543. doi: 10.1038/sj.emboj.7600267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Prigozhina NL, Waterman-Storer CM. Protein kinase D-mediated anterograde membrane trafficking is required for fibroblast motility. Curr Biol. 2004;14:88–98. doi: 10.1016/j.cub.2004.01.003. [DOI] [PubMed] [Google Scholar]
- 15.Matthews SA, Liu P, Spitaler M, Olson EN, McKinsey TA, Cantrell DA, Scharenberg AM. Essential role for protein kinase D family kinases in the regulation of class II histone deacetylases in B lymphocytes. Mol Cell Biol. 2006;26:1569–1577. doi: 10.1128/MCB.26.4.1569-1577.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Marklund U, Lightfoot K, Cantrell D. Intracellular location and cell context-dependent function of protein kinase D. Immunity. 2003;19:491–501. doi: 10.1016/s1074-7613(03)00260-7. [DOI] [PubMed] [Google Scholar]
- 17.Medeiros RB, Dickey DM, Chung H, Quale AC, Nagarajan LR, Billadeau DD, Shimizu Y. Protein kinase D1 and the beta 1 integrin cytoplasmic domain control beta 1 integrin function via regulation of Rap1 activation. Immunity. 2005;23:213–226. doi: 10.1016/j.immuni.2005.07.006. [DOI] [PubMed] [Google Scholar]
- 18.Storz P. Mitochondrial ROS--radical detoxification, mediated by protein kinase D. Trends Cell Biol. 2007;17:13–18. doi: 10.1016/j.tcb.2006.11.003. [DOI] [PubMed] [Google Scholar]
- 19.Storz P, Doppler H, Johannes FJ, Toker A. Tyrosine phosphorylation of protein kinase D in the pleckstrin homology domain leads to activation. J Biol Chem. 2003;278:17969–17976. doi: 10.1074/jbc.M213224200. [DOI] [PubMed] [Google Scholar]
- 20.Waldron RT, Rozengurt E. Oxidative stress induces protein kinase D activation in intact cells. Involvement of Src and dependence on protein kinase C. J Biol Chem. 2000;275:17114–17121. doi: 10.1074/jbc.M908959199. [DOI] [PubMed] [Google Scholar]
- 21.Storz P, Doppler H, Toker A. Protein kinase D mediates mitochondrion-to-nucleus signaling and detoxification from mitochondrial reactive oxygen species. Mol Cell Biol. 2005;25:8520–8530. doi: 10.1128/MCB.25.19.8520-8530.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Altschmied J, Haendeler J. A new kid on the block: PKD1: a promising target for antiangiogenic therapy? Arterioscler Thromb Vasc Biol. 2008;28:1689–1690. doi: 10.1161/ATVBAHA.108.174250. [DOI] [PubMed] [Google Scholar]
- 23.Ha CH, Jin ZG. Protein kinase D1, a new molecular player in VEGF signaling and angiogenesis. Mol Cells. 2009;28:1–5. doi: 10.1007/s10059-009-0109-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McKinsey TA. Derepression of pathological cardiac genes by members of the CaM kinase superfamily. Cardiovasc Res. 2007;73:667–677. doi: 10.1016/j.cardiores.2006.11.036. [DOI] [PubMed] [Google Scholar]
- 25.Haworth RS, Avkiran M. Inhibition of protein kinase D by resveratrol. Biochem Pharmacol. 2001;62:1647–1651. doi: 10.1016/s0006-2952(01)00807-3. [DOI] [PubMed] [Google Scholar]
- 26.Wong C, Jin ZG. Protein kinase C-dependent protein kinase D activation modulates ERK signal pathway and endothelial cell proliferation by vascular endothelial growth factor. J Biol Chem. 2005;280:33262–33269. doi: 10.1074/jbc.M503198200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ha CH, Wang W, Jhun BS, Wong C, Hausser A, Pfizenmaier K, McKinsey TA, Olson EN, Jin ZG. Protein kinase D-dependent phosphorylation and nuclear export of histone deacetylase 5 mediates vascular endothelial growth factor-induced gene expression and angiogenesis. J Biol Chem. 2008;283:14590–14599. doi: 10.1074/jbc.M800264200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ha CH, Jhun BS, Kao HY, Jin ZG. VEGF stimulates HDAC7 phosphorylation and cytoplasmic accumulation modulating matrix metalloproteinase expression and angiogenesis. Arterioscler Thromb Vasc Biol. 2008;28:1782–1788. doi: 10.1161/ATVBAHA.108.172528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang S, Li X, Parra M, Verdin E, Bassel-Duby R, Olson EN. Control of endothelial cell proliferation and migration by VEGF signaling to histone deacetylase 7. Proc Natl Acad Sci U S A. 2008;105:7738–7743. doi: 10.1073/pnas.0802857105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vega RB, Harrison BC, Meadows E, Roberts CR, Papst PJ, Olson EN, McKinsey TA. Protein kinases C and D mediate agonist-dependent cardiac hypertrophy through nuclear export of histone deacetylase 5. Mol Cell Biol. 2004;24:8374–8385. doi: 10.1128/MCB.24.19.8374-8385.