

1. Introduction
It is well established that exposure of DNA to high energy radiation results in a variety of physical and chemical changes in DNA including strand breakage, mutation, and DNA damage.1-16 Initially high energy radiation randomly ionizes or excites DNA components (base, sugar, and phosphate backbone) as well as the surrounding water molecules which are an integral part of the DNA structure. Holes produced quickly shed excess energy and result in ground state cation radicals.12-15,17,18 Secondary electrons with kinetic energy are produced in a large quantity (4 × 104 per MeV energy deposited)19 along the tracks of the ionizing radiation and have been recently shown to produce single- and double-strand breaks in DNA.20-26 Only a small fraction of the secondary electrons are able to cause DNA damage. Most secondary electrons undergo collisional loss of energy with the medium and thermalize within picoseconds. They then either recombine with holes or are captured by the pyrimidines (thymine (T) and cytosine (C)) to form DNA radical anions, T•− and C•−.27 Holes produced during the initial ionizing event in DNA for the most part transfer to the base with the lowest ionization potential. Guanine (G) has the lowest ionization potentials of the four DNA bases (adenine (A), T, G and C)28-31 and as a consequence guanine becomes the locus for hole trapping in DNA.32-34 Ionization of the sugar phosphate backbone initiates two competitive reactions for the hole formed: (i) deprotonation from sugar ring carbon sites to form neutral sugar radicals34,35-39 and (ii) hole transfer to a neighboring DNA base that after base-to-base hole transfer would end up on guanine.37,38,39b Figure 1 gives an overview of the processes that lead from radiation induced hole and secondary electron generation in DNA, to hole and electron transfer, proton transfer processes, and subsequent molecular product formation such as 8-oxo-G from G•+.32-34 Proton coupled electron and hole transfer is an important feature of the radiation damage process. An example is the equilibrium shown in Figure 1 left side in which protonation of the cytosine anion radical at N3 results in transfer of electrons from thymine to cytosine. Coupling of these prototropic equilibria to charge transfer of radiation produced ion radicals is the focus of this review.
Figure 1.

Schematic diagram showing major oxidation and reduction processes that occur in DNA during high energy radiation interaction with DNA. (HT = hole transfer; ET = electron transfer). Processes involving excited states and low energy electrons are not shown. The sketch is based on the works from refs. 3,7,9,10-17, 31-42.
One electron oxidation or reduction of a molecule profoundly affects the acid/base properties of the molecule. On loss of one electron, DNA bases greatly increase in acidity whereas on gain of one electron DNA bases become substantially more basic in comparison to the neutral base. Steenken40-42 considered the proton transfer reactions in base pair ion radicals where hydrogen bonded protons likely transfer between the base pairs. In his pioneering work, he showed that acidity of the one electron oxidized purine base and the basicity of one electron reduced pyrimidine base would affect the extent of such inter base pair proton transfer reactions. Proton coupling with the hole or electron transfer processes in DNA plays a vital role in controlling the charge transfer process in DNA.40 It is well known that proton coupled electron transfer (PCET) or proton coupled hole transfer (PCHT) reactions are crucial for biological processes such as photosynthesis,43-48 respiration49,50, enzyme reactions51 and in oxidized DNA duplexes.52,53 From the early 1990s to present, several reviews considering the experimental aspects of PCET from the groups of Shafirovich,52 Meyer,54,55 Thorp,53,55-57, Mayer,58,59 Babcock,60 Stubbe,61 Robert,62 Costentin63 and their coworkers have appeared in the literature while a series of excellent theoretical reviews on the PCET have become available from the groups of Cukier,64 Hammes-Schiffer65-70 and Nocera.64,71 Application of quantum chemical methods to explore the redox enzymes including PCET have also appeared.72,73
While PCET is critical to charge transfer in DNA, only a few reviews have covered PCET aspects of charge transfer process in DNA.61-63 In this review we first treat hole and electron transfer processes in DNA which lead to ion radical localization in DNA. A brief description of theoretical treatments of PCET is then presented. Finally, the present review reports on experimental and theoretical examples from the work of number of groups, including our own, which illustrates the role of PCET in long range hole/electron transfer in DNA, formation of oxidized and reduced DNA bases and base pairs in ground and excited states as well as resulting sugar radical formation.
2. Background of Charge Transfer in DNA
The hypothesis that π-orbital overlap of paired bases in DNA, can serve as a pathway for charge migration in DNA, was proposed in 1962 by Eley, Spivey and Leslie74,75 from conductivity measurements of DNA75 and its bases (A, T, G and C).74 This led to suggestions that DNA could be a conductor,76 a semiconductor77 or even an insulator78,79 Since then an enormous interest in DNA-mediated charge transfer has grown in order to understand its role in biological cellular process80,81 and its use in DNA-based electrochemical devices82-86 which have broadened our views of the complex nature of DNA charge transfer.
While the generation of hole and excess electron sites in DNA can be accomplished by physical means such as γ-irradiation or photoexcitation, chemically induced one electron oxidation or reduction, also, may lead to holes and excess electron sites in DNA. After formation of holes or excess electrons, charge transfer through DNA ensues to more thermodynamically favorable sites. Thus, DNA-mediated charge transfer can employ either oxidative hole transfer or reductive electron transfer processes. In fact, hole transfer is always accompanied with the transfer of an electron in the opposite direction. Therefore, hole transfer and electron transfer are both electron transfer reactions, see Figure 2. However, for the purposes of this review hole transfer is employed to describe transfers originating from an electron loss center, whereas, electron transfer will refer to those processes begun by addition of an excess electron to the system. Hole transfer chiefly migrates through the highest occupied molecular orbital (HOMOs) of the system whereas electron transfer migrates through the lowest unoccupied molecular orbital (LUMOs). Despite its obvious biological and technological significance, the mechanism of long distance hole migration in DNA resulting from oxidative damage has been controversial and continues to be an exciting area of research.87-89
Figure 2.

(a) One electron oxidation of G in DNA is followed by (b) hole transfer to a distant GGG (mutational hot spot). Figure based in part from Ref. 101. Hole localization is mainly at the 5′-G based on electron spin resonance measurements Ref. 119.
2.1. Oxidatively Induced Hole Transfer
In late 1990’s a number of experiments76,90-93 demonstrated that one electron oxidized guanine, generated by selective oxidation of DNA, could induce a hole migration to a distant guanine through the stacked base pairs of DNA; in these experiments oxidation (charge injection) of DNA was induced by photoexcitation. In the photoexcitation process, an electron is transferred from DNA to the photoexcited chromaphor injecting a hole in DNA which transfers from the charge donor to the final acceptor. Barton and coworkers76,90,91,95-98 inserted donor and acceptor type intercalators in DNA oligomers to investigate charge transfer within DNA. In their experiments the efficiency of the charge transfer depends on several factors, such as: (i) coupling of the redox probes (intercalators) to the π-stacked base pairs, (ii) coupling between the bases in duplex DNA, (iii) presence of base-pair mismatch in the intervening DNA bridge, and (iv) DNA dynamics. Giese and coworkers99-102 used another approach for charge injection into DNA which is achieved by the photolytic generation of C4′ sugar radical via Norrish type I photo-cleavage from 4′-tert-butyl ketones at specific sites in deoxynucleotides which rearrange to an enol-ether radical cation via β-phosphate elimination resulting in a strand break. The enol ether radical cation, subsequently oxidizes a nearby guanine in the DNA duplex.99,103 A competition exists between hole transfer and water addition to G•+ the later of which results in further oxidation in 8-oxo-G and other oxidatively modified guanines.32,34,104,105 These products allow for selective DNA strand cleavage by base or enzymatic treatment and are detected by gel electrophoresis.99 In this approach, the relative charge transfer rate in DNA may be obtained by measuring the product yield of oxidatively modified guanine as a function of distance between guanines and the site of the enol ether radical cation. Lewis106-109 used covalently attached stilbene in DNA hairpins along with time resolved spectroscopy to follow the charge transfer process. Schuster110-112 employed anthraquinones attached to the DNA strand. Photoexcitation of anthraquinone covalently attached with DNA oxidizes a guanine to form the anthraquinone radical anion and G•+ in an overall triplet state.110 Hole transfer was monitored by 8-oxo-G formation. Some one electron oxidants used in the above experiments are shown in Figure 3.
Figure 3.

Structures of one electron oxidants (a) tethered ruthenium II derivative95 (b) anthraquinone derivative110 and (c) one electron oxidation of G by photolysis of tert-butyl ketone attached to C4′ site of sugar.99
The hole generated in DNA on a single G (see Figure 2(a)), by any one of the techniques described above, transfers to sites having GG or GGG sequences in DNA (see Figure 2(b)) because of their lower oxidation potential than single G.113-118 Based on theoretical calculations,113-115 Ratner and coworkers118 proposed the ionization potential (IP) of GG and GGG to be lower than G by 0.5 and 0.7 eV, respectively, concluding that GG and GGG are deeper hole trapping sites. However, using experiment and theory, Lewis116 and Conwell117, reported much smaller hole trapping energies ranging from 0.051 to 0.081 eV, respectively. In the sequences 5′-GG-3′ and 5′-GGG-3′, 5′-site was observed to be the preferred site for oxidative damage followed by a small contributions from neighboring guanines.90,113,114,119 These one electron oxidized [GG] or [GGG] sequences in DNA serves as the mutational hot spot and can be detected biochemically and used as a marker of hole transfer.
2.2. Reductive Electron Transfer
An excess electron added to the DNA from a photoexcited donor or γ-irradiated DNA samples can migrate within DNA to the final electron acceptor and this process is known as reductive electron transfer or electron transfer. In contrast to hole transfer, which is mediated via the DNA bases with lowest oxidation potentials, i.e., the purines, electron transfer is guided by pyrimidines, which have the lowest reduction potentials and highest electron affinities of the DNA bases. Compared to hole transfer, there is significantly less knowledge about processes involved in excess electron transfer within DNA. Electron transfer rates in DNA from base radical anions to electron acceptor intercalators have been studied using pulse radiolysis experiments.120,121 Also, Sevilla and coworkers122,123 used electron spin resonance (ESR) spectroscopy to investigate electron transfer in DNA using electron acceptor intercalators such as mitoxantrone, ethidium bromide, and phenanthroline dissolved in LiBr aqueous glassy medium at low temperatures. The samples were γ-irradiated to produce excess electrons which added to DNA. Subsequent, electron transfer from DNA base anion radicals to randomly placed intercalators within DNA was detected by the loss of DNA-radical-anion ESR signal and the buildup in the intercalator-electron-adduct signal with time at 77 K.
Electron injections in DNA, have also been carried out using photochemical methods. Carell and coworkers124,125 used flavine derivatives and incorporated it into oligonucleotides for electron injection in DNA. Lewis et al.126 used stilbene diether derivatives as DNA hairpins which upon excitation transfers an excess electron to the pyrimidines in DNA. Giese et al.127 employed a 5-substituted thymidine which when inserted into DNA double stranded oligomers and photoexcited formed a substituted thymine radical anion via a Norrish I-type reaction. This injects an electron into the DNA duplex and caused thymine dimer cleavage at long distances. Pyrene-modified pyridine nucleosides were used by Wagenknecht et al.94 One electron reductants used to inject an electron into the DNA are shown in Figure 4.
Figure 4.

Structures of one electron reductants (a) riboflavin coenzyme nucleobase,124 (b) stilbenediether (SE) linkers126a and (c) Pyrene-modified nucleosides.94
2.3. Mechanism of Charge Transport (Superexchange vs. Hopping)
The distance dependent charge transfer rate kCT can be simply expressed by128-130 eq. (1). For tunneling
| (1a) |
for hopping
| (1b) |
In eq (1a), kCT is the charge transfer rate, Δr is the distance between donor and acceptor and β is related to the height of the barrier through which the charge tunnels. In eq. (1b), N is the number of hopping steps and kih is the rate for interbase charge transfer. Both processes are active at ambient temperatures and these equations are thus simplifications. More theoretically satisfying treatments have been presented in which these processes are not separated.131 However, these two extremes provide a convenient way of visualizing the ongoing processes. For tunneling, exponential fall off with distance is expected to limit charge transfer to only a few base pairs unless β is quite low. Thus when experiments demonstrated long range (ca. 200 Å) hole migration in DNA, Barton et al.76,90,132-134 proposed DNA as a molecular wire having delocalized molecular orbitals (MOs) on the stacked DNA bases (see Figure 5(a)) and reported fast charge transfer rate ca. 10−10 s−1 with an appreciably small β value 0.1 Å−1. The apparent small β value has been confirmed in other charge transfer experiments using similar systems but alternative interpretations have been proposed for the charge transfer reactions in DNA. The photoinduced electron transfer studies proposed the multistep hopping model101,108,110,112,118,128 where the hole hops from one base to the adjacent base as shown in Figure 5(b). This mechanism follows a diffusional rate process which falls off much more slowly than the tunneling mechanism giving the low apparent β value. If the hole hops through intervening guanines, it is known as “G-hopping” and, if adenines are the charge carrier, the process is known as “A-hopping”. The situation is different when two guanines are separated by several adenines. Giese and coworkers135 demonstrated experimentally that if two guanines are separated by several adenines; the rate of charge transfer decreases rapidly for short distances (≤ 13.6 Å) showing a typical tunneling distance dependence, however, for large distance (> 13.6 Å) the rate of charge transfer is only weakly distance dependent as expected for a diffusional (hopping) process. This important result shows that for short distances charge transfers through tunneling (superexchange) while long range charge transfer is mediated by thermally induced hopping between adenine bases (A-hopping).135 Thus tunneling components would have β values between 0.6 and 1.2; whereas, when hopping is operative it would make these values appear far lower. For example, Giese and coworkers,93,101,136 reported β values of 0.7± 0.1 Å−1 and 1.0 Å−1. Lewis and coworkers108,137 observed the β values for DNA-capped stilbenes in their excited (ES) and ground state (GS) as 0.7 ± 0.1 Å−1 (ES), 0.63 ± 0.1 Å−1 (ES) and 0.61 ± 0.1 Å−1 (GS). Fukui and Tanakat138 reported the β value 1.42 Å−1 for the intercalated acridine-DNA system in the excited state. The theoretically calculated β ~ 1.2 - 1.6 Å−1 from the work of Priyadarshy et al.77,78 are higher than experimental β values. Siebbeles and coworkers,139 also, calculated the β values for donor and acceptor separated by several AT bridges using the tight binding approach and Miller-Abrahams model of incoherent hopping model. The calculated β value varies with the height of the barrier and ranges from 0.1 to 1.0 Å−1.
Figure 5.