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fielitz J, Kim MS, Shelton JM, Qi X, Hill JA, Richardson JA, Bassel-Duby R, Olson EN. Requirement of protein kinase D1 for pathological cardiac remodeling. Proc Natl Acad Sci U S A. 2008;105:3059–3063. doi: 10.1073/pnas.0712265105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Harrison BC, Kim MS, van Rooij E, Plato CF, Papst PJ, Vega RB, McAnally JA, Richardson JA, Bassel-Duby R, Olson EN, McKinsey TA. Regulation of cardiac stress signaling by protein kinase d1. Mol Cell Biol. 2006;26:3875–3888. doi: 10.1128/MCB.26.10.3875-3888.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sharlow ER, Giridhar KV, LaValle CR, Chen J, Leimgruber S, Barrett R, Bravo-Altamirano K, Wipf P, Lazo JS, Wang QJ. Potent and selective disruption of protein kinase D functionality by a benzoxoloazepinolone. J Biol Chem. 2008;283:33516–33526. doi: 10.1074/jbc.M805358200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Monovich L, Vega RB, Meredith E, Miranda K, Rao C, Capparelli M, Lemon DD, Phan D, Koch KA, Chapo JA, Hood DB, McKinsey TA. A novel kinase inhibitor establishes a predominant role for protein kinase D as a cardiac class IIa histone deacetylase kinase. FEBS Lett. 2009 doi: 10.1016/j.febslet.2009.12.014. [DOI] [PubMed] [Google Scholar]
- 35.Harikumar KB, Kunnumakkara AB, Ochi N, Tong Z, Deorukhkar A, Sung B, Kelland L, Jamieson S, Sutherland R, Raynham T, Charles M, Bagherazadeh A, Foxton C, Boakes A, Farooq M, Maru D, Diagaradjane P, Matsuo Y, Sinnett-Smith J, Gelovani J, Krishnan S, Aggarwal BB, Rozengurt E, Ireson CR, Guha S. A novel small-molecule inhibitor of protein kinase D blocks pancreatic cancer growth in vitro and in vivo. Mol Cancer Ther. 2010;9:1136–1146. doi: 10.1158/1535-7163.MCT-09-1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Johannes FJ, Prestle J, Eis S, Oberhagemann P, Pfizenmaier K. PKCu is a novel, atypical member of the protein kinase C family. J Biol Chem. 1994;269:6140–6148. [PubMed] [Google Scholar]
- 37.Valverde AM, Sinnett-Smith J, Van Lint J, Rozengurt E. Molecular cloning and characterization of protein kinase D: a target for diacylglycerol and phorbol esters with a distinctive catalytic domain. Proc Natl Acad Sci U S A. 1994;91:8572–8576. doi: 10.1073/pnas.91.18.8572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sturany S, Van Lint J, Muller F, Wilda M, Hameister H, Hocker M, Brey A, Gern U, Vandenheede J, Gress T, Adler G, Seufferlein T. Molecular cloning and characterization of the human protein kinase D2. A novel member of the protein kinase D family of serine threonine kinases. J Biol Chem. 2001;276:3310–3318. doi: 10.1074/jbc.M008719200. [DOI] [PubMed] [Google Scholar]
- 39.Hayashi A, Seki N, Hattori A, Kozuma S, Saito T. PKCnu, a new member of the protein kinase C family, composes a fourth subfamily with PKCmu. Biochim Biophys Acta. 1999;1450:99–106. doi: 10.1016/s0167-4889(99)00040-3. [DOI] [PubMed] [Google Scholar]
- 40.Wang QJ. PKD at the crossroads of DAG and PKC signaling. Trends Pharmacol Sci. 2006;27:317–323. doi: 10.1016/j.tips.2006.04.003. [DOI] [PubMed] [Google Scholar]
- 41.Iglesias T, Rozengurt E. Protein kinase D activation by mutations within its pleckstrin homology domain. J Biol Chem. 1998;273:410–416. doi: 10.1074/jbc.273.1.410. [DOI] [PubMed] [Google Scholar]