Proposed DNA-mediated charge transfer mechanisms; (a) Superexchange, the overlapping molecular orbitals (MOs) on the well stacked DNA bases provide a path for fast hole transfer, (b) Hopping, the MOs are localized on the DNA bases and the hole hops from one base to another, shown by arrows and (c) polaron formation occurs as solvent polarizes around DNA holes. This limits hole delocalization and transfer rates.
Using density functional theory (DFT) and Hartree-Fock (HF) methods, Olofsson and Larsson140 also calculated β values for several sequences in the range 0.68 – 1.68 Å−1, which is in good agreement with experiment. Thus, results to date clearly show holes and electrons travel through DNA by a combination of tunneling and hopping mechanisms, which might classify DNA as a semiconductor but a semiconductor whose properties can be easily manipulated by base composition. Indeed this classification is too simplistic and more complexities must be taken into account such as polaron formation,141-144 and structural and solvent dynamics.145,146 Charge injection into a molecule induces the surrounding environment (water) to polarize by partially delocalizing the charge on adjacent bases (see Figure 5(c)) and lowering the energy of the system.141 Using theoretical calculations, Conwell has shown that polaron formation may be delocalized over 2 - 5 base pairs, depending on the sequence.142-144 A few reviews147-150 covering this topic in detail have appeared in the literature.
Here we simply emphasize radical generation in DNA which can occur from one electron oxidative and reductive pathways as shown in Figure 1. Once these ion radicals are formed, they migrate within the DNA to the most stable trapping sites. It has also been known that one electron oxidation or reduction of a nucleobase changes the pKa drastically and DNA bases become substantially more acidic on oxidation and more basic on reduction. Protonation/deprotonation reactions then take place either within the hydrogen bonded base pair or from the surrounding medium, i.e., hydration shell. As a result, hole and excess electron transfer most often become coupled to proton transfers and limit the charge transfer process in DNA. The occurrence of such PCET or PCHT in DNA also depends on the rate of charge transfer vs. the rate of proton transfer. In such situations, proton transfer must be fast (ps)40 in order to compete with the hole and electron transfer rate which lies in the range 108 – 1012 s−1.137 Such PCET or PCHT are the focus of our review which will be discussed in sections 4 and 5.
3. Proton Coupled Electron Transfer (Conceptual Background)
Proton coupled electron transfer (PCET) reactions are important pathways in mediating a variety of processes in biology and chemistry.43-53 In contrast to simple electron transfer (ET) or proton (H+) transfer (PT) reactions, PCET is more complex as both electron and proton must transfer and their coupling strongly influences the process thermodynamically and kinetically. In PCET, the transfer of an electron and a proton may be sequential (stepwise) or concerted.54, 64 In sequential transfer, either electron or proton transfers first and is termed as sequential PCET or ET-PT or PT-ET. In the concerted mechanism, electron and proton transfer simultaneously and it is termed CPET.54,64 Since electrons and protons both behave quantum mechanically with wave and particle nature, these two processes (sequential and concerted) can be difficult to distinguish from one another.54,55,61,64,66,151 Hydrogen atom transfer (HAT) involves a concerted process in which an electron and a proton transfer as a near single neutral entity.66 Finally, hydride (H−) transfer can also be considered a PCET as it involves two electrons and a proton transfer.66
The schematic diagram for sequential and concerted pathways for PCET is shown in Figure 6. In Figure 6, the arrows at the edges of the parallelogram show the direction of electron (blue color) and proton (green color) transfer while the four corners (ia, ib, ic and id) of the parallelogram correspond to possible states of the system starting with (ia) proton and electron donor (D-H) hydrogen bonded to the acceptor A (D-H---A). The processes then follow as proton donation to (ic) followed by electron transfer to (id) or electron transfer to (ib) followed by proton transfer to (id). In this representation, arrows, shown at the edges of the parallelogram, correspond to the sequential transfer of electron and proton (ET-PT or PT-ET), the diagonal arrow shows the concerted electron and proton transfer (CPET) or hydrogen atom transfer (HAT) and PCET includes the entire area of the parallelogram,151,152 shown in purple color. In CPET the reaction involves only one transition state (TS1) and no intermediate states are formed (see Figure 6) but electron and proton events can be asynchronous or synchronous. In the sequential mechanism, two transition states (TS1 and TS2), joined by an intermediate, are formed, as shown in Figure 6. Since, in this case, electron and proton transfer events are separated, the overall reaction rate constant (k) is given by eq. (2)
| (2) |
where kET and kPT are electron and proton transfer rate constants, respectively.151,152 From Figure 6, it is evident that PCET (inside of the parallelogram) and stepwise (edge) mechanisms are difficult to distinguish from each other, if PCET is kinetically fast. To aid the investigations of PCET reactions, a number of theoretical methods have been developed by Cukier,152-157 Hammes-Schiffer158-163 and their coworkers as well as by others.164-166 The rate constant expression for fixed proton donor and acceptor distance R for nonadiabatic PCET reactions in solution is given by159
| (3) |
In eq. (3), Pμ is the Boltzmann probability for the reactant state μ, Vel is the electronic coupling and is the proton vibrational wavefunction overlap. is the free energy of reaction for vibronic states μ and ν and λμν is the total reorganizational energy. KB is the Boltzmann constant and T is the temperature. The term in eq (3) gives the mixing of electronic coupling with vibrational wavefunction overlap of the proton in its initial and final states. The square of gives the measure of the extent of which reactant and product coexist along the proton transfer coordinate. Generally, the vibrational overlap of the proton is small because the proton mass is about ca. 1840 times the mass of the electron. From the de Broglie wavelength λ = h/(2mE)1/2 the wavelength of a proton is about 40 times smaller than the electron for a fixed energy. Thus, the proton’s vibrational wavefunction decreases more rapidly with distance in comparison to electronic wavefunctions. A small change in proton transfer distance can therefore greatly affect the vibrational overlap and the overall PCET reaction.
Figure 6.

Schematic diagram showing stepwise (sequential) electron transfer and proton transfer (ET-PT or PT-ET) processes which proceed along the sides of the parallelogram. In ET-PT the reaction path has two transition states TS1 and TS2, right lower figure. The entire area of the parallelogram represents the proton coupled electron transfer (PCET) mechanism and the reaction has only one transition state (TS1), right upper figure. D-H is electron and proton donor and A is the acceptor. Figure based on Refs. 61, 151 and 164.
From experiments it is also found that the electron and proton can transfer to different acceptor sites and fall into the category of PCET.54,55,151 Depending on the nature and location of the electron and proton acceptor sites, PCET can be classified as (i) collinear and (ii) orthogonal. In collinear PCET, electron and proton transfer to the same acceptor site while in orthogonal PCET, the electron and the proton transfer to different acceptor sites as shown in Figure 7. The orthogonal PCET is also termed as bidirectional or multisite electron and proton transfer (MSEPT).54 From theoretical calculations, it is also found that proton motion can affect the electron transfer even though they do not transfer to the same acceptor site (orthogonal PCET). Also, in certain circumstances the same electron and proton need not be coupled during their entire transformation. During transfer the electron may actually encounter different protons in a transport chain. Therefore, kinetic and thermodynamic measurements provide evidence for any coupling between a moving electron with a specific proton or a set of protons at any given time. It is also emphasized that any motion of the coupled proton from its initial position affects the PCET kinetics. Thus, the complete transfer of the proton is not necessary for PCET. An experimental design for a PCET study, in which an electron donor and acceptor are separated by a hydrogen-bonded interface, is shown in Figure 8. Since carboxylic acids have the ability to form cyclic dimers in low dielectric constant solvents, they were used as the hydrogen-bonded interface in the experiment.167 The two carboxylic acids were, respectively, substituted by electron donor (Ru(II) polypyridines or Zn(II) porphyrin) and an organic electron acceptor such as 3,4 dinitrobenzoic acid.167 In this study, the donor was photoexcited to initiate the PCET reaction. The pronounced kinetic isotope effect (KIE) of kH/kD = 1.7 and 1.6 for the charge separation and recombination rates provided the evidence of PCET reaction in the hydrogen-bonded network.
Figure 7.

Collinear and orthogonal PCET. In collinear PCET, proton and electron transfer to the same acceptor site (A). In orthogonal or bidirectional PCET, proton and electron transfer to two different acceptor sites, X (electron acceptor) and A (proton acceptor).
Figure 8.

Donor (D) and acceptor (A) compounds for PCET experiment. The photoinduced electron transfer from D to A induces symmetric double proton transfer between D and A. The symmetric hydrogen-bonded interface produces minor charge rearrangement due to double proton exchange.167
3.1. PCET Studies by Quantum Chemical Methods: Model Systems
3.1.1. Phenoxyl Radical-Phenol and Benzyl Radical-Toluene Complexes
Quantum chemical methods such as Hartree-Fock (HF), density functional theory (DFT) and complete active space self-consistent-field (CASSCF) have been used to study the PCET reactions.65,168,169 These methods can provide good estimates of relevant kinetic and thermodynamic properties in PCET reactions. As proton and electron transfer from a donor to an acceptor site in a system, the charge, spin and molecular orbitals (MOs) localized on donor and acceptor sites change simultaneously. Thus, the calculation of the charge, spin and plots of MOs along a reaction coordinate (transferring hydrogen or proton) provides useful insight in understanding the PCET process. Mayer et al.169 used B3LYP density functional and 6-31G* and 6-311G(2d,2p) basis sets to study PCET and HAT in (i) phenoxyl radical-phenol and (ii) benzyl radical-toluene. Based on the analysis of the singly occupied molecular orbital (SOMO) at a transition state (TS), the authors169 identified phenoxyl radical-phenol reaction as PCET while benzyl radical-toluene was identified as HAT. For phenoxyl radical-phenol system, the SOMO is localized on the 2p orbitals on donor and acceptor oxygen atoms that are perpendicular to the axis joining the two oxygen atoms (see Figure 9a), while proton transfers along the hydrogen bond involving σ-MO. Since the electron and proton transfer through two different types of orbitals, the reaction was identified as PCET. In the benzyl radical-toluene system, the SOMO is localized along the direction joining donor and acceptor (C- --H---C) axis, and this was characterized as HAT reaction (see Figure 9b). Using state-averaged CASSCF(3,6) calculation Hammes-Schiffer168 also studied the phenoxyl radical-phenol and benzyl radical-toluene systems and supported the conclusions of Mayer et al.169 Thus, based on an examination of the plot of the SOMO, the two types of reactions PCET and HAT were identified.
Figure 9.

The singly occupied molecular orbital (SOMO) plot of (a) Phenoxyl radical-phenol (b) Benzyl radical-toluene at the transition state structure. Reactions (a) and (b) are identified as PCET and HAT, respectively. (Figure reproduced with permission from ref. 169. Copyright 2002 American Chemical Society.)
3.1.2. Thymine-Acrylamide Radical Anion Complex
Acrylamide has a structure which is well suited to form hydrogen bonds with thymine in DNA. Studies on γ-irradiated acrylamide-DNA complexes at 77 K, showed acrylamide is a poor electron scavenger; however, on thermal annealing to 130 K, electron transfer from DNA to acrylamide takes place with the apparent simultaneous formation of a neutral radical [CH3-CH(•)-CONH2) by proton transfer from the hydrogen bonded thymine. The reaction clearly shows the involvement of PCET, as shown in Figure 10. The reaction of radical anion of thymine:acrylamide complex was theoretically studied by Sevilla and coworkers170 using B3LYP/6-31+G* density functional method. The optimized structures of the thymine:acrylamide radical anion complex and unpaired spin density distributions before and after proton transfer from thymine to acrylamide are shown in Figure 10. Before proton transfer, the spin density is localized on both thymine and acrylamide (Figure 10 A). After proton transfer, the unpaired spin density is totally localized on acrylamide, which shows the involvement of PCET as suggested by ESR experiment.170
Figure 10.

Optimized structures of thymine:acrylamide radical anion complexes, before and after proton transfer and their spin density distributions. (A) DFT spatial spin distribution for species I (before proton transfer). Note that the spin is shared over both the thymine and acrylamide structures at the isodensity of 0.002 e(Å)−3. (B) DFT spatial spin distribution for species II (after PCET) at the isodensity of 0.002 e(Å)−3. The pink circle highlights the transferring proton. (Figure 2 of ref. 170 reproduced with permission. Copyright 2001 American Chemical Society.)
In a subsequent work, the radical anion of thymine:acrylamide and DNA:acrylamide complexes were studied by Hammes-Schiffer and coworkers171 using the complete active space self-consistent-field (CASSCF) method using a frequency-resolved cavity model for the solvent based on the multistate continuum theory. The study showed that for solvated thymine-acrylamide radical anion complex, electron transfer process dominates while for solvated DNA:acrylamide complex PCET is involved. This difference was attributed to a decrease in the solvent accessibilities in the DNA, which changes the relative free energies of electron transfer and PCET product states.
4. PCET in One Electron Oxidized DNA Bases and Base Pairs
4.1. Guanine
The reversible deprotonation of one-electron oxidized guanine (G•+) is a process of critical importance to the mediation of hole transfer within DNA. Experiments employing pulse radiolysis and 193 nm laser photolysis showed that one-electron oxidized deoxyguanosine (dG•+) has a pKa of 3.9 and deprotonates from its N1-H site (see Figure 11) in an aqueous environment.40,172,173 In aqueous solutions of DNA, production of guanine radical (G(N1-H)•) from guanine is possible either through a stepwise (ET-PT) or a concerted PCET reaction pathway as shown in Figure 11. In the PCET pathway, the reaction proceeds directly to the formation of thermodynamically stable product (G(N1-H)•). The kinetic solvent isotope effects on the electron transfer kinetics associated with the oxidation of guanine in 5′-dGMP, DNA and oligonucleotides by 2-aminopurine (2AP), 2-aminopurine ribose (2APr) and aromatic pyrenyl radical cations (BPT•+) (structures shown in Figure 12), employing laser flash photolysis transient absorption spectroscopy, have been studied by Shafirovich and coworkers.52,174-179
Figure 11.

ET-PT (stepwise) and PCET reaction pathways for the oxidation of guanine.
Figure 12.

Structures of 2-aminopurine (2AP), 2-aminopurine ribose (2APr) and aromatic pyrenyl (BPT).174,179
Photoexcitation of 2AP or 2APr in aqueous buffer solution (pH 7.0) with 308 nm XeCl excimer laser pulses results in the two-photon ionization of 2AP or 2APr52,174
| (4) |
The radical cation (2AP•+ or 2APr•+) rapidly deprotonates in the aqueous solution to give 2AP(−H)• or 2APr(−H•) neutral radicals.
| (5) |
The reaction of these neutral radicals (2AP(−H)• or 2APr(−H•)) with 5′-dGMP results in the formation of 5′-dGMP(−H)• neutral radical, shown in eq. 6
| (6) |
A pronounced kinetic isotope effect was observed on the kinetics of the oxidation of 5′-dGMP by 2AP(−H)• (or 2APr(−H)•) in the H2O and D2O solutions from transient absorption measurements. The values of k(H2O)/k(D2O) in the range 1.5 – 2.0 is evidence that the involvement of PCET through electron transfer from 5′-dGMP to 2AP(−H)• (or 2APr(−H)•) radicals are coupled to the proton transfer from 5′-dGMP.52,74 The oxidation of 5′-dGMP by BPT•+ was also carried out in a similar fashion as above and in this case too the PCET mechanism was proposed from the KIE value of ~1.5.175
Subsequently, the electron transfer reactions between guanine-guanine (GG) doublet (donor) and 2AP radical (acceptor) separated by intervening thymine or adenine bases in oligonucleotides and in DNA were studied using two-photon laser excitation.176,177 The sequences used in the study are shown in Figure 13. The photoexcitation of 2AP modified oligonucleotides (single- and double-stranded, see Figure 13) with 308 nm laser pulses, results in the site-selective two-photon photoionization of the 2AP residue, which subsequently deprotonates to give 2AP(−H)• radical, see eqs. 4, 5. The oxidation of guanine by 2AP radicals was monitored by the evolution of the transient absorption spectra of 2AP radicals and guanine radicals. The oxidation of guanines by 2AP(−H)• and the formation of guanine radical (G(−H)•) occurs within 0.1 – 500 μs and was proposed to proceed through a PCET mechanism as shown in scheme 1.
Figure 13.