- 42.Waldron RT, Rozengurt E. Protein kinase C phosphorylates protein kinase D activation loop Ser744 and Ser748 and releases autoinhibition by the pleckstrin homology domain. J Biol Chem. 2003;278:154–163. doi: 10.1074/jbc.M208075200. [DOI] [PubMed] [Google Scholar]
- 43.Matthews SA, Rozengurt E, Cantrell D. Characterization of serine 916 as an in vivo autophosphorylation site for protein kinase D/Protein kinase Cmu. J Biol Chem. 1999;274:26543–26549. doi: 10.1074/jbc.274.37.26543. [DOI] [PubMed] [Google Scholar]
- 44.Jacamo R, Sinnett-Smith J, Rey O, Waldron RT, Rozengurt E. Sequential protein kinase C (PKC)-dependent and PKC-independent protein kinase D catalytic activation via Gq-coupled receptors: differential regulation of activation loop Ser(744) and Ser(748) phosphorylation. J Biol Chem. 2008;283:12877–12887. doi: 10.1074/jbc.M800442200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Storz P, Doppler H, Toker A. Protein kinase Cdelta selectively regulates protein kinase D-dependent activation of NF-kappaB in oxidative stress signaling. Mol Cell Biol. 2004;24:2614–2626. doi: 10.1128/MCB.24.7.2614-2626.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.von Blume J, Knippschild U, Dequiedt F, Giamas G, Beck A, Auer A, Van Lint J, Adler G, Seufferlein T. Phosphorylation at Ser244 by CK1 determines nuclear localization and substrate targeting of PKD2. Embo J. 2007;26:4619–4633. doi: 10.1038/sj.emboj.7601891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rey O, Yuan J, Young SH, Rozengurt E. Protein kinase C nu/protein kinase D3 nuclear localization, catalytic activation, and intracellular redistribution in response to G protein-coupled receptor agonists. J Biol Chem. 2003;278:23773–23785. doi: 10.1074/jbc.M300226200. [DOI] [PubMed] [Google Scholar]
- 48.Rey O, Sinnett-Smith J, Zhukova E, Rozengurt E. Regulated nucleocytoplasmic transport of protein kinase D in response to G protein-coupled receptor activation. J Biol Chem. 2001;276:49228–49235. doi: 10.1074/jbc.M109395200. [DOI] [PubMed] [Google Scholar]
- 49.Zugaza JL, Waldron RT, Sinnett-Smith J, Rozengurt E. Bombesin, vasopressin, endothelin, bradykinin, and platelet-derived growth factor rapidly activate protein kinase D through a protein kinase C-dependent signal transduction pathway. J Biol Chem. 1997;272:23952–23960. doi: 10.1074/jbc.272.38.23952. [DOI] [PubMed] [Google Scholar]
- 50.Matthews SA, Pettit GR, Rozengurt E. Bryostatin 1 induces biphasic activation of protein kinase D in intact cells. J Biol Chem. 1997;272:20245–20250. doi: 10.1074/jbc.272.32.20245. [DOI] [PubMed] [Google Scholar]
- 51.Rozengurt E, Sinnett-Smith J, Zugaza JL. Protein kinase D: a novel target for diacylglycerol and phorbol esters. Biochem Soc Trans. 1997;25:565–571. doi: 10.1042/bst0250565. [DOI] [PubMed] [Google Scholar]
- 52.Guha S, Rey O, Rozengurt E. Neurotensin induces protein kinase C-dependent protein kinase D activation and DNA synthesis in human pancreatic carcinoma cell line PANC-1. Cancer Res. 2002;62:1632–1640. [PubMed] [Google Scholar]
- 53.Zhukova E, Sinnett-Smith J, Rozengurt E. Protein kinase D potentiates DNA synthesis and cell proliferation induced by bombesin, vasopressin, or phorbol esters in Swiss 3T3 cells. J Biol Chem. 2001;276:40298–40305. doi: 10.1074/jbc.M106512200. [DOI] [PubMed] [Google Scholar]
- 54.Rennecke J, Rehberger PA, Furstenberger G, Johannes FJ, Stohr M, Marks F, Richter KH. Protein-kinase-Cmu expression correlates with enhanced keratinocyte proliferation in normal and neoplastic mouse epidermis and in cell culture. Int J Cancer. 1999;80:98–103. doi: 10.1002/(sici)1097-0215(19990105)80:1<98::aid-ijc19>3.0.co;2-d. [DOI] [PubMed] [Google Scholar]
- 55.Shapiro BA, Ray S, Jung E, Allred WT, Bollag WB. Putative conventional protein kinase C inhibitor Godecke 6976 [12-(2-cyanoethyl)-6,7,12,13-tetrahydro-13-methyl-5-oxo-5H-indolo(2,3-a)py rrolo(3,4-c)-carbazole] stimulates transglutaminase activity in primary mouse epidermal keratinocytes. J Pharmacol Exp Ther. 2002;302:352–358. doi: 10.1124/jpet.302.1.352. [DOI] [PubMed] [Google Scholar]
- 56.Chen J, Deng F, Singh SV, Wang QJ. Protein kinase D3 (PKD3) contributes to prostate cancer cell growth and survival through a PKCepsilon/PKD3 pathway downstream of Akt and ERK 1/2. Cancer Res. 2008;68:3844–3853. doi: 10.1158/0008-5472.CAN-07-5156. [DOI] [PubMed] [Google Scholar]
- 57.Trauzold A, Schmiedel S, Sipos B, Wermann H, Westphal S, Roder C, Klapper W, Arlt A, Lehnert L, Ungefroren H, Johannes FJ, Kalthoff H. PKCmu prevents CD95-mediated apoptosis and enhances proliferation in pancreatic tumour cells. Oncogene. 2003;22:8939–8947. doi: 10.1038/sj.onc.1207001. [DOI] [PubMed] [Google Scholar]
- 58.Sinnett-Smith J, Zhukova E, Hsieh N, Jiang X, Rozengurt E. Protein kinase D potentiates DNA synthesis induced by Gq-coupled receptors by increasing the duration of ERK signaling in swiss 3T3 cells. J Biol Chem. 2004;279:16883–16893. doi: 10.1074/jbc.M313225200. [DOI] [PubMed] [Google Scholar]
- 59.Sinnett-Smith J, Zhukova E, Rey O, Rozengurt E. Protein kinase D2 potentiates MEK/ERK/RSK signaling, c-Fos accumulation and DNA synthesis induced by bombesin in Swiss 3T3 cells. J Cell Physiol. 2007;211:781–790. doi: 10.1002/jcp.20984. [DOI] [PubMed] [Google Scholar]
- 60.Van Lint J, Rykx A, Maeda Y, Vantus T, Sturany S, Malhotra V, Vandenheede JR, Seufferlein T. Protein kinase D: an intracellular traffic regulator on the move. Trends Cell Biol. 2002;12:193–200. doi: 10.1016/s0962-8924(02)02262-6. [DOI] [PubMed] [Google Scholar]
- 61.Wang Y, Waldron RT, Dhaka A, Patel A, Riley MM, Rozengurt E, Colicelli J. The RAS effector RIN1 directly competes with RAF and is regulated by 14-3-3 proteins. Mol Cell Biol. 2002;22:916–926. doi: 10.1128/MCB.22.3.916-926.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhang W, Zheng S, Storz P, Min W. Protein kinase D specifically mediates apoptosis signal-regulating kinase 1-JNK signaling induced by H2O2 but not tumor necrosis factor. J Biol Chem. 2005;280:19036–19044. doi: 10.1074/jbc.M414674200. [DOI] [PubMed] [Google Scholar]
- 63.Biliran H, Jan Y, Chen R, Pasquale EB, Ruoslahti E. Protein kinase D is a positive regulator of Bit1 apoptotic function. J Biol Chem. 2008;283:28029–28037. doi: 10.1074/jbc.M803139200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Endo K, Oki E, Biedermann V, Kojima H, Yoshida K, Johannes FJ, Kufe D, Datta R. Proteolytic cleavage and activation of protein kinase C [micro] by caspase-3 in the apoptotic response of cells to 1-beta -D-arabinofuranosylcytosine and other genotoxic agents. J Biol Chem. 2000;275:18476–18481. doi: 10.1074/jbc.M002266200. [DOI] [PubMed] [Google Scholar]
- 65.Vantus T, Vertommen D, Saelens X, Rykx A, De Kimpe L, Vancauwenbergh S, Mikhalap S, Waelkens E, Keri G, Seufferlein T, Vandenabeele P, Rider MH, Vandenheede JR, Van Lint J. Doxorubicin-induced activation of protein kinase D1 through caspase-mediated proteolytic cleavage: identification of two cleavage sites by microsequencing. Cell Signal. 2004;16:703–709. doi: 10.1016/j.cellsig.2003.11.009. [DOI] [PubMed] [Google Scholar]
- 66.Doppler H, Storz P, Li J, Comb MJ, Toker A. A phosphorylation state-specific antibody recognizes Hsp27, a novel substrate of protein kinase D. J Biol Chem. 2005;280:15013–15019. doi: 10.1074/jbc.C400575200. [DOI] [PubMed] [Google Scholar]
- 67.Ding G, Sonoda H, Yu H, Kajimoto T, Goparaju SK, Jahangeer S, Okada T, Nakamura S. Protein kinase D-mediated phosphorylation and nuclear export of sphingosine kinase 2. J Biol Chem. 2007;282:27493–27502. doi: 10.1074/jbc.M701641200. [DOI] [PubMed] [Google Scholar]
- 68.Hassan S, Biswas MH, Zhang C, Du C, Balaji KC. Heat shock protein 27 mediates repression of androgen receptor function by protein kinase D1 in prostate cancer cells. Oncogene. 2009;28:4386–4396. doi: 10.1038/onc.2009.291. [DOI] [PubMed] [Google Scholar]