Design of 2AP modified oligonucleotides and DNA duplexes used in refs. 176, 177.
Scheme 1.
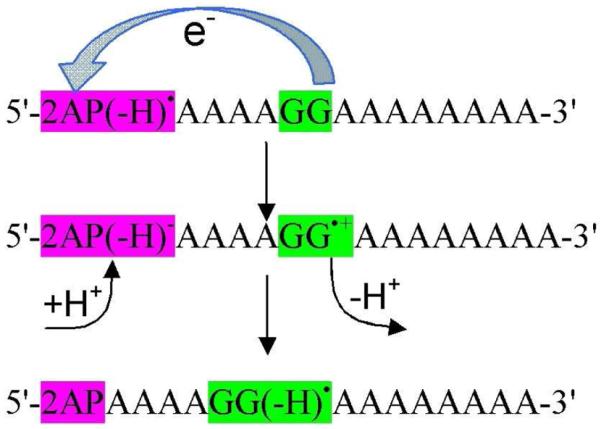
Oxidation of guanine by 2AP(−H)• subsequently proceeds through protonation and deprotonation from solvent and result in G(−H) • formation through PCET.
The KIE associated with the oxidation of guanine by 2AP in DNA duplexes were measured in H2O and D2O, respectively. The rate constants of the formation of G(−H)• in H2O was found to be larger than in D2O and the measured k(H2O)/k(D2O) lie in the range 1.3 – 1.7. From KIE values, the reaction was proposed to involve PCET. However, Huynh and Meyer54 in their review suggested that a simple PCET is not a feasible pathway in this reaction because electron transfers a long distance (>10 Å) in DNA and could not couple with the proton of the guanine because of its short-range nature. They54 suggested that this reaction may occur by one electron and two proton (1e−/2H+) MSEPT pathway. In this pathway, the long range electron transfer from guanine to 2AP(−H)• is coupled by two spatially separated protons which occurs from the aqueous solvent, see scheme 1. We note that in duplex DNA the initial deprotonation of G•+ is to cytosine not the aqueous solution, and this adds additional complexity to the process.
Thorp and coworkers53,180,181 used stopped-flow spectrophotometry and electrochemical methods to study kinetics of the oxidation of guanine in 5′-dGMP, herring-testes DNA (double-stranded), 2′-deoxyguanosine-5′-triphosphate and the oligonucleotides d[5′-GCA GTA GCA TGT GAC GAG TCG] hybridized to its Watson-Crick complement and complexed with Ru(bpy) 33+ ((bpy) 2,2′-bipyridine)) in phosphate buffer at pH = 7. The oxidation reaction of guanine by Ru(bpy) 33+ which leads to G(−H)• formation is given in eqs. 7 and 8.
| (7) |
| (8) |
If eq. 7 is rate limiting, the slope of the Marcus plot RT ln kET vs E1/2 (III/II (redox potential)) should be 0.5, but, if eq. 8 is rate limiting, the corresponding slope should be 1.0.181 In their study, RT ln kET and E1/2 for reaction of guanine with different metal complexes X(bpy)33+/2+ (X=Fe, Ru and OS)53,180 was plotted and a slope of 0.8 ± 0.2 was obtained and PCET was invoked involving reactions 7 and 8. The rate of reaction of guanine oxidation was also measured in H2O and D2O and a KIE = 2.1 and 1.4 was observed for DNA and mononucleotides, respectively. The study was extended to 7-deaza analogues of guanine and adenine in mononucleotide triphosphate forms and slopes of the plot RT ln kET vs E1/2 corresponding to 1.1 in each case was obtained. The KIEs with different metal complexes ranged from 2.2 to 10, respectively. Thus, the slope of > 0.5 clearly indicates the involvement of PCET mechanism in each of these systems. In this reaction, solvent acts as a proton acceptor and thus the reaction is likely a MSEPT.
The deprotonation preferred site of the guanine radical cation in aqueous solution is also considered in a few studies.40,172,182-184 Pulse radiolysis experiments suggest the N1-H site for deprotonation in 2′-deoxyguanosine radical cation (2′-dG•+), while, in 1-methylguanosine radical cation deprotonation must occur from the NH2 group.172,173,182 In aqueous solution, 2′-dG•+ and 1-methylguanosine radical cation have pKa values 3.9 and 4.7.172 The pKa of 1-methylguanosine radical cation is higher than 2′-dG•+ by 0.8 pH units. Thus while both the sites of G•+ are in competition for deprotonation in aqueous solution the N1-H site is likely favored over N2-H. Recently, one electron oxidized 2′-dG by γ-irradiation at 77 K, ESR measurements and theoretical calculations confirmed that N1-H site is the preferred site for deprotonation of 2′-dG•+ in aqueous solution.184
4.1.1. Photooxidation of Guanine by Metal-Ligand Charge Transfer (MLCT) Complexes
[Ru(TAP)2(dppz)]2+ (TAP = 1,4,5,8-tetraazaphenanthrene, dppz = dipyrido[3,2-a:2′,3′-c]phenazine), shown in Figure 14, is a DNA intercalator and in it’s excited state becomes a highly oxidizing agent.185,186 It not only oxidizes guanine in guanosine-5′-monophosphate (5′-GMP), but also guanine-containing polynucleotides [poly(dG-dC)]2. Excitation of [Ru(TAP)2(dppz)]2+ is of ππ* type and results in a singlet excited state. Within picoseconds intersystem (singlet-to-triplet) crossing (ISC) takes place in aqueous solution at pH 7, as observed from UV/visible absorption data.186 This triplet excited state arises due to metal-ligand (Ru-TAP) charge transfer (MLCT) and has a lifetime of several nanoseconds in aqueous solution. The oxidation of guanine by [Ru(TAP)2(dppz)]2+ in an excited triplet state is shown in Figure 15.
Figure 14.
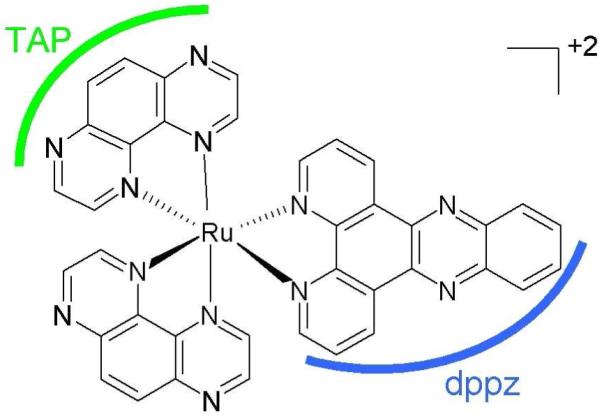
Structure of [Ru(TAP)2(dppz)]2+ ref. 186.
Figure 15.

Formation of the MLCT excited state of [Ru(TAP)2(dppz)]2+ and ET from G to form G(−H)• via PCET. See refs. 185, 186.
The reaction was carried out in both H2O and D2O, giving KIE values of 1.3 and 1.6 for forward and back electron transfer (BET), shown in Figure 15, and it was proposed that oxidation of guanine in the excited state is due to PCET or MSEPT. In the case of the G-C base pair, proton transfers from G to C, while in 5′-GMP proton transfer is to the solvent. In this study, it was also proposed that a proton from G•+ may also transfer to the reduced parent complex [Ru(TAP−•)(TAP)(dppz)]+ + G(−H)• + H+ → [Ru(TAP•(H))(TAP)(dppz)]2+ + G(−H)•.
4.1.2. PCET in Guanine-Cytosine (G-C) Base Pair
One electron oxidation of G-C base pair results in a transfer of a proton from N1 of G to N3 of cytosine as shown in Figure 16. In this context, proton transfer reactions in one-electron oxidized double-stranded DNA oligonucleotides containing G, GG and GGG sequences have been investigated using nanosecond pulse radiolysis experiments.187,188 One electron oxidation of guanine in double-stranded DNA was produced by SO4•−.172,187,188 It is also noted that the spectra of dG•+ and N1-deprotonared dG(−H)• are quite similar and absorb around 380 and 480 nm.172,173,185,187,188 A weaker absorbance at ca. 625 nm was characterized as due to deprotonation of G•+ to G(−H)•.187,188 Recent work in our laboratory suggests the absorption at 625 nm is likely from G(−H)• with the proton loss to solvent from N2. This we discuss further below. The kinetics of one electron oxidation of guanine by SO4•− in deoxyguanosine (dG) and different DNA sequences was measured in H2O and D2O, respectively. The change in the absorbance at 625 nm after pulse radiolysis in H2O and D2O of dG and double-stranded oligonucleotide (5′-AAAAAGGGAAAAA-3′) is shown in Figure 17 and the corresponding KIE= 1.7 and 3.5 were measured.
Figure 16.

Scheme showing prototropic equilibria of proton transfer in one electron oxidized G-C base pair. One electron oxidation of G-C base pair and proton transfer from G to C can occur from step wise or concerted PCET within the DNA duplex.
Figure 17.
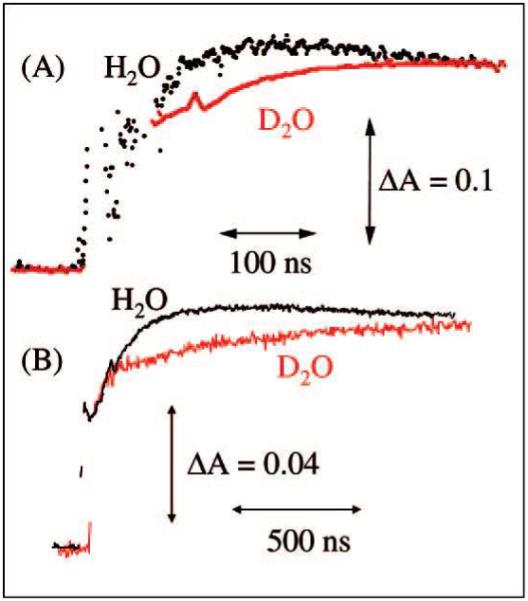
Absorbance changes at 625 nm after pulse radiolysis of dG (5.6 mM) (A) and double-stranded DNA (5′-AAAAAGGGAAAAA-3′) (2.1 mM) (B) in the presence of ammonium persulfate (20 mM), NaCl (0.1 M), and tert-butyl alcohol (0.1 M) in 20 mM sodium phosphate in H2O at pH 7 (black) or in D2O at pD 7 (red). (Figure reproduced with permission from ref. 188. Copyright 2008 American Chemical Society.)
Double stranded oligodeoxynucleotides containing 5-bromocytosine (pKa=2.8) and 5-methylcytosine (pKa=4.7) substitutions in the sequences were also investigated as they change the pKa of cytosine (pKa= 4.3) and were found to affect the rates of production of G(−H)• as expected. With 5-bromocytosine having the lowest rate at 1.2 × 106 s−1, cytosine 8.7 × 106 s−1 and 5-methylcytosine having the highest rate at 2.0 × 107 s1. However, all dsDNA oligos with and without substitutions were found to have large KIE for G(−H)• formation of 3 to 3.5 whereas for the deoxynucleoside, dG, only 1.7 was found.188 These large KIEs in oligonucleotides provide strong evidence that the deprotonation reaction of G•+ is a PCET. Further, in this study, the proton transfer from G•+ to C is strongly supported by the results found for of substituted cytosines in dsDNA. A further step involving transfer to the solvent is also proposed suggesting MSEPT.188
The protonation state and hole localization in one electron oxidized double-stranded DNA sequences were investigated very recently using ESR. The site of hole localization was determined by use of oligos with deuterium substitution at the C8 position of guanine (8-deuteroguanine (Gd)) at selected sites in the DNA sequences.119 The work demonstrated that ESR spectra of one electron oxidized 8-deutero-2′-deoxyguanosine (2′-dGd+•) and its deprotonated species (2′-dG(−H)d•) recorded at 77 K are distinguishable from each other, see Figure 18. The ESR spectra of 2′-dGd+• and 2′-dG(−H)d• were used as benchmark spectra for ESR analysis of one electron oxidized deuterated double-stranded DNA oligomers to characterize the protonation state of one electron oxidized guanine in the DNA oligomers. The ESR measurements for one electron oxidized double-stranded d[GdCGdCGdCGdC]2 showed that one electron oxidized guanine exists as a neutral radical G(N1-H)• in DNA, see Figure 18. Thus at low temperatures only G(N1-H)• is observed, however, this study pointed out that while proton transfer from guanine to cytosine in DNA is thermodynamically favored at ambient temperatures, owing to the small free energy difference, an equilibrium between the two forms (see Figure 16) is expected as predicted earlier by Steenken.40
Figure 18.

ESR spectra of (A) Gd•+ (black) and Gd(−H)• (red) obtained from glassy (7.5 M LiCl in D2O) samples of Gd [Gd = 8-D-dGuo, 96% D] (3 mg/mL). (B) Spectrum of the one-electron oxidized ds DNA oligomer d[GdCGdCGdCGdC]2 (2 mg/mL) with Gd(−H)• from A in red superimposed. The match of green and red spectra in (B) clearly shows that one-electron oxidized guanine in ds DNA oligomer exists as Gd(−H)•. One-electron oxidation of the monomer and the DNA-oligomers were carried out via thermal annealing at 155 K. All spectra were recorded at 77 K. (Modified Figure from Figure 2 of ref. 119. Copyright 2009 American Chemical Society.)
Another interesting feature of this work is that by investigating ds DNA oligos with Gd substitutions at specific sites the site of hole localization could be ascertained. For example the oligo, TGdGGCCCA showed that most of the localization of the hole was at the C5′-end of the GGG stack as had been predicted in earlier studies which relied on product analysis as well as by theoretical calculations. Although some hole localization at other sites (15 to 20%) was found, this was attributed to the small thermodynamic differences in energy between the various sites which only slightly favor the C5′-site. No evidence for charge delocalization was found.
A number of other studies involving DNA systems are of note. Hole transfer rates in double-stranded DNA was also measured in H2O and D2O.189 A decrease in charge transfer rate by a factor of 3 in D2O was observed, which clearly shows the slower deuteron shift from G•+ to C compare to the corresponding proton shift. This KIE effect supports the coupling of hole transfer with intra-strand proton transfer in DNA duplex.189 A small KIEs = 1.2 and 1.3 for double-stranded DNA sequence for hole transfer between Naphthalimide (NI) and phenothiazine (PTZ) in its excited state is observed by Majima and coworkers.190,191 Oxidation of guanine in DNA by Ru(phen)2(bpy)33+ using flash-quench technique has been studied and neutral guanine radical (G(−H)•) was detected by transient absorption spectroscopy and EPR.147,192,193 Oxidation of guanine in DNA duplex by SeO3•− and SO4•− ions generated by pulse radiolysis and the rapid formation of cytosine radical has been observed by transient absorption spectroscopy.194 Using first-principles quantum-mechanical and molecular mechanics (QMMM) approach, molecular dynamics simulation on a fully hydrated 38-base-pair B-DNA d(5′-ACGCACGTCGCATAATATTACGTGGGTATTATATTAGC-3′) in radical cation state showed that double proton transfer from guanine to cytosine can be coupled with the charge transfer.195
4.1.3. Thermodynamic Stability of G•+-C and G(N1-H)•-(+H+)C
A number of theoretical investigations of the proton transfer reaction in one electron oxidized guanine-cytosine base pair have been attempted.195-199 In these computational studies, the transition state for the proton transfer from G•+ to C in the G•+-C base pair and the relative stabilities of G•+-C (reactant) and G(N1-H)•-(+H+)C (product) (see Figure 16) were calculated in the gas phase and the reaction was found to be endergonic by a few kcal/mol which disfavors the proton transfer from G•+ to C in disagreement with experiment. In a more recent study,200 the proton transfer reaction in G•+-C was modeled in the presence of 11 water molecules surrounding the G-C base pair (see Figure 19) and the reaction was found to be exothermic by 1.2 kcal/mol after zero point energy (ZPE) correction. The calculated ZPE-corrected free energy (ΔG) −0.65 kcal/mol at 298 K is in excellent agreement with experimental estimation of ΔG= −0.55 kcal/mol, based on the aqueous-phase pKa’s of the G•+ and that of the C base.42,199 The calculated spin densities are localized on the guanine moiety in agreement with experiment, see Figure 19. The greater thermodynamically stability of G(N1-H)•-(+H+)C over G•+-C adds support to the involvement of PCET in any hole transfer by hopping within DNA.
Figure 19.
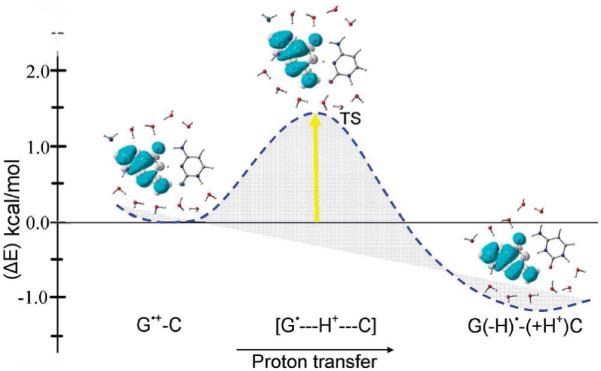
The B3LYP/6-31+G** calculated potential energy surface (PES) of proton transfer (PT) in G•+-C in the presence of 11 waters with zero point energy (ZPE) correction. Energy is given in kcal/mol. Spin density distributions during proton transfer are also shown. (Figure reproduced with permission from ref. 200. Copyright 2009 American Chemical Society.)
4.1.4. PCET in the Excited State of the G-C Base Pair
A PCET excited state deactivation mechanism for a G-C base pair within double-stranded DNA has been proposed from experiment and theory.201-207 Sobolewski and Domcke201,202 proposed a coupled electron proton transfer mechanism for G-C base pair which is illustrated in Figure 20. In the ground state of G-C base pair the highest occupied molecular orbital (HOMO) is localized on guanine, while the lowest unoccupied molecular orbital (LUMO) is localized on cytosine. Photoexcitation of G-C base pair, initiates an electron transfer from HOMO to LUMO, which corresponds to a charge transfer (CT) excited state, designated as 1ππ*(CT) (blue curve), see Figure 20. In this charge transfer excited state, the N1-H proton of G transfers spontaneously to C without a barrier and crosses to the ground state curve (pink color). Thus the populated excited state returns to the ground state and normal G-C structure is restored. This PCET mechanism is supported by molecular dynamics simulations and experiment.203-207
Figure 20.