- 69.De Kimpe L, Janssens K, Derua R, Armacki M, Goicoechea S, Otey C, Waelkens E, Vandoninck S, Vandenheede JR, Seufferlein T, Van Lint J. Characterization of cortactin as an in vivo protein kinase D substrate: interdependence of sites and potentiation by Src. Cell Signal. 2009;21:253–263. doi: 10.1016/j.cellsig.2008.10.015. [DOI] [PubMed] [Google Scholar]
- 70.Watkins JL, Lewandowski KT, Meek SE, Storz P, Toker A, Piwnica-Worms H. Phosphorylation of the Par-1 polarity kinase by protein kinase D regulates 14-3-3 binding and membrane association. Proc Natl Acad Sci U S A. 2008;105:18378–18383. doi: 10.1073/pnas.0809661105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Eiseler T, Doppler H, Yan IK, Kitatani K, Mizuno K, Storz P. Protein kinase D1 regulates cofilin-mediated F-actin reorganization and cell motility through slingshot. Nat Cell Biol. 2009;11:545–556. doi: 10.1038/ncb1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Nishita M, Tomizawa C, Yamamoto M, Horita Y, Ohashi K, Mizuno K. Spatial and temporal regulation of cofilin activity by LIM kinase and Slingshot is critical for directional cell migration. J Cell Biol. 2005;171:349–359. doi: 10.1083/jcb.200504029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Eiseler T, Doppler H, Yan IK, Goodison S, Storz P. Protein kinase D1 regulates matrix metalloproteinase expression and inhibits breast cancer cell invasion. Breast Cancer Res. 2009;11:R13. doi: 10.1186/bcr2232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lee JW, Kwak HJ, Lee JJ, Kim YN, Lee JW, Park MJ, Jung SE, Hong SI, Lee JH, Lee JS. HSP27 regulates cell adhesion and invasion via modulation of focal adhesion kinase and MMP-2 expression. Eur J Cell Biol. 2008;87:377–387. doi: 10.1016/j.ejcb.2008.03.006. [DOI] [PubMed] [Google Scholar]
- 75.Roy R, Yang J, Moses MA. Matrix metalloproteinases as novel biomarkers and potential therapeutic targets in human cancer. J Clin Oncol. 2009;27:5287–5297. doi: 10.1200/JCO.2009.23.5556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 77.Qin L, Zeng H, Zhao D. Requirement of protein kinase D tyrosine phosphorylation for VEGF-A165-induced angiogenesis through its interaction and regulation of phospholipase Cgamma phosphorylation. J Biol Chem. 2006;281:32550–32558. doi: 10.1074/jbc.M604853200. [DOI] [PubMed] [Google Scholar]
- 78.Martin M, Kettmann R, Dequiedt F. Class IIa histone deacetylases: regulating the regulators. Oncogene. 2007;26:5450–5467. doi: 10.1038/sj.onc.1210613. [DOI] [PubMed] [Google Scholar]
- 79.Bowden ET, Barth M, Thomas D, Glazer RI, Mueller SC. An invasion-related complex of cortactin, paxillin and PKCmu associates with invadopodia at sites of extracellular matrix degradation. Oncogene. 1999;18:4440–4449. doi: 10.1038/sj.onc.1202827. [DOI] [PubMed] [Google Scholar]
- 80.Farmer P, Bonnefoi H, Becette V, Tubiana-Hulin M, Fumoleau P, Larsimont D, Macgrogan G, Bergh J, Cameron D, Goldstein D, Duss S, Nicoulaz AL, Brisken C, Fiche M, Delorenzi M, Iggo R. Identification of molecular apocrine breast tumours by microarray analysis. Oncogene. 2005;24:4660–4671. doi: 10.1038/sj.onc.1208561. [DOI] [PubMed] [Google Scholar]
- 81.Perou CM, Sorlie T, Eisen MB, van de Rijn M, Jeffrey SS, Rees CA, Pollack JR, Ross DT, Johnsen H, Akslen LA, Fluge O, Pergamenschikov A, Williams C, Zhu SX, Lonning PE, Borresen-Dale AL, Brown PO, Botstein D. Molecular portraits of human breast tumours. Nature. 2000;406:747–752. doi: 10.1038/35021093. [DOI] [PubMed] [Google Scholar]
- 82.van de Vijver MJ, He YD, van’t Veer LJ, Dai H, Hart AA, Voskuil DW, Schreiber GJ, Peterse JL, Roberts C, Marton MJ, Parrish M, Atsma D, Witteveen A, Glas A, Delahaye L, van der Velde T, Bartelink H, Rodenhuis S, Rutgers ET, Friend SH, Bernards R. A gene-expression signature as a predictor of survival in breast cancer. N Engl J Med. 2002;347:1999–2009. doi: 10.1056/NEJMoa021967. [DOI] [PubMed] [Google Scholar]
- 83.Turashvili G, Bouchal J, Baumforth K, Wei W, Dziechciarkova M, Ehrmann J, Klein J, Fridman E, Skarda J, Srovnal J, Hajduch M, Murray P, Kolar Z. Novel markers for differentiation of lobular and ductal invasive breast carcinomas by laser microdissection and microarray analysis. BMC Cancer. 2007;7:55. doi: 10.1186/1471-2407-7-55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Poola I, DeWitty RL, Marshalleck JJ, Bhatnagar R, Abraham J, Leffall LD. Identification of MMP-1 as a putative breast cancer predictive marker by global gene expression analysis. Nat Med. 2005;11:481–483. doi: 10.1038/nm1243. [DOI] [PubMed] [Google Scholar]
- 85.Stylli SS, Kaye AH, Lock P. Invadopodia: at the cutting edge of tumour invasion. J Clin Neurosci. 2008;15:725–737. doi: 10.1016/j.jocn.2008.03.003. [DOI] [PubMed] [Google Scholar]
- 86.Janssens K, De Kimpe L, Balsamo M, Vandoninck S, Vandenheede JR, Gertler F, Van Lint J. Characterization of EVL-I as a protein kinase D substrate. Cell Signal. 2009;21:282–292. doi: 10.1016/j.cellsig.2008.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Krause M, Dent EW, Bear JE, Loureiro JJ, Gertler FB. Ena/VASP proteins: regulators of the actin cytoskeleton and cell migration. Annu Rev Cell Dev Biol. 2003;19:541–564. doi: 10.1146/annurev.cellbio.19.050103.103356. [DOI] [PubMed] [Google Scholar]
- 88.Rhodes DR, Yu J, Shanker K, Deshpande N, Varambally R, Ghosh D, Barrette T, Pandey A, Chinnaiyan AM. ONCOMINE: a cancer microarray database and integrated data-mining platform. Neoplasia. 2004;6:1–6. doi: 10.1016/s1476-5586(04)80047-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Chicoine MR, Silbergeld DL. Assessment of brain tumor cell motility in vivo and in vitro. J Neurosurg. 1995;82:615–622. doi: 10.3171/jns.1995.82.4.0615. [DOI] [PubMed] [Google Scholar]
- 90.Krause M, Bear JE, Loureiro JJ, Gertler FB. The Ena/VASP enigma. J Cell Sci. 2002;115:4721–4726. doi: 10.1242/jcs.00218. [DOI] [PubMed] [Google Scholar]
- 91.Bear JE, Svitkina TM, Krause M, Schafer DA, Loureiro JJ, Strasser GA, Maly IV, Chaga OY, Cooper JA, Borisy GG, Gertler FB. Antagonism between Ena/VASP proteins and actin filament capping regulates fibroblast motility. Cell. 2002;109:509–521. doi: 10.1016/s0092-8674(02)00731-6. [DOI] [PubMed] [Google Scholar]
- 92.Kennett SB, Roberts JD, Olden K. Requirement of protein kinase C micro activation and calpain-mediated proteolysis for arachidonic acid-stimulated adhesion of MDA-MB-435 human mammary carcinoma cells to collagen type IV. J Biol Chem. 2004;279:3300–3307. doi: 10.1074/jbc.M305734200. [DOI] [PubMed] [Google Scholar]
- 93.Palmantier R, George MD, Akiyama SK, Wolber FM, Olden K, Roberts JD. Cis-polyunsaturated fatty acids stimulate beta1 integrin-mediated adhesion of human breast carcinoma cells to type IV collagen by activating protein kinases C-epsilon and -mu. Cancer Res. 2001;61:2445–2452. [PubMed] [Google Scholar]
- 94.Jezierska A, Motyl T. Matrix metalloproteinase-2 involvement in breast cancer progression: a mini-review. Med Sci Monit. 2009;15:RA32–40. [PubMed] [Google Scholar]
- 95.Peterburs P, Heering J, Link G, Pfizenmaier K, Olayioye MA, Hausser A. Protein kinase D regulates cell migration by direct phosphorylation of the cofilin phosphatase slingshot 1 like. Cancer Res. 2009;69:5634–5638. doi: 10.1158/0008-5472.CAN-09-0718. [DOI] [PubMed] [Google Scholar]
- 96.Philip PA. Targeted therapies for pancreatic cancer. Gastrointest Cancer Res. 2008;2:S16–19. [PMC free article] [PubMed] [Google Scholar]