Sketch of the ultrafast excited state deactivation pathway of G-C base pair through proton coupled electron transfer mechanism.201, 202
Very recently, Kohler and co-workers207 investigated the deuterium isotope effect on the excited state dynamics of G-C containing DNA duplexes using transient absorption spectroscopy. This study demonstrated a pronounced isotope effect on the excited state lifetimes in an alternating G-C oligonucleotide. For a d(GC)9.d(GC)9 oligonucleotide, the transient absorption signals were recorded at 266 nm with probe wavelengths of 250 and 270 nm in H2O and D2O. The results evidence a faster ground state recovery for d(GC)9.d(GC)9 in H2O than D2O, see Figure 21. However, for 5′-GMP and 5′-CMP the KIE is very modest, see inset of Figure 21. This study also comes to the conclusion that the formation of exciplex states with significant CT character enable intrabase pair proton transfer in DNA which decay with a PCET mechanism as described in Figure 20.
Figure 21.
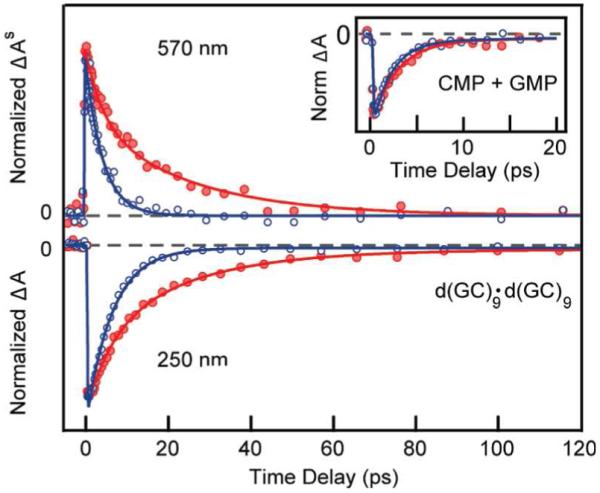
Normalized transient absorption signals showing (top) excited-state absorption and (bottom) ground-state bleach recovery of d(GC)9·d(GC)9 in H2O (blue) and D2O (red). The inset shows the 250 nm transient for an equimolar mixture of the monomers CMP and GMP in H2O (blue) and D2O (red). (Figure reproduced with permission from ref. 207. Copyright 2009 American Chemical Society.)
4.1.5. Repair of Guanyl Radical (G(N1-H)•) Through PCET
A series of studies have employed γ-irradiated aqueous solutions of plasmid DNA samples in the presence of thiocyanate to generate guanyl radicals (G(N1-H)•). The sites of guanine radical formation are detected after formation of products, 8-oxo-G and fapyG (2,6-diamino-4-hydroxy-5-formamidopyrimidine). Escherichia coli base excision repair endonucleases are used to convert these stable end products to single and double strand breaks in the plasmids which are readily detected and distinguished using gel electrophoresis.208-210 In these works,208-210 it has been shown that the strand breaks can be strongly attenuated if micromolar concentrations of substituted phenols or indoles or anilines are present. These phenols are able to reduce the guanyl radical which subsequently accepts a proton from phenol. Based on the energetics involved for stepwise ET and PT pathways and the concerted PCET pathway (see Figure 6 in section 3), it was concluded that the repair of guanyl radical occurs by PCET as shown in eq. 9.
 |
The repair mechanism of deprotonated DNA bases (B•+ → B(−H)• + H+; B=A, T, G and C) by thiols were investigated by theoretical calculations.211 The results showed that the repair of A(−H)• and C(−H)• should be favored via PCET at any pH while for G(−H)• and T(−H)•, a PCET process is preferred at acidic to neutral pHs. In the pH range 9 to 11, ET pathway would dominate. The repair of G(N1-H)• and its anion (G(N1-H)−) by amino acids (cysteine and tyrosine) was also proposed to involve hydrogen atom transfer and PCET.
4.2. Cytosine and Thymine
Deprotonation reactions of one electron oxidized cytosine (C•+) and thymine (T•+) (shown in Figure 22) have been studied in a number of publications.213-217 The radical cation of 5-methylcytosine produced in aqueous glasses by UV photolysis at 77 K was found to deprotonate from the methyl group upon warming at 190 K, as evidenced from ESR measurements.213,214 Similarly, the oxidation of thymine at 77 K by photoionization and ESR spectra showed the formation of similar deprotonated species, UCH2• (methyl deprotonated thymine cation radical).215 In another study,216 photoexcitation of anthraquinone-2,6-disulfonic acid (2,6-AQDS) with cytosine by 308 nm XeCl excimer laser oxidizes cytosine by electron transfer from cytosine to 2,6-AQDS in triplet state via interstate (singlet-to-triplet) crossing. The oxidized cytosine deprotonates from its N1 site.216 If cytosine is replaced by 1-methylcytosine, deprotonation occurs from the NH2 group of the cytosine. The radicals were directly detected by time-resolved Fourier-transform EPR in H2O and D2O at 10° C on nanosecond timescale,216 while, the formation of the radical cation was not detected. The reaction mechanism along with the structure of 2,6-AQDS is shown in scheme 2.
Figure 22.

Typical deprotonation reactions of one electron oxidized cytosine and thymine.
Scheme 2.

One electron oxidation of cytosine via PCET. See Ref. 216.
Very recently, one electron oxidation in the single crystals of cytosine monohydrate doped with a very small amount of 2-thiocytosine was carried out by X-irradiation.217 The radicals formed between 10 and 150 K after oxidation in normal and partially deuterated samples were investigated by EPR spectroscopy. A huge KIE (> 102 - 103) at 100 K indicates that radical formation occurs via proton coupled electron/hole transfer (PCET/PCHT) mechanism. The hydrogen-bonds between cytosines in the crystals provide a ready minimum energy path for proton transfer. In the X-irradiated doped crystals both anion and cation radicals of cytosine are initially formed. The one electron oxidized cytosine (C•+) subsequently transfers a hole to 2-thiocytosine because of its lower ionization potential than cytosine.217 Thus, 2-thiocytosine acts as a hole acceptor at 10 K in these crystals. Finally, one electron oxidized 2-thiocytosine deprotonates to a hydrogen-bonded cytosine. Theoretical calculations show that only when hole transfer is combined with a proton transfer (PT1) does the reaction become exothermic. The schematic diagram for the PCHT reaction is shown in Scheme 3. The hole, electron recombination reaction activated at 60 K also was suggested to involve PCET.
Scheme 3.
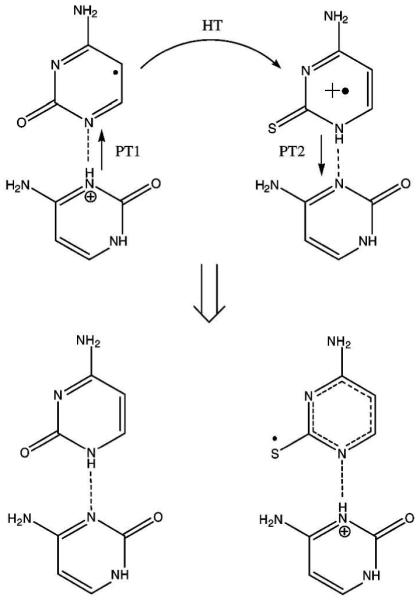
Formation of the thiocytosine deprotonated cation by PCHT. (Figure reproduced with permission from ref. 217. Copyright 2008 American Chemical Society.)
4.3. Adenine
The absorption spectra of one electron oxidation of adenine in 2′-deoxyadenosine (dA) by SO4•− in aqueous solution was studied by Steenken.40 The resulting absorption spectra of dA•+, measured between pH 0 and 6, showed the existence of dA(−H)• produced by deprotonation at the NH2 group of the adenine cation radical.40 From the absorption spectra, it was concluded that the pKa of dA•+ in aqueous solution is < 1. The dA(−H)• formation from dA•+ was also supported from ESR and electron nuclear double resonance (ENDOR) studies of X-irradiated dAdo crystals at 10 K,218 pulse radiolysis experiments in aqueous solution219 and ESR studies of γ-irradiated dA.220 While dA•+ is experimentally found to produce the neutral radical (dA(−H)•) in aqueous environment, the oxidation of adenine stacks in DNA oligomers, was observed to be metastable to deprotonation.220 This observation was reported in a recent ESR study of one electron oxidation by Cl2•− of adenine in dA and in stacked DNA oligomers (dA)6 as a function of pH.220 The stability of one electron oxidized adenine stacks in DNA was attributed to a charge delocalization mechanism which inhibits the deprotonation of A•+ and may facilitate the observed long range charge transfer process through A stacks in DNA. These experiments strongly suggest that deprotonation would strongly attenuate large range hole transfer and greatly slow PCET.
4.3.1. Adenine-Thymine (A-T) Base Pair
Steenken40 considered the proton transfer on one electron oxidation of an A-T base pair from the NH2 group of adenine to O4 atom on thymine (see Figure 24) and he suggested from experimental data that such transfer should be unfavorable. A theoretical treatment of this proton transfer reaction for the ground state of hydrogen bonded adenine-thymine (A-T) base pair radical cation has been reported for the gas phase.198 From the calculation of the relative stabilities of A•+-T (reactant) and A(−H)•-(H+)T (product), the reaction was found to be endothermic by 1.2 kcal/mol,198 which disfavors the proton transfer as predicted earlier by Steenken.40 The inclusion of aqueous media would be helpful in future efforts. The excited states of A-T base pair using the CC2 (simplified singles and doubles coupled-cluster) method were treated recently.221 The calculated excited state potential energy surface (PES) for proton transfer from adenine to thymine in the 1ππ*(CT) (charge transfer excited state) showed that deactivation process of excited A-T base pair is driven by PCET mechanism221 as shown in section 4.1.4 for G-C base pair.
Figure 24.

Scheme showing prototropic equilibria of proton transfer in one electron oxidized A-T base pair.
4.4. Sugar Radical Formation Through PCET
In addition to DNA base radicals, sugar radicals are also formed due to ionization and γ-irradiation of DNA,35-39 which ESR experiments suggest account for about 7-15% of trapped radicals at low temperatures.39b These sugar radicals are known to produce unaltered base release and associated single strand breaks in DNA. The formation of sugar radicals through the abstraction of hydrogen atom from the sugar ring by OH• and the direct oxidation of sugar-phosphate backbone followed by deprotonation is well documented in the literature.35,37 The direct formation of sugar radical by photoexcitation of radical cations of guanine (G•+) and adenine (A•+) in model systems of deoxyribonucleotides, ribonucleotides, and DNA and RNA oligos has been proposed recently in a number of studies.36,39,222-226 The underlying mechanism of sugar radical formation proposed in these studies36,39,222-226 is that photoexcitation induces hole transfer from one electron oxidized DNA base to the sugar ring which is followed by rapid deprotonation (−H+) at specific carbon sites on the sugar ring, which prevents the back transfer of hole to base.37,38,227 The hole transfer from base to sugar is, of course, accompanied by an electron transfer from the sugar to the one electron oxidized base. Thus the overall sugar radical formation process is clearly a proton coupled hole transfer (PCHT) (or proton coupled electron transfer (PCET)) as depicted in Scheme 4.228
The involvement of PCET/PCHT for the sugar radical formation from one electron oxidized 2′-deoxyguanosine (dG•+) in the presence of seven water molecules was explored using the density functional theory.228 In these calculations, the PES for the C5′ radical formation was calculated by stretching the C5′-H bond in dG•+ in the presence of water molecules employing the B3LYP/6-31G* level of theory. At each step of C5′-H bond elongation (proton transfer) on the PES, the spin densities were also calculated to observe the electron transfer process, see Figure 25. The study clearly demonstrates that only a slight increase in C5′-H bond length of 0.13 Å is sufficient to reach the transition state and completely transfers the spin initially localized on guanine base in dG•+ (step number 1, in Figure 25) to the C5′ site on the deoxyribose (sugar) group (step number 2 in Figure 25). Beyond the TS the proton ultimately transfers to N7 of guanine (step number 6 in Figure 25) through the intervening waters without any significant barrier. The proton transferred (dG(•C5′, N7-H+) + 7H2O) (product, step no. 6) is found to be ca. 13 kcal/mol more stable than the reactant dG•+ + 7H2O (reactant, step number 1). Very small deuterium isotope effects of 1.5 ± 0.3 and 1.3 ± 0.3 were observed for the formation of C5′• and C3′•, respectively, from 2′-deoxyguanosine radical cation at 143 K223 which according to Hammond’s postulate229 shows that the geometries of the TS and reactant are likely to be close to each other. The theoretical calculations strongly support this conclusion. Clearly the direct formation of sugar radicals from G•+ necessitates the involvement of PCET.
Figure 25.