- 97.Harikumar KB, Kunnumakkara AB, Ochi N, Tong Z, Deorukhkar A, Sung B, Kelland L, Jamieson S, Sutherland R, Raynham T, Charles M, Bagherazadeh A, Foxton C, Boakes A, Farooq M, Maru D, Diagaradjane P, Matsuo Y, Sinnett-Smith J, Gelovani J, Krishnan S, Aggarwal BB, Rozengurt E, Ireson CR, Guh S. A Novel Small-Molecule Inhibitor of Protein Kinase D Blocks Pancreatic Cancer Growth In vitro and In vivo. Mol Cancer Ther. doi: 10.1158/1535-7163.MCT-09-1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Yuan J, Rozengurt E. PKD, PKD2, and p38 MAPK mediate Hsp27 serine-82 phosphorylation induced by neurotensin in pancreatic cancer PANC-1 cells. J Cell Biochem. 2008;103:648–662. doi: 10.1002/jcb.21439. [DOI] [PubMed] [Google Scholar]
- 99.Berkowitz P, Hu P, Warren S, Liu Z, Diaz LA, Rubenstein DS. p38MAPK inhibition prevents disease in pemphigus vulgaris mice. Proc Natl Acad Sci U S A. 2006;103:12855–12860. doi: 10.1073/pnas.0602973103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Rousseau S, Houle F, Landry J, Huot J. p38 MAP kinase activation by vascular endothelial growth factor mediates actin reorganization and cell migration in human endothelial cells. Oncogene. 1997;15:2169–2177. doi: 10.1038/sj.onc.1201380. [DOI] [PubMed] [Google Scholar]
- 101.Kostenko S, Moens U. Heat shock protein 27 phosphorylation: kinases, phosphatases, functions and pathology. Cell Mol Life Sci. 2009;66:3289–3307. doi: 10.1007/s00018-009-0086-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Ciocca DR, Calderwood SK. Heat shock proteins in cancer: diagnostic, prognostic, predictive, and treatment implications. Cell Stress Chaperones. 2005;10:86–103. doi: 10.1379/CSC-99r.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Jemal A, Siegel R, Ward E, Hao Y, Xu J, Thun MJ. Cancer statistics, 2009. CA Cancer J Clin. 2009;59:225–249. doi: 10.3322/caac.20006. [DOI] [PubMed] [Google Scholar]
- 104.Shirai T. Significance of chemoprevention for prostate cancer development: experimental in vivo approaches to chemoprevention. Pathol Int. 2008;58:1–16. doi: 10.1111/j.1440-1827.2007.02182.x. [DOI] [PubMed] [Google Scholar]
- 105.Jaggi M, Rao PS, Smith DJ, Hemstreet GP, Balaji KC. Protein kinase C mu is down-regulated in androgen-independent prostate cancer. Biochem Biophys Res Commun. 2003;307:254–260. doi: 10.1016/s0006-291x(03)01161-6. [DOI] [PubMed] [Google Scholar]
- 106.Mak P, Jaggi M, Syed V, Chauhan SC, Hassan S, Biswas H, Balaji KC. Protein kinase D1 (PKD1) influences androgen receptor (AR) function in prostate cancer cells. Biochem Biophys Res Commun. 2008;373:618–623. doi: 10.1016/j.bbrc.2008.06.097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Jaggi M, Chauhan SC, Du C, Balaji KC. Bryostatin 1 modulates beta-catenin subcellular localization and transcription activity through protein kinase D1 activation. Mol Cancer Ther. 2008;7:2703–2712. doi: 10.1158/1535-7163.MCT-08-0119. [DOI] [PubMed] [Google Scholar]
- 108.Jaggi M, Rao PS, Smith DJ, Wheelock MJ, Johnson KR, Hemstreet GP, Balaji KC. E-cadherin phosphorylation by protein kinase D1/protein kinase C{mu} is associated with altered cellular aggregation and motility in prostate cancer. Cancer Res. 2005;65:483–492. [PubMed] [Google Scholar]
- 109.Syed V, Mak P, Du C, Balaji KC. Beta-catenin mediates alteration in cell proliferation, motility and invasion of prostate cancer cells by differential expression of E-cadherin and protein kinase D1. J Cell Biochem. 2008;104:82–95. doi: 10.1002/jcb.21603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Du C, Jaggi M, Zhang C, Balaji KC. Protein kinase D1-mediated phosphorylation and subcellular localization of beta-catenin. Cancer Res. 2009;69:1117–1124. doi: 10.1158/0008-5472.CAN-07-6270. [DOI] [PubMed] [Google Scholar]
- 111.Sarker D, Reid AH, Yap TA, de Bono JS. Targeting the PI3K/AKT pathway for the treatment of prostate cancer. Clin Cancer Res. 2009;15:4799–4805. doi: 10.1158/1078-0432.CCR-08-0125. [DOI] [PubMed] [Google Scholar]
- 112.Nelson EC, Evans CP, Mack PC, Devere-White RW, Lara PN., Jr Inhibition of Akt pathways in the treatment of prostate cancer. Prostate Cancer Prostatic Dis. 2007;10:331–339. doi: 10.1038/sj.pcan.4500974. [DOI] [PubMed] [Google Scholar]