BHandHLYP/6-31G*//B3LYP/6-31G* calculated spin density distribution during proton transfer from C5′ on the deoxyribose group to the N7 site on guanine in dG•+ + 7H2O (step No. 1). The stretching of the C5′-H bond from its equilibrium bond length (1.099) to 1.23 Å (TS) (Step Nos. 1 and 2) results in the complete transfer of the hole from guanine to the C5′ site which is equivalent to electron transfer from the C5′ site to guanine. The pink circle highlights the transferring proton when not obscured by the spin distribution. (Figure reproduced with permission from ref. 228. Copyright 2009 American Chemical Society.)
5. PCET in One Electron Reduced DNA Bases and Base Pairs
The electron attachment process involves one electron reduction of a molecule (M) and leads to molecular radical anion formation (M•−). The energetic stability of M•− is given by the electron affinity (EA) of the molecule. Electron transmission spectroscopy (ETS)230 and anion photoelectron spectroscopy231 experiments showed that the vertical and adiabatic EAs of conventional DNA bases (A, T, G and C) in the gas phase are negative (< 0) or near zero. In competition with valence anion formation in the gas phase, bases such as T and U with significant dipole moments, can bind the free electron near the molecular framework with a weak interaction typically less than 0.1 eV. Such species are called dipole bound anions and are not considered relevant to DNA in condensed media. However, in aqueous environment the solvation energy increases the electron affinity of the bases by several eV and stable valence radical anions are formed for all the DNA bases. These valence DNA base anion radicals are quite basic and usually react by protonation at heteroatom sites (N and O) (reversibly) as well as carbon sites (irreversibly) to form stable neutral radicals. These species are observed from ESR, pulse radiolysis and photoelectron spectroscopy experiments. Below we give several examples of these studies.
5.1 Guanine
Single crystals of N7 protonated guanine in guanine hydrochloride dehydrate X-irradiated at 20K, 65K and 150K were studied by ESR/ENDOR techniques.232,233 The study showed one electron reduction of the guanine:HCl results in a neutral species which undergoes protonation at O6 to restore the one electron reduced guanine to its original charge state at 20 K. The species starts as a molecular cation (N7 protonated guanine) and on one electron reduction the neutral intermediate formed which protonates at O6 restoring the species to the initial charge state, as a radical cation. The authors232 found that this is a general mechanism for one electron reduced species which undergo protonation to restore the original charge state and one electron oxidized species which undergo deprotonation to the original charge state within the crystalline lattice.233 This mechanism was initially proposed by Bernhard.234 The crystalline coulombic stabilization is likely a factor; however, in aqueous solutions the same processes take place because the electron adducts and one electron oxidized species become far more basic and acidic, respectively.
Wang and Sevilla235 investigated various A, T, G and C systems and observed irreversible protonations at carbon at every base under specific conditions. For example they observed C8 protonated guanine radical anion (G(C8H)• by ESR of γ-irradiated frozen aqueous solutions of dGMP•dCMP, PolyG•polyC and poly[dG]•poly[dC] samples. However for DNA systems with A and T present no protonation of the guanine radical anion is found.
The reactions of hydrated electrons with guanosine (Guo), 2′-deoxyguanosine (2′-dG) and 1-methylguanosine were studied by pulse radiolysis in aqueous solution with optical and conductometric detection.236 At pH 7, the reduction of dG was completed in < 0.1 microseconds and protonation initially occurred at N7 of dG•−, which was observed by an increase in the absorption spectra at 300 nm. Tautomerization, from N7 to C8, resulting in the irreversibly protonated neutral radical of dG•−, was proposed to result as shown in eqs. 10-12.
| (10) |
| (11) |
The OH− produced in eq. (11) is neutralized by H+ to form H2O as confirmed from the conductance measurement with k=1.4 × 1011 M−1 s−1. At pH < 6, dG(C8H)• is further protonated to give the radical cation, eq. (12)
| (12) |
The rate of nitrogen protonation at 300 nm (eq. 11) was also monitored in D2O and KIE = 8.0 was observed, which demonstrated the rate determining step was reaction 11 for the proton transfer to C8 of guanine.
The valence bound radical anion of guanine was also studied by Bowen and his collaborators, using photoelectron spectroscopy and theory.237-239 Figure 26 shows the photoelectron spectrum of G•− measured with 3.493 eV photons. The spectrum of G•−, shown in Figure 26, has two maxima, one ranging from 0.8 – 1.1 eV and the other steeply increases from 2.4 until 3.2 eV. This broad band nature of the spectra suggested the presence of several tautomers of G•− that arise as a result of intramolecular proton transfer to carbon sites.237 The presence of tautomers was supported by theoretical calculations at the B3LYP and CCSD(T) level of theories.237-239 This study provides good evidence for electron induced internal proton transfer reactions. The calculated photoelectron energies at 1.6 and 2.4 eV are for the two tautomer structures of G•− shown in Figure 26.
Figure 26.
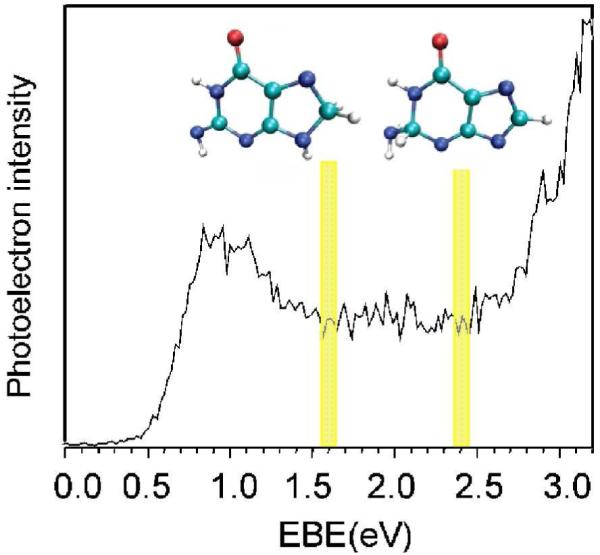
Photoelectron spectrum of G•− measured with 3.493 eV photons. The electron induced proton transferred structures (tautomers of G•−) present at 1.6 and 2.4 eV are shown. At 1.6 eV, proton transfers from NH2 group to C8 and at 2.4 eV proton transfers from N9 to C2 of guanine. For numbering, see Figure 11. (Figure 1 of ref. 237 reproduced with permission. Copyright 2007 American Chemical Society. Structural figures taken from Figure 2 of ref. 237).
Very recently, one electron reduction of 8-bromoisoguanosine and 8-bromoxanthosine anion were investigated in aqueous media using pulse radiolysis techniques and theory.240 From theory and experiments, it was proposed that electron induced protonation of 8-bromoisoguanosine and 8-bromoxanthosine (monoanion) occurs through ET-PT (stepwise) and concerted PCET mechanisms.
5.1.1. Proton Transfer in One Electron Reduced G-C Base Pair
Intra-base pair proton transfer, induced by electron attachment to G-C base pairs, has been observed by pulse radiolysis241 in solution and photoelectron spectroscopy242 gas phase experiments. These experimental works were strongly supported by a number of theoretical196,199,243-246 studies.
Tagawa and coworkers241 measured the dynamics of one electron reduced single- and double-stranded oligonucleotides containing G and C sequences spectroscopically via nanosecond pulse radiolysis in aqueous solution. The addition of a hydrated electron (eaq−) to the oligonucleotide results in transfer to cytosine in a G-C base pair. The cytosine anion radical (C•−) rapidly protonates at N3 by transfer from N1 of the hydrogen bonded guanine. The equilibrium constant for this reaction was estimated to be 3.16 × 103 suggesting a near complete proton transfer.
The gas phase photoelectron spectra of the radical anion of 9-methylguanine-1-methylcytosine (MGMC•−) was recorded using 3.94 eV photons by Bowen and coworkers,242 see Figure 27. The photoelectron spectra of MGMC•− shows a broad peak in the energy range ca. 1.4 to 2.5 eV having maximum intensity near 2 eV, see Figure 27. The broad nature of the spectra shows the presence of several tautomers of MGMC•−. From theory, the presence of two types of MGMC•− were characterized: (i) normal MGMC•− in the Watson-Crick conformation, shown left in Figure 27. and (ii) proton transferred from N1 atom of guanine to N3 atom of cytosine, shown on the right in Figure 27. This experiment establishes the involvement of electron induced proton transfer events in the G-C base pair.
Figure 27.

Photoelectron spectrum of the 9-methylguanine-1-methylcytosine radical anion (MGMC•−) recorded with 3.49 eV photons. Left structure is MGMC•− in Watson-Crick conformation. Right structure is due to proton transfer from N1 guanine to N3 site of cytosine. (Figure 3 of ref. 242 reproduced with permission. Copyright 2009 American Chemical Society. Structural Figures taken from Figure 5 of ref. 242).
A number of detailed theoretical investigations have treated the proton transfer reaction in the one electron reduced G-C base pair.196,199,243-246 In each of these studies, the PES for N1-H proton transfer form guanine to N3 of cytosine was calculated and the proton transfer reaction was found to be exothermic by several kcal/mol. The PES for proton transfer along with the SOMO plots, calculated by B3LYP/DZP++ method by Schaefer and coworkers244 is shown in Figure 28. Experiment and theory, thus support the involvement of electron induced proton transfer (PCET) in one electron reduced G-C base pair.
Figure 28.

Plots of the singly occupied molecular orbitals (SOMOs) for the nucleoside pair dG-dC•− and proton transferred dG(−H)−-dC(+H)•. The dG(−H)−-dC(+H)• is 2.3 kcal/mol more stable than the dG-dC•− calculated by B3LYP/DZP++ method.244 (Figure 1 (in part) and Figure 4 (SOMO plots) reproduced with permission from ref. 244. Copyright 2007, American Institute of Physics).
5.2. Cytosine and Thymine
Neutral radicals of cytosine and thymine (shown in Figure 29) are formed on protonation of one electron reduced cytosine and thymine bases and their 2′-nucleosides and 2′-nucleotides. Each of these species have been detected by ESR,213 - 215,247-249 and pulse radiolysis41,241,250,251 experiments. Pulse radiolysis41,241 and conductance measurements250 have shown that in the pH range 6 – 13, one electron reduction of cytosine results in the N3 protonated cytosine radical (C(N3H)•). The N3 protonated species has a pKa near 13; so it is not surprising that the formation of C(N3H)• from C•− is rapid ≤ 20 nanoseconds as shown in Figure 29. EPR/ENDOR spectroscopy248b of X-irradiated cytosine monohydrate (C:H2O) single crystals at 10 K has also shown the presence of C(N3H)•. In addition, the formation of the C6 protonated species has been observed in ESR experiments which likely stems from C(N H)•.235
Figure 29.

Neutral radical formation from one electron reduced cytosine and thymine bases (R=H, deoxyribose (phosphate)). The water acts as a proton (H+) donor. The site of protonation is shown by pink circle.
T(O4H)• and T(C6H)• radicals have been detected by ESR213-215,247,248a and pulse radiolysis41,241 experiments on protonation of one electron reduced thymine, dT and dTMP as shown in Figure 29. The protonation at O4 is reversible,41,241 whereas the protonation at C6 is irreversible.213-215,247,248a The spectra recorded at 1 microseconds after the pulse241 showed the presence of T(O4H)• and T•− at pHs 3.5 and 10.5. The transiently formed T•− was directly protonated by the water and at pH 7, the rate constant for the O4 protonation was estimated to be 4.3 × 105 s−1.241 Proton transfer from water in the X-irradiated thymine monohydrated (T:H2O) crystal was also proposed from electron paramagnetic resonance (EPR) spectroscopy.252 These early studies based on the pulse radiolysis and ESR/EPR experiments also support that protonation of cytosine and thymine is induced by the attachment of an excess electron to the bases. Both cytosine and thymine anion radicals undergo irreversible protonation at carbon (shown for thymine only) as a slower step than the reversible protonation at the heteratom.235
5.2.1. PCET in Excited State of Pyrene-modified Pyrimidine Nucleosides
Steady state fluorescence and femtosecond time resolved transient absorption spectroscopy253 have been used to investigate the DNA-mediated reductive electron transfer employing 5-(Pyren-1-yl)-2′-deoxyuridine (PydU) and 5-(Pyren-1-yl)-2′-deoxycytidine (PydC), structure shown in Figure 30, as model nucleosides for DNA. The excitation of the pyrene moiety in PydU and PydC leads to electron transfer (charge transfer excited state) from the pyrene moiety to uracil and cytosine yielding Py•+ and U•− and C•− formation.253,254 The excitation was carried out in the presence of acetonitrile, MeCN and in water at different pH to investigate the protonation state of the dU•− and dC•−. The result showed that dC•− was protonated spontaneously, see scheme 5, even in the basic aqueous solution within picoseconds as expected from its strongly basic pKa of 13. The fast protonation of dC directly from the local excited (LE) state (Py*dC + H+) occurs through concerted electron and proton transfer (PCET) process. However, the thermodynamically unstable CT excited state (Py•+dC•− + H+) occurs at high pH by deprotonation of Py•+dC(H)• and clearly lies higher in energy than the initial LE state.
Figure 30.

Structures of 5-(Pyren-1-yl)-2′-deoxyuridine (PydU) and 5-(Pyren-1-yl)-2′-deoxycytidine (PydC).253
Scheme 5.
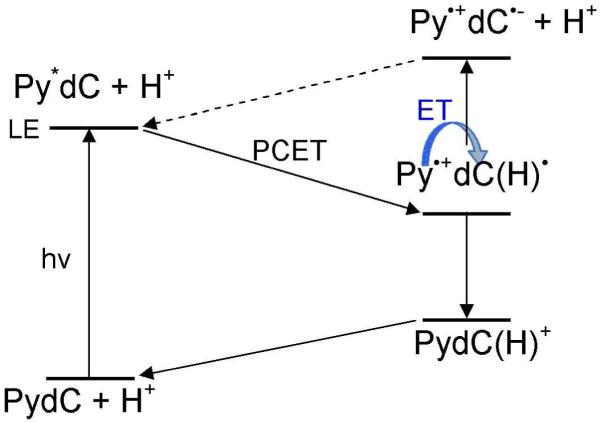
PCET process in excited state of PydC. Scheme redrawn from scheme 2 of ref. 253.
The effect of base sequence on excess electron transfer (ET) in various polynucleotide duplexes and salmon sperm DNA were studied in frozen glassy aqueous solutions (7 M LiBr in D2O) of the duplexes polydAdT•polydAdT and polydIdC•polydIdC (dI= inosine) randomly intercalated with mitoxantrone (MX) by Sevilla and coworkers.122,255 In this study only a modest kinetic isotope effect was observed for DNA on the electron transfer rate 0.83 ± 0.1 Å−1 (H2O) vs 0.92 ± 0.1 Å−1 (D2O). However, the rate of electron transfer in polydAdT•polydAdT 0.7 Å−1 was substantially faster than in polydIdC•polydIdC 1.4 Å−1. This was attributed to the fact that proton transfer readily occurs from I to C anion radical and greatly slows the electron transfer process; however, for the A-T polynucleotide no such transfer process occurs and the electron transfer rate is unhindered. This result was supported by theoretical calculations that show the proton transfer in I-C base pair anion radical from I to C is barrierless.199
Proton transfer reactions in radical anions of 1-methylcytosine and uracil in gas phase and in solvation were also investigated by Harańczyk et al.256-258 using theory. In these studies, the relative stabilities of various tautomers of 1-methylcytosine and uracil radical anions were investigated using DFT, MP2 and CCSD(T) methods. On the basis of stabilities, the presence of the most stable tautomer, formed by intramolecular proton transfer, was proposed to be present. For radical anion of uracil complexed with alcohols, it was found that the excess electron attachment to the complex (uracil:alcohol) induces a barrier free proton transfer from the OH group of alcohol to the O4 atom of uracil. This theoretical prediction was supported by the photoelectron spectra.
5.2.2. Protonation of One Electron Reduced Adenine and A-T Base Pair
Pulse radiolysis and optical dc-conductivity measurements found that attachment of an excess hydrated electron (eaq−) to adenosine (Ado) and dAMP is followed by proton transfer to the adenine from water within a nanosecond, as shown in Scheme 6.259,260 At pH 7, the nitrogen protonated dA(N7H)• is converted into carbon protonated dA(C8H)• by an irreversible tautomerization with a rate constant 2 × 106 M−1 s−1.260 This result shows that electron attachment to adenine is coupled with the intermolecular proton transfer from solvent that could be described as PCET.
Scheme 6.