- 113.Wong CS, Strange RC, Lear JT. Basal cell carcinoma. Bmj. 2003;327:794–798. doi: 10.1136/bmj.327.7418.794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Ristich VL, Bowman PH, Dodd ME, Bollag WB. Protein kinase D distribution in normal human epidermis, basal cell carcinoma and psoriasis. Br J Dermatol. 2006;154:586–593. doi: 10.1111/j.1365-2133.2005.07073.x. [DOI] [PubMed] [Google Scholar]
- 115.Kim M, Jang HR, Kim JH, Noh SM, Song KS, Cho JS, Jeong HY, Norman JC, Caswell PT, Kang GH, Kim SY, Yoo HS, Kim YS. Epigenetic inactivation of protein kinase D1 in gastric cancer and its role in gastric cancer cell migration and invasion. Carcinogenesis. 2008;29:629–637. doi: 10.1093/carcin/bgm291. [DOI] [PubMed] [Google Scholar]
- 116.Lee HJ, Yang HK, Ahn YO. Gastric cancer in Korea. Gastric Cancer. 2002;5:177–182. doi: 10.1007/s101200200031. [DOI] [PubMed] [Google Scholar]
- 117.Paolucci L, Rozengurt E. Protein kinase D in small cell lung cancer cells: rapid activation through protein kinase C. Cancer Res. 1999;59:572–577. [PubMed] [Google Scholar]
- 118.Kovalevska LM, Yurchenko OV, Shlapatska LM, Berdova GG, Mikhalap SV, Van Lint J, Sidorenko SP. Immunohistochemical studies of protein kinase D (PKD) 2 expression in malignant human lymphomas. Exp Oncol. 2006;28:225–230. [PubMed] [Google Scholar]
- 119.Gschwendt M, Dieterich S, Rennecke J, Kittstein W, Mueller HJ, Johannes FJ. Inhibition of protein kinase C mu by various inhibitors. Differentiation from protein kinase c isoenzymes. FEBS Lett. 1996;392:77–80. doi: 10.1016/0014-5793(96)00785-5. [DOI] [PubMed] [Google Scholar]
- 120.Stewart JR, Christman KL, O’Brian CA. Effects of resveratrol on the autophosphorylation of phorbol ester-responsive protein kinases: inhibition of protein kinase D but not protein kinase C isozyme autophosphorylation. Biochem Pharmacol. 2000;60:1355–1359. doi: 10.1016/s0006-2952(00)00450-0. [DOI] [PubMed] [Google Scholar]
- 121.Aggarwal BB, Bhardwaj A, Aggarwal RS, Seeram NP, Shishodia S, Takada Y. Role of resveratrol in prevention and therapy of cancer: preclinical and clinical studies. Anticancer Res. 2004;24:2783–2840. [PubMed] [Google Scholar]
- 122.Gschwendt M, Kittstein W, Johannes FJ. Differential effects of suramin on protein kinase C isoenzymes. A novel tool for discriminating protein kinase C activities. FEBS Lett. 1998;421:165–168. doi: 10.1016/s0014-5793(97)01530-5. [DOI] [PubMed] [Google Scholar]
- 123.Torres-Marquez E, Sinnett-Smith J, Guha S, Kui R, Waldron RT, Rey O, Rozengurt E. CID755673 enhances mitogenic signaling by phorbol esters, bombesin and EGF through a protein kinase D-independent pathway. Biochem Biophys Res Commun. 391:63–68. doi: 10.1016/j.bbrc.2009.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.LaValle CR, Bravo-Altamirano K, Giridhar KV, Chen J, Sharlow ER, Lazo JS, Wipf P, Wang QJ. Novel protein kinase D inhibitors cause potent arrest in prostate cancer cell growth and motility. BMC Chemical Biology. 2010 doi: 10.1186/1472-6769-10-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Raynham TM, Hammonds TR, Gillatt JH, Charles MD, Pave GA, Foxton CH, Carr JL, Mistry NS. WIPO. Amino-Ethyl-Amino-Aryl (AEAA) Compounds and Their Use. 2007. [Google Scholar]
- 126.Raynham TM, Hammonds TR, Charles MD, Pave GA, Foxton CH, Blackaby WP, Stevens AP, Ekwuru CT. WIPO. Pyridine Benzamides and Pyrazine Benzamides Used as PKD Inhibitors. 2008. [Google Scholar]
- 127.Singh R, Li H, Zhao H, Payan DG, Kolluri R, Tso K, Ramphal J, Gu S. WIPO. Cyclic Amine Substituted Pyrimidinediamines as PKC Inhibitors. 2009. [Google Scholar]
- 128.van Montfort RL, Workman P. Structure-based design of molecular cancer therapeutics. Trends Biotechnol. 2009;27:315–328. doi: 10.1016/j.tibtech.2009.02.003. [DOI] [PubMed] [Google Scholar]