Hydrated electron (eaq−) induced protonation of the adenine base (A) in adenosine at nitrogen and carbon sites.260
Recently, barrier free proton transfer induced by an excess electron attachment to monomers of adenine, adenosine-5′-monophosphate and 2′-deoxyadenosine-5′-monophosphate and adenine complexed with formic acid was investigated using photoelectron spectroscopy in the gas phase along with theory.261-264 Further, the proton transfer from adenine to thymine in one electron reduced A-T base pair was proposed from the photoelectron spectra and the theoretical calculation,265 though the predicted base pair structure does not correspond to the canonical Watson-Crick base pair. This is not surprising considering these are gas phase systems.
5.2.3 Electron Induced DNA-Strand Break Formation in Excited States
Recent advances in several laboratories have shown that the interaction of low energy electrons (LEE) with DNA is of significant biological importance. Since the discovery by Sanche and coworkers that LEEs < 4 eV (below the ionization threshold of DNA) can induce single- and double-strand breaks,20,266,267 the problem of elucidating the mechanism of strand breaks in DNA has been extensively treated in theoretical investigations268-273 and by experiment.20-27,266,267 From experiments and theoretical calculations, two mechanisms were proposed. In the first, LEEs are initially captured by the base into their π• MOs to create a shape resonance (transient negative ion (TNI)) and the electron is transferred to the sugar-phosphate region leading to strand breaks. The second mechanism is direct attachment to the sugar phosphate backbone in higher energy empty (virtual) orbitals leading to dissociative electron attachment (DEA) and associated strand breaks.
The ground state theoretical calculations, considering 5′-thymidine monophosphate (5′-dTMPH) radical anion as a model of DNA, calculated the PES for the strand breaks (C5′-O5′ bond). These works report the barrier height for C5′-O5′ bond dissociation in the range 10 – 19 kcal/mol, showing a quite small rate (1010 to 10−4 s−1)269 is expected for C5′-O5′ bond cleavage. Recently, the excited state of the TNI of 5′-dTMPH were calculated using BHandHLYP/6-31G* method272 and the excited state potential energy surfaces for C5′-O5′ bond dissociation were calculated as shown in Figure 31.
Figure 31.

Lower curve: Potential energy surface (PES) of the 5′-dTMPH transient negative ion (TNI); calculated in the neutral optimized geometry of 5′-dTMPH with C5′-O5′ bond elongation. SOMO is shown at selected points. Upper curves: calculated vertical excitation energies of the radical anion at each point along the PES, MOs involved in excitations are also shown. Energies and distances are given in eV and Å, respectively. The lowest ππ*state (triangles) and lowest πσ* states (square) are shown. (Figure reproduced with permission from ref. 272. Copyright 2008 American Chemical Society.)
In the calculation, the negative charge of the phosphate group of 5′-dTMPH was neutralized by protonating one of the oxygens of the phosphate group. From Figure 31, it is evident that at excitation energy 1.4 eV, electron transfers from thymine to PO4 region and this transition is πσ* in nature as suggested by the first mechanism proposed above. This transition is dissociative in nature and is predicted to lead to facile formation of strand breaks by cleavage of the C5′-O5′ bond, see Figure 31.
A more complex mechanism involving an excess electron attachment to base followed by a proton attachment to the base and the subsequent attachment of another electron inducing strand breaks and base release is also proposed both by Dąbkowska et al.231, 274 and Gu et al.231, 275 using MPW1K/6-31+G** and B3LYP/DZP++ level of theories. 3′-dCMP and 5′-dCMP were considered as models for the calculations.
6. Overview and Conclusion
The exposure of high energy radiation to DNA initially forms ion radicals within DNA that quickly undergo proton transfer reactions. The resultant species undergo transfer within DNA by either (i) proton coupled hole or (ii) proton coupled electron transfer. These species transfer to sites where products are subsequently formed. The initial proton transfers occur because formation of holes or addition of an electron to nucleobase strongly affects the pKa’s of the nucleobases changing them by orders of magnitude. Oxidized DNA bases become substantially more acidic while reduced DNA bases become more basic often by more than 5 pKa units.40 This acquired acid/base character of DNA bases gives rise to protonation/deprotonation reactions. Radiation induced proton transfer often provides the energetic driving force for electron or hole transfer within DNA and between hydrogen bonded base pairs in DNA. Remarkably, as described in this review, the reactions observed in aqueous solutions, crystalline solids and in the gas phase are often quite similar owing to the large driving forces involved after ion radical formation. After the PCET process is complete the new stabilized species is clearly resistant to subsequent transfer unless the proton transfer process is reversible or a subsequent more exergonic reaction takes place. For example, within DNA, proton transfer processes may hinder both hole and electron transfer. For the hole which localizes predominantly on guanine in the GC radical cation base pair a equilibrium between G•+-C and G(N1-H)•-C(+H+) is established with a slight favoring of the proton transferred species (G(N1-H)•-C(+H+)) as the free energy difference between the two forms is small. At room temperature, an equilibrium between near equal amounts of G•+-C and G(N1-H)•-C(+H+) is established, which allows hole to transfer. Excess electrons in DNA localize at both T and C but proton transfer only occurs in the G-C−• base pair and strongly favors the proton transfer state, G(N1-H)−-CH•. This results in a redistribution of the initial distribution of the excess electron from both T and C to predominantly on C. This should create a strong base sequence dependence on electron transfer favoring A-T sequences and limits rapid transfer rates in G-C sequences to those processes and distances which can take place before proton transfer. As described in this review, these and other proton coupled electron transfer processes are common in DNA after radical ion formation.
Finally, we note that that there is a growing awareness that excited states combined with ion radicals are potent initiators of chemical events which include PCET. Excited states themselves create driving forces for hole and electron transfer which when combined with existing holes and excess electrons potentiate rapid reactive events such as hole transfer from the DNA base guanine to the sugar phosphate backbone discussed earlier. For the excess electron pathway, excited states are significant as well. For example low energy electron interactions with DNA can create in effect an excited state anion radical if the attachment is in an MO above the LUMO. Calculations described above show that excited ion radical species are especially prone to inducing rapid cleavage of the DNA backbone.
Clearly PCET events initiated by radiation damage to DNA have provided and will continue to provide a fertile ground for research seeking to understand the initial complex processes which are critical to understanding of the ultimate biological effects of radiation.
Figure 23.

Deprotonation of one electron oxidized adenine.
Scheme 4.
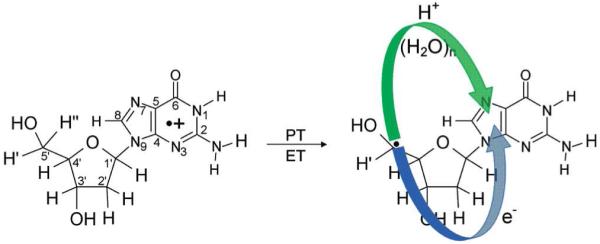
Formation of the Neutral Sugar Radical (C5′•) through a Proton Coupled Electron Transfer Mechanism in 2′-Deoxyguanosine radical cation (dG•+).a
aThe proton transfers from C5′ to N7 of guanine through waters and results in electron transfer from C5′ to guanine yielding the product, (dG(•C5′, N7-H+) + 7H2O). (Figure reproduced with permission from ref. 228. Copyright 2009 American Chemical Society.)
7. Acknowledgments
This work was supported by the NIH NCI Grant No. R01CA045424. The authors thank Dr. Amitava Adhikary for helpful discussions and suggestions.
8. Abbreviations
- 2AP
2-aminopurine
- 2Apr
2-aminopurine ribose
- 2′-dGd
8-deutero-2′-deoxyguanosine
- 2,6-AQDS
Anthraquinone-2,6-disulfonic acid
- 5′-dTMP
5′-thymidine monophosphate
- A
Adenine
- A-T
Adenine-Thymine Base Pair
- BET
Back electron transfer
- bpy
2,2′-bipyridine
- C
Cytosine
- C(N3H)•
N3 protonated cytosine radical
- CASSCF
Complete active space self-consistent-field
- CT
Charge transfer
- DFT
Density functional theory
- dG
Deoxyguanosine
- dG•+
One electron oxidized 2′-deoxyguanosine or 2′-deoxyguanosine radical cation
- dI
Inosine
- dppz
Dipyrido[3,2-a:2′,3′-c]phenazine
- EA
Electron affinity
- eaq−
Hydrated electron
- ENDOR
Electron nuclear double resonance
- EPR
Electron paramagnetic resonance
- ESR
Electron spin resonance
- ET
Electron transfer
- ETS
Electron transmission spectroscopy
- fapyG
2,6-diamino-4-hydroxy-5-formamidopyrimidine
- G
Guanine
- G•+
Guanine Radical Cation
- G-C
Guanine-Cytosine Base Pair
- G•+-C
One electron oxidized Guanine-Cytosine Base Pair
- Gd
8-deuteroguanine
- G(N1-H)•
Guanyl radical
- H−
Hydride
- HAT
Hydrogen atom transfer
- HF
Hartree-Fock
- HOMO
Highest occupied molecular orbital
- IP
Ionization potential
- KIE
Kinetic isotope effect
- LEE
Low energy electrons
- LUMO
Lowest unoccupied molecular orbital
- MGMC
9-methylguanine-1-methylcytosine
- MLCT
Metal-Ligand Charge Transfer
- MO
Molecular orbital
- MSEPT
Multisite electron and proton transfer
- MX
Mitoxantrone
- NI
Naphthalimide
- PCET
Proton coupled electron transfer
- PCHT
Proton coupled hole transfer
- PES
Potential energy surface
- ps
Picosecond
- PT
Proton transfer
- PTZ
Phenothiazine
- PydC
5-(Pyren-1-yl)-2′-deoxycytidine
- PydU
5-(Pyren-1-yl)-2′-deoxyuridine
- SOMO
Singly occupied molecular orbital
- T
Thymine
- TAP
1,4,5,8-tetraazaphenanthrene
- T(C6H)•
C6 protonated thymine radical
- T(O4H)•
O4 protonated thymine radical
- TNI
Transient negative ion
- TS
Transition state
- UCH2•
Methyl deprotonated thymine radical cation
Biographies

Michael D. Sevilla is currently Distinguished Professor of Chemistry at Oakland University. He received a BS in Chemistry from San Jose State College, a Ph.D. in Physical Chemistry from the University of Washington in 1967. After a postdoctoral year at University of Washington and two years with Atomics International Corporation he joined Oakland University in 1970. He has been Chair of the Chemistry Department at Oakland University, 1997-2003, Associate Editor, Radiation Research, 2003-2006 and President of the Radiation Research Society 2005-2006. Professor Sevilla is a well known radiation chemist and has focused his research on free radical reactions induced by radiation damage to DNA and other biomolecules employing experimental approaches such as electron spin resonance spectroscopy and theoretical approaches such as density functional theory. Recent efforts in collaboration with Dr. Kumar have elucidated the role of excited states of DNA ion radicals in the formation of DNA damage. He is a member of the AAAS, the ACS and the Radiation Research Society.

Anil Kumar received his Ph. D. in Physics from the Department of Physics, Banaras Hindu University, Varanasi, India. His Ph. D. dissertation investigated the structure, properties and specific molecular interactions of DNA and its constituents using quantum chemical methods. He was awarded a position as Pool Scientist from Council of Scientific and Industrial Research (CSIR), New Delhi, India, in 1997. Later he did his postdoctoral work at the Department of Chemistry, University of Hannover, Germany. He was a visiting scientist at the Department of Molecular Biophysics, German Cancer Research Institute, DKFZ, Heidelberg, Germany. Presently, he is working as a research associate in the group of Prof. M. D. Sevilla, at the Department of Chemistry, Oakland University. His research interests are to explore the mechanism of DNA damage, DNA ion radical formation, charge transfer process and involvement of excited states for producing DNA strand breaks employing computational chemistry approaches.
9. References
- (1).Russell P. iGenetics. Benjamin Cummings; New York: 2001. [Google Scholar]
- (2).Saenger W. Principles of Nucleic Acid Structure. Springer-Verlag; New York: 1984. [Google Scholar]
- (3).von Sonntag C. The chemical basis of radiation biology. Taylor and Francis; London, New York, Philadelphia: 1987. [Google Scholar]
- (4).Swiderek P. Angew. Chem. Int. Ed. 2006;45:4056. doi: 10.1002/anie.200600614. and references therein. [DOI] [PubMed] [Google Scholar]
- (5).Turecek F. Adv. Quantum. Chem. 2007;52:89. [Google Scholar]
- (6).Lukin M, de los Santos C. Chem. Rev. 2006;106:607. doi: 10.1021/cr0404646. [DOI] [PubMed] [Google Scholar]
- (7).Becker D, Sevilla MD. Adv. Radiat. Biol. 1993;17:121. [Google Scholar]
- (8).Tomkinson AE, Vijayakumar S, Pascal JM, Ellenberger T. Chem. Rev. 2006;106:687. doi: 10.1021/cr040498d. [DOI] [PubMed] [Google Scholar]
- (9).Close DM. In: Radiation Induced Molecular Phenomena in Nucleic Acids. Shukla MK, Leszczynski J, Leszczynski J, editors. Vol. 5. Springer Science + Business Media B.V.; 2008. pp. 493–529. under Challenges and Advances in Computational Chemistry and Physics. [Google Scholar]
- (10).Swarts SG, Sevilla MD, Becker D, Tokar CJ, Wheeler KT. Radiat. Res. 1992;129:333. [PubMed] [Google Scholar]
- (11).Li X, Sevilla MD. Adv. Quantum. Chem. 2007;52:59. [Google Scholar]
- (12).Becker D, Adhikary A, Sevilla MD. In: Charge Migration in DNA. Chakraborty T, editor. Springer-Verlag; Berlin, Heidelberg: 2007. pp. 139–175. [Google Scholar]
- (13).Kumar A, Sevilla MD. In: Radiation Induced Molecular Phenomena in Nucleic Acids. Shukla MK, Leszczynski J, Leszczynski J, editors. Vol. 5. Springer Science + Business Media B.V.; 2008. pp. 577–617. under Challenges and Advances in Computational Chemistry and Physics. [Google Scholar]
- (14).Kumar A, Sevilla MD. In: Radical and Radical Ion Reactivity in Nucleic Acid Chemistry. Greenberg M, editor. John Wiley & Sons, Inc.; 2010. pp. 1–40. [Google Scholar]
- (15).Becker D, Sevilla MD. In: Royal Society of Chemistry Specialist Periodical Report. Gilbert BC, Davies MJ, Murphy DM, editors. Vol. 21. 2008. p. 33. Electron Paramagnetic Resonance. 21. [Google Scholar]
- (16).Yokoya A, Shikazono N, Fujii K, Urushibara A, Akamatsu K, Watanabe R. Radiat. Phys. Chem. 2008;77:1280. [Google Scholar]
- (17).Sevilla MD, Becker D, Yan M, Summerfield SR. J. Phys. Chem. 1991;95:3409. [Google Scholar]
- (18).Faraggi M, Ferradini C, JayGerin JP. New J. Chem. 1995;19:1203. [Google Scholar]
- (19).International Commission on Radiation Units and Measurements . ICRU Report No. 31. ICRU; Washington, DC: 1979. [Google Scholar]
- (20).Boudaïffa B, Cloutier P, Hunting D, Huels MA, Sanche L. Science. 2000;287:1658. doi: 10.1126/science.287.5458.1658. [DOI] [PubMed] [Google Scholar]
- (21).Sanche L. Chem. Phys. Lett. 2009;474:1. [Google Scholar]
- (22).Sanche L. In: Radical and Radical Ion Reactivity in Nucleic Acid Chemistry. Greenberg M, editor. John Wiley & Sons, Inc.; 2010. pp. 239–293. [Google Scholar]
- (23).Zheng Y, Cloutier P, Hunting DJ, Sanche L, Wagner JR. J. Am. Chem. Soc. 2005;127:16592. doi: 10.1021/ja054129q. [DOI] [PubMed] [Google Scholar]
- (24).Ptasińska S, Sanche L. Phys. Chem. Chem. Phys. 2007;9:1730. doi: 10.1039/b616619a. [DOI] [PubMed] [Google Scholar]
- (25).Sulzer P, Ptasińska S, Zappa F, Mielewska B, Milosavljevic AR, Scheier P, Märk TD, Bald I, Gohlke S, Huels MA, Illenberger E. J. Chem. Phys. 2006;125:044304. doi: 10.1063/1.2222370. [DOI] [PubMed] [Google Scholar]
- (26).(a) Bald I, Dąbkowska I, Illenberger E. Angew. Chem. Int. Ed. 2008;47:8518. doi: 10.1002/anie.200803382. [DOI] [PubMed] [Google Scholar]; (b) Baccarelli I, Gianturco FA, Grandi A, Sanna N, Lucchese RR, Bald I, Kopyra J, Illenberger E. J. Am. Chem. Soc. 2007;129:6269. doi: 10.1021/ja070542h. [DOI] [PubMed] [Google Scholar]
- (27).Bernhard WA. J. Phys. Chem. 1989;93:2187. [Google Scholar]
- (28).Hush NS, Cheung AS. Chem. Phys. Lett. 1975;34:11. [Google Scholar]
- (29).Orlov VM, Smirnov AN, Varshavsky Ya. M. Tetrahedron Lett. 1976;17:4315. [Google Scholar]
- (30).Yang X, Wang X-B, Vorpagel ER, Wang L-S. Proc. Natl. Acad. Sci. USA. 2004;101:17588. doi: 10.1073/pnas.0405157101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Steenken S, Jovanovic SV. J. Am. Chem. Soc. 1997;119:617. [Google Scholar]
- (32).Burrows CJ, Muller JG. Chem. Rev. 1998;98:1109. doi: 10.1021/cr960421s. [DOI] [PubMed] [Google Scholar]
- (33).(a) Cadet J, Douki T, Ravanat J-L. Acc. Chem. Res. 2008;41:1075. doi: 10.1021/ar700245e. [DOI] [PubMed] [Google Scholar]; (b) Cadet J, Douki T, Gasparutto D, Ravanat J-L. Mutation Research. 2003;531:5. doi: 10.1016/j.mrfmmm.2003.09.001. [DOI] [PubMed] [Google Scholar]
- (34).Swarts SG, Gilbert DC, Sharma KK, Razskazovskiy Y, Purkayastha S, Naumenko KA, Bernhard WA. Radiat. Res. 2007;168:367. doi: 10.1667/RR1058.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Pogozelski WK, Tullius TD. Chem. Rev. 1998;98:1089. doi: 10.1021/cr960437i. [DOI] [PubMed] [Google Scholar]
- (36).(a) Becker D, Bryant-Friedrich A, Trzasko C, Sevilla MD. Radiat. Res. 2003;160:174. doi: 10.1667/rr3037. [DOI] [PubMed] [Google Scholar]; (b) Shukla LI, Pazdro R, Huang J, DeVreugd C, Becker D, Sevilla MD. Radiat. Res. 2004;161:582. doi: 10.1667/rr3167. [DOI] [PubMed] [Google Scholar]; (c) Shukla LI, Pazdro R, Becker D, Sevilla MD. Radiat. Res. 2005;163:591. doi: 10.1667/rr3347. [DOI] [PubMed] [Google Scholar]
- (37).Adhikary A, Kumar A, Sevilla MD. Radiat. Res. 2006;165:479. doi: 10.1667/rr3563.1. [DOI] [PubMed] [Google Scholar]
- (38).Bernhard WA. In: Radical and Radical Ion Reactivity in Nucleic Acid Chemistry. Greenberg M, editor. John Wiley & Sons, Inc.; 2010. pp. 41–68. [Google Scholar]
- (39).(a) Sharma KK, Purkayastha S, Bernhard WA. Radiat. Res. 2007;167:501. doi: 10.1667/RR0847.1. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Purkayastha S, Milligan JR, Bernhard WA. Radiat. Res. 2006;166:1. doi: 10.1667/RR3585.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Steenken S. Chem. Rev. 1989;89:503. [Google Scholar]
- (41).Steenken S, Telo JP, Novais HM, Candeias LP. J. Am. Chem. Soc. 1992;114:4701. [Google Scholar]
- (42).Steenken S. Biol. Chem. 1997;378:1293. [PubMed] [Google Scholar]
- (43).Tommos C, Tang X-S, Warncke K, Hoganson CW, Styring S, McCracken J, Diner BA, Babcock GT. J. Am. Chem. Soc. 1995;117:10325. doi: 10.1007/BF00020428. [DOI] [PubMed] [Google Scholar]
- (44).Okamura MY, Feher G. Annual Rev. Biochem. 1992;61:861. doi: 10.1146/annurev.bi.61.070192.004241. [DOI] [PubMed] [Google Scholar]
- (45).Hoganson CW, Babcock GT. Science. 1997;277:1953. doi: 10.1126/science.277.5334.1953. [DOI] [PubMed] [Google Scholar]
- (46).Diner BA, Babcock GT. In: Advances in Photosynthesis: The Light Reactions. Ort DR, Yocum CF, editors. Vol. 4. Kluwer Academic Publishers; Dordrecht, The Netherlands: 1996. p. 213. [Google Scholar]
- (47).Blomberg MRA, Siegbahn PEM, Styring S, Babcock GT, Akermark B, Korall P. J. Am. Chem. Soc. 1997;119:8285. [Google Scholar]
- (48).Hoganson CW, Lydakis-Simantiris N, Tang X-S, Tommos C, Warncke K, Babcock GT, Diner BA, McCracken J, Styring S. Photosynthesis Res. 1995;47:177. doi: 10.1007/BF00020428. [DOI] [PubMed] [Google Scholar]
- (49).Babcock GT, Wikstrom M. Nature. 1992;356:301. doi: 10.1038/356301a0. [DOI] [PubMed] [Google Scholar]
- (50).Malmstrom BG. Acc. Chem. Res. 1993;26:332. [Google Scholar]
- (51).Siegbahn PEM, Eriksson L, Himo F, Pavlov M. J. Phys. Chem. B. 1998;102:10622. [Google Scholar]
- (52).Shafirovich V, Geacintov NE. Top. Curr. Chem. 2004;237:129. [Google Scholar]
- (53).Thorp HH. Top. Curr. Chem. 2004;237:159. [Google Scholar]
- (54).Huynh My H. V., Meyer TJ. Chem. Rev. 2007;107:5004. doi: 10.1021/cr0500030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Meyer TJ, Huynh My H. V., Thorp HH. Angew. Chem. Int. Ed. 2007;46:5284. doi: 10.1002/anie.200600917. [DOI] [PubMed] [Google Scholar]
- (56).Thorp HH. Chem. Abstr. Inorg. Chem. 1991;3:171. [Google Scholar]
- (57).Lui W, Thorp HH. Excited-State Proton Transfer Reactions of Multiply-Bonded Ligands. Advances in Transition Metal Coordination Chemistry. 1:187. [Google Scholar]
- (58).Roth JP, Yoder JC, Won T–J, Mayer JM. Science. 2001;294:2524. doi: 10.1126/science.1066130. [DOI] [PubMed] [Google Scholar]
- (59).Mayer JM, Rhile IJ. Biochim. Biophys. Acta Bioenerg. 2004;1655:51. doi: 10.1016/j.bbabio.2003.07.002. [DOI] [PubMed] [Google Scholar]
- (60).Tommos C, Babcock GT. Acc. Chem. Res. 1998;31:18. [Google Scholar]
- (61).Stubbe J, Nocera DG, Yee CS, Chang MCY. Chem. Rev. 2003;103:2167. doi: 10.1021/cr020421u. [DOI] [PubMed] [Google Scholar]
- (62).Boussicault F, Robert M. Chem. Rev. 2008;108:2622. doi: 10.1021/cr0680787. [DOI] [PubMed] [Google Scholar]
- (63).Costentin C. Chem. Rev. 2008;108:2145. doi: 10.1021/cr068065t. [DOI] [PubMed] [Google Scholar]
- (64).Cukier RI, Nocera DG. Annu. Rev. Phys. Chem. 1998;49:337. doi: 10.1146/annurev.physchem.49.1.337. [DOI] [PubMed] [Google Scholar]
- (65).Hammes-Schiffer S, Iordanova N. Biochim. Biophys. Acta. 2004;1655:29. doi: 10.1016/j.bbabio.2003.07.009. [DOI] [PubMed] [Google Scholar]
- (66).Hammes-Schiffer S. ChemPhysChem. 2002;3:33. doi: 10.1002/1439-7641(20020118)3:1<33::AID-CPHC33>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- (67).Hammes-Schiffer S. Acc. Chem. Res. 2009;42:1881. doi: 10.1021/ar9001284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Hammes-Schiffer S. Acc. Chem. Res. 2001;34:273. doi: 10.1021/ar9901117. [DOI] [PubMed] [Google Scholar]
- (69).Hammes-Schiffer S. Acc. Chem. Res. 2006;39:93. doi: 10.1021/ar040199a. [DOI] [PubMed] [Google Scholar]
- (70).Hammes-Schiffer S. Proton-coupled electron transfer. In: Balzani V, editor. Electron Transfer in Chemistry. Vol. I. Principles, Theories, Methods and Techniques. I. Wiley-VCH; Weinheim: 2001. p. 189. [Google Scholar]
- (71).Chang CJ, Chang MCY, Damrauer NH, Nocera DG. Biochim. Biophys. Acta Bioenerg. 2004;1655:13. doi: 10.1016/j.bbabio.2003.08.010. [DOI] [PubMed] [Google Scholar]
- (72).Himo F, Siegbahn PEM. Chem. Rev. 2003;103:2421. doi: 10.1021/cr020436s. [DOI] [PubMed] [Google Scholar]
- (73).Lovell T, Hino F, Han WG, Noodleman L. Coord. Chem. Rev. 2003;238:211. [Google Scholar]
- (74).Eley DD, Spivey DI. Trans. Farad. Soc. 1962;58:411. [Google Scholar]
- (75).Eley DD, Leslie RB. Nature. 1963;197:898. [Google Scholar]
- (76).Murphy CJ, Arkin MR, Jenkins Y, Ghatlia ND, Bossmann SH, Turro NJ, Barton JK. Science. 1993;262:1025. doi: 10.1126/science.7802858. [DOI] [PubMed] [Google Scholar]
- (77).Priyadarshy S, Risser SM, Beratan DN. J. Phys. Chem. 1996;100:17678. [Google Scholar]
- (78).Beratan DN, Priyadarshy S, Risser SM. Chem. Biol. 1997;4:3. doi: 10.1016/s1074-5521(97)90230-1. [DOI] [PubMed] [Google Scholar]
- (79).(a) Debije MG, Milano MT, Bernhard WA. Angew. Chem. Int. Ed. 1999;38:2752. [PMC free article] [PubMed] [Google Scholar]; (b) Debije MG, Bernhard WA. J. Phys. Chem. B. 2000;104:7845. [Google Scholar]
- (80).Boon EM, Livingston AL, Chmiel NH, David SS, Barton JK. Proc. Natl. Acad. Sci. USA. 2003;100:12543. doi: 10.1073/pnas.2035257100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (81).DeRosa MC, Sancar A, Barton JK. Proc Natl Acad Sci USA. 2005;102:10788. doi: 10.1073/pnas.0503527102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (82).Boon EM, Ceres DM, Drummond TG, Hill MG, Barton JK. Nature Biotechnol. 2000;18:1096. doi: 10.1038/80301. [DOI] [PubMed] [Google Scholar]
- (83).Porath D, Bezryadin A, de Vries S, Dekker C. Nature. 2000;403:635. doi: 10.1038/35001029. [DOI] [PubMed] [Google Scholar]
- (84).Okamoto A, Tanaka K, Saito I. J. Am. Chem. Soc. 2004;126:9458. doi: 10.1021/ja047628k. [DOI] [PubMed] [Google Scholar]
- (85).Porath D, Cuniberti G, Felice R. Di Top. Curr. Chem. 2004;237:183. [Google Scholar]
- (86).Gorodetsky AA, Buzzeo MC, Barton JK. Bioconjugate Chem. 2008;19:2285. doi: 10.1021/bc8003149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (87).Diederichsen U. Angew. Chem., Int. Ed. 1997;36:2317. [Google Scholar]
- (88).Ratner M. Nature. 1999;397:480. doi: 10.1038/17232. [DOI] [PubMed] [Google Scholar]
- (89).Grinstaff MW. Angew. Chem., Int. Ed. 1999;38:3629. doi: 10.1002/(sici)1521-3773(19991216)38:24<3629::aid-anie3629>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- (90).Hall DB, Holmlin RE, Barton JK. Nature. 1996;382:731. doi: 10.1038/382731a0. [DOI] [PubMed] [Google Scholar]
- (91).Núñez ME, Hall DB, Barton JK. Chem. Biol. 1999;6:85. doi: 10.1016/S1074-5521(99)80005-2. [DOI] [PubMed] [Google Scholar]
- (92).Henderson PT, Jones D, Hampikian G, Kan YZ, Schuster GB. Proc. Natl. Acad. Sci. USA. 1999;96:8353. doi: 10.1073/pnas.96.15.8353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (93).Meggers E, Michel-Beyerle ME, Giese B. J. Am. Chem. Soc. 1998;120:12950. [Google Scholar]
- (94).Wagenknecht H-A. Angew. Chem., Int. Ed. 2003;42:2454. doi: 10.1002/anie.200301629. [DOI] [PubMed] [Google Scholar]
- (95).Pascaly M, Yoo J, Barton JK. J. Am. Chem. Soc. 2002;124:9083. doi: 10.1021/ja0202210. [DOI] [PubMed] [Google Scholar]
- (96).Boon EM, Barton JK. Curr. Opin. Struct. Biol. 2002;12:320. doi: 10.1016/s0959-440x(02)00327-5. [DOI] [PubMed] [Google Scholar]
- (97).Núñez ME, Barton JK. Curr. Opin. Chem. Biol. 2000;4:199. doi: 10.1016/s1367-5931(99)00075-7. [DOI] [PubMed] [Google Scholar]
- (98).Erkkila KE, Odom DT, Barton JK. Chem. Rev. 1999;99:2777. doi: 10.1021/cr9804341. [DOI] [PubMed] [Google Scholar]
- (99).Meggers E, Kusch D, Spichty M, Wille U, Giese B. Angew. Chem. Int. Ed. 1998;37:459. doi: 10.1002/(SICI)1521-3773(19980302)37:4<460::AID-ANIE460>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- (100).Giese B. Curr. Opin. Chem. Biol. 2002;6:612. doi: 10.1016/s1367-5931(02)00364-2. [DOI] [PubMed] [Google Scholar]
- (101).Giese B. Acc. Chem. Res. 2000;33:631. doi: 10.1021/ar990040b. [DOI] [PubMed] [Google Scholar]
- (102).Giese B. Annu. Rev. Biochem. 2002;71:51. doi: 10.1146/annurev.biochem.71.083101.134037. [DOI] [PubMed] [Google Scholar]
- (103).Bernhard K, Geimer J, Canle-Lopez M, Reynisson J, Beckert D, Gleiter R, Steenken S. Chem. Eur. J. 2001;7:4640. doi: 10.1002/1521-3765(20011105)7:21<4640::aid-chem4640>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- (104).Shukla LI, Adhikary A, Pazdro R, Becker D, Sevilla MD. Nucleic Acids Res. 2004;32:6565. doi: 10.1093/nar/gkh989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (105).Munk BH, Burrows CJ, Schlegel HB. Chem. Res. Toxicol. 2007;20:432. doi: 10.1021/tx060187t. [DOI] [PubMed] [Google Scholar]
- (106).Lewis FD, Liu X, Miller SE, Hayes RT, Wasielewski MR. J. Am. Chem. Soc. 2002;124:11280. doi: 10.1021/ja026751d. [DOI] [PubMed] [Google Scholar]
- (107).Lewis FD, Liu X, Liu J, Miller SE, Hayes RT, Wasielewski MR. Nature. 2000;406:51. doi: 10.1038/35017524. [DOI] [PubMed] [Google Scholar]
- (108).Lewis FD, Letsinger RL, Wasielewski MR. Acc. Chem. Res. 2001;34:159. doi: 10.1021/ar0000197. [DOI] [PubMed] [Google Scholar]
- (109).Lewis FD, Liu X, Liu J, Hayes RT, Wasielewski MR. J. Am. Chem. Soc. 2000;122:12037. [Google Scholar]
- (110).Schuster GB. Acc. Chem. Res. 2000;33:253. doi: 10.1021/ar980059z. [DOI] [PubMed] [Google Scholar]
- (111).Henderson PT, Jones D, Hampikian G, Kan Y, Schuster GB. Proc. Natl. Acad. Sci. U.S.A. 1999;96:8353. doi: 10.1073/pnas.96.15.8353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (112).Ly D, Kan Y, Armitage B, Schuster GB. J. Am. Chem. Soc. 1996;118:8747. [Google Scholar]
- (113).Saito I, Nakamura T, Nakatani K, Yoshioka Y, Yamaguchi K, Sugiyama H. J. Am. Chem. Soc. 1998;120:12686. [Google Scholar]
- (114).Sugiyama H, Saito I. J. Am. Chem. Soc. 1996;118:7063. [Google Scholar]
- (115).Prat F, Houk KN, Foote CS. J. Am. Chem. Soc. 1998;120:845. [Google Scholar]
- (116).Lewis FD, Liu X, Liu J, Hayes RT, Wasielewski MR. J. Am. Chem. Soc. 2000;122:12037. [Google Scholar]
- (117).Conwell EM, Basko DM. J. Am. Chem. Soc. 2001;123:11441. doi: 10.1021/ja015947v. [DOI] [PubMed] [Google Scholar]
- (118).Berlin YA, Burin AL, Ratner MA. J. Am. Chem. Soc. 2001;123:260. doi: 10.1021/ja001496n. [DOI] [PubMed] [Google Scholar]
- (119).Adhikary A, Khanduri D, Sevilla MD. J. Am. Chem. Soc. 2009;131:8614. doi: 10.1021/ja9014869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (120).Anderson RF, Wright GA. Phys. Chem. Chem. Phys. 1999;1:4827. [Google Scholar]
- (121).Anderson RF, Patel KB. J. Chem. Soc. Faraday Trans. 1991;87:3739. [Google Scholar]
- (122).Messer A, Carpenter K, Forzley K, Buchanan J, Yang S, Razskazovskii Y, Cai Z, Sevilla MD. J. Phys. Chem. B. 2000;104:1128. [Google Scholar]
- (123).Cai Z, Sevilla MD. Top. Curr. Chem. 2004;237:103. [Google Scholar]
- (124).Schwögler A, Burgdorf LT, Carell T. Angew.Chem. Int. Ed. 2000;39:3918. doi: 10.1002/1521-3773(20001103)39:21<3918::AID-ANIE3918>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- (125).Behrens C, Burgdorf LT, Schwögler A, Carell T. Angew.Chem. Int. Ed. 2002;41:1763. doi: 10.1002/1521-3773(20020517)41:10<1763::aid-anie1763>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- (126).(a) Lewis FD, Liu X, Wu Y, Miller SE, Wasielewski MR, Letsinger RL, Sanishvili R, Joachimiak A, Tereshko V, Egli M. J. Am. Chem. Soc. 1999;121:9905. [Google Scholar]; (b) Lewis FD, Liu X, Miller SE, Hayes RT, Wasielewski MR. J. Am. Chem. Soc. 2002;124:11 280. doi: 10.1021/ja026751d. [DOI] [PubMed] [Google Scholar]
- (127).(a) Giese B, Carl B, Carl T, Carell T, Behrens C, Hennecke U, Schiemann O, Feresin E. Angew. Chem. Int. Ed. 2004;43:1848. doi: 10.1002/anie.200353264. [DOI] [PubMed] [Google Scholar]; (b) Schiemann O, Feresin E, Carl T, Giese B. ChemPhysChem. 2004;5:270. doi: 10.1002/cphc.200300896. [DOI] [PubMed] [Google Scholar]
- (128).Jortner J, Bixon M, Langenbacher T, Michel-Beyerle ME. Proc. Natl. Acad. Sci. USA. 1998;95:12759. doi: 10.1073/pnas.95.22.12759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (129).Marcus RA, Sutin N. Biochim. Biophys. Acta. 1985;811:265. [Google Scholar]
- (130).Bixon M, Jortner J. Chem. Phys. 2002;281:393. [Google Scholar]
- (131).Grozema FC, Berlin YA, Siebbeles LDA. J. Am. Chem. Soc. 2000;122:10903. [Google Scholar]
- (132).Purugganan MD, Kumar CV, Turro NJ, Barton JK. Science. 1988;241:1645. doi: 10.1126/science.241.4873.1645. [DOI] [PubMed] [Google Scholar]
- (133).Kelley SO, Barton JK. Science. 1999;283:375. doi: 10.1126/science.283.5400.375. [DOI] [PubMed] [Google Scholar]
- (134).Turro NJ, Barton JK. J. Biol. Inorg. Chem. 1998;3:201. [Google Scholar]
- (135).Giese B, Amaudrut J, Kohler A-K, Spormann M, Wessely S. Nature. 2001;412:318. doi: 10.1038/35085542. [DOI] [PubMed] [Google Scholar]
- (136).Meggers E, Kusch D, Spichty M, Wille U, Giesse B. Angew. Chem. Int. Ed. Engl. 1998;37:460. doi: 10.1002/(SICI)1521-3773(19980302)37:4<460::AID-ANIE460>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- (137).Lewis FD, Wu T, Zhang Y, Letsinger RL, Greenfield SR, Wasielewski MR. Science. 1997;277:673. doi: 10.1126/science.277.5326.673. [DOI] [PubMed] [Google Scholar]
- (138).Fukui K, Tanaka K. Angew. Chem. Int. Ed. Engl. 1998;37:158. [Google Scholar]
- (139).Grozema FC, Berlin YA, Siebbeles LDA. Int. J. Quant. Chem. 1999;75:1009. [Google Scholar]
- (140).Olofsson J, Larsson S. J. Phys. Chem. B. 2001;105:10398. [Google Scholar]
- (141).Conwell EM, McLaughlin PM, Bloch SM. J. Phys. Chem. B. 2008;112:2268. doi: 10.1021/jp077344x. [DOI] [PubMed] [Google Scholar]
- (142).Conwell EM. Proc. Natl. Acad. Sci. U.S.A. 2005;102:8795. doi: 10.1073/pnas.0501406102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (143).Conwell EM, Bloch SM. J. Phys. Chem. B. 2006;110:5801. doi: 10.1021/jp0553986. [DOI] [PubMed] [Google Scholar]
- (144).Conwell EM, Bloch SM, McLaughlin PM, Basko DM. J. Am. Chem. Soc. 2007;129:9175. doi: 10.1021/ja0691472. [DOI] [PubMed] [Google Scholar]
- (145).Kanvah S, Joseph J, Schuster GB, Barnett RN, Cleveland CL, Landman U. Acc. Chem. Res. 2010;43:280. doi: 10.1021/ar900175a. [DOI] [PubMed] [Google Scholar]
- (146).Barnett RN, Cleveland CL, Joy A, Landman U, Schuster GB. Science. 2001;294:567. doi: 10.1126/science.1062864. [DOI] [PubMed] [Google Scholar]
- (147).Genereux JC, Barton JK. Chem. Rev. DOI: 10.1021/cr900228f. [Google Scholar]
- (148).Prunkl C, Berndl S, Wanninger-Weiß C, Barbaric J, Wagenknecht H-A. Phys. Chem. Chem. Phys. 2010;12:32. doi: 10.1039/b914487k. [DOI] [PubMed] [Google Scholar]
- (149).Wagenknecht HA, editor. Charge Transfer in DNA. Wiley-VCH; Weinheim, Germany: 2005. [Google Scholar]
- (150).Schuster GB, editor. Long-Range Charge Transfer in DNA, I and II. 236 and 237. Springer; New York: 2004. [Google Scholar]
- (151).Reece SY, Nocera DG. Annu. Rev. Biochem. 2009;78:673. doi: 10.1146/annurev.biochem.78.080207.092132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (152).Cukier RI. J. Phys. Chem. B. 2002;106:1746. [Google Scholar]
- (153).Cukier RI. J. Phys. Chem. 1994;98:2377. [Google Scholar]
- (154).Cukier RI. J. Phys. Chem. 1995;99:16101. [Google Scholar]
- (155).Zhao XG, Cukier RI. J. Phys. Chem. 1995;99:945. [Google Scholar]
- (156).Cukier RI. J. Phys. Chem. 1996;100:15428. [Google Scholar]
- (157).Cukier RI. Biochim. Biophys. Acta. 2004;1655:37. doi: 10.1016/j.bbabio.2003.06.011. [DOI] [PubMed] [Google Scholar]
- (158).Soudackov A, Hammes-Schiffer S. J. Chem. Phys. 1999;111:4672. [Google Scholar]
- (159).Soudackov A, Hammes-Schiffer S. J. Chem. Phys. 2000;113:2385. [Google Scholar]
- (160).Soudackov A, Hatcher E, Hammes-Schiffer S. J. Chem. Phys. 2005;122:014505. doi: 10.1063/1.1814635. [DOI] [PubMed] [Google Scholar]
- (161).Ludlow MK, Skone JH, Hammes-Schiffer S. J. Phys. Chem. B. 2008;112:336. doi: 10.1021/jp0753474. [DOI] [PubMed] [Google Scholar]
- (162).Hammes-Schiffer S, Soudackov A. J. Phys. Chem. B. 2008;112:14108. doi: 10.1021/jp805876e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (163).Edwards SJ, Soudackov AV, Hammes-Schiffer S. J. Phys. Chem. B. 2009;113:14545. doi: 10.1021/jp907808t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (164).Mayer JM. Annu. Rev. Phys. Chem. 2004;55:363. doi: 10.1146/annurev.physchem.55.091602.094446. [DOI] [PubMed] [Google Scholar]
- (165).Costentin C, Robert M, Saveant J-M. J. Am. Chem. Soc. 2006;128:4552. doi: 10.1021/ja060527x. [DOI] [PubMed] [Google Scholar]
- (166).Georgievskii Y, Stuchebrukhov AA. J. Chem. Phys. 2000;113:10438. doi: 10.1063/1.1796751. [DOI] [PubMed] [Google Scholar]
- (167).Turró C, Chang CK, Leroi GE, Cukier RI, Nocera DG. J. Am. Chem. Soc. 1992;114:4013. [Google Scholar]
- (168).Skone JH, Soudackov AV, Hammes-Schiffer S. J. Am. Chem. Soc. 2006;128:16655. doi: 10.1021/ja0656548. [DOI] [PubMed] [Google Scholar]
- (169).Mayer JM, Hrovat DA, Thomas JL, Borden WT. J. Am. Chem. Soc. 2002;124:11142. doi: 10.1021/ja012732c. [DOI] [PubMed] [Google Scholar]
- (170).Taylor J, Eliezer I, Sevilla MD. J. Phys. Chem. B. 2001;105:1614. [Google Scholar]
- (171).Carra C, Iordanova N, Hammes-Schiffer S. J. Phys. Chem. B. 2002;106:8415. [Google Scholar]
- (172).Candeias LP, Steenken S. J. Am. Chem. Soc. 1989;111:1094. [Google Scholar]
- (173).Candeias LP, Steenken S. J. Am. Chem. Soc. 1992;114:699. [Google Scholar]
- (174).Shafirovich V, Dourandin A, Luneva NP, Geacintov NE. J. Phys. Chem. B. 2000;104:137. [Google Scholar]
- (175).Kuzmin VA, Dourandin A, Shafirovich V, Geacintov NE. Phys. Chem. Chem. Phys. 2000;2:1531. [Google Scholar]
- (176).Shafirovich V, Dourandin A, Geacintov NE. J. Phys. Chem. B. 2001;105:8431. [Google Scholar]
- (177).Shafirovich V, Dourandin A, Huang WD, Luneva NP, Geacintov NE. Phys. Chem. Chem. Phys. 2000;2:4399. [Google Scholar]
- (178).Shafirovich V, Cadet J, Gasparutto D, Dourandin A, Huang WD, Geacintov NE. J. Phys. Chem. B. 2001;105:586. doi: 10.1021/tx000204t. [DOI] [PubMed] [Google Scholar]
- (179).Shafirovich V. Ya., Courtney SH, Ya N, Geacintov NE. J. Am. Chem. Soc. 1995;117:4920. [Google Scholar]
- (180).Weatherly SC, Yang IV, Armistead PA, Thorp HH. J. Phys. Chem. B. 2003;107:372. [Google Scholar]
- (181).Weatherly SC, Yang IV, Thorp HH. J. Am. Chem. Soc. 2001;123:1236. doi: 10.1021/ja003788u. [DOI] [PubMed] [Google Scholar]
- (182).Chatgilialoglu C, Caminal C, Guerra M, Mulazzani QG. Angew. Chem., Int. Ed. 2005;44:6030. doi: 10.1002/anie.200501087. [DOI] [PubMed] [Google Scholar]
- (183).Mundy CJ, Colvin ME, Quong AA. J. Phys. Chem. A. 2002;106:10063. [Google Scholar]
- (184).Adhikary A, Kumar A, Becker D, Sevilla MD. J. Phys. Chem. B. 2006;110:24171. doi: 10.1021/jp064361y. see references therein. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (185).Ortmans I, Elias B, Kelly JM, Moucheron C, Kirsch-DeMesmaeker A. Dalton Trans. 2004:668. doi: 10.1039/b313213g. [DOI] [PubMed] [Google Scholar]
- (186).Elias B, Creely C, Doorley GW, Feeney MM, Moucheron C, Kirsch-DeMesmaeker A, Dyer J, Grills DC, George MW, Matousek P, Parker AW, Towrie M, Kelly JM. Chem. Eur. J. 2008;14:369. doi: 10.1002/chem.200700564. [DOI] [PubMed] [Google Scholar]
- (187).Kobayashi K, Tagawa S. J. Am. Chem. Soc. 2003;125:10213. doi: 10.1021/ja036211w. [DOI] [PubMed] [Google Scholar]
- (188).Kobayashi K, Yamagami R, Tagawa S. J. Phys. Chem. B. 2008;112:10752. doi: 10.1021/jp804005t. [DOI] [PubMed] [Google Scholar]
- (189).Giese B, Wessely S. Chem. Commun. 2001:2108. doi: 10.1039/b106059g. [DOI] [PubMed] [Google Scholar]
- (190).Takada T, Kawai K, Fujitsuka M, Majima T. Chem. Eur. J. 2005;11:3835. doi: 10.1002/chem.200500052. [DOI] [PubMed] [Google Scholar]
- (191).Kawai K, Osakada Y, Majima T. ChemPhysChem. 2009;10:1766. doi: 10.1002/cphc.200900148. [DOI] [PubMed] [Google Scholar]
- (192).Stemp EDA, Arkin M, Barton JK. J. Am. Chem. Soc. 1997;119:2921. [Google Scholar]
- (193).Schiemann O, Turro NJ, Barton JK. J. Phys. Chem. B. 2000;104:7214. [Google Scholar]
- (194).Anderson RF, Shinde SS, Maroz A. J. Am. Chem. Soc. 2006;128:15966. doi: 10.1021/ja0658416. [DOI] [PubMed] [Google Scholar]
- (195).Gervasio FL, Boero M, Parrinello M. Angew. Chem., Int. Ed. 2006;45:5606. doi: 10.1002/anie.200602106. [DOI] [PubMed] [Google Scholar]
- (196).Colson AO, Besler B, Sevilla MD. J. Phys. Chem. 1992;96:9787. [Google Scholar]
- (197).Hutter M, Clark T. J. Am. Chem. Soc. 1996;118:7574. [Google Scholar]
- (198).Bertran J, Oliva A, Rodriguez-Santiago L, Sodupe M. J. Am. Chem. Soc. 1998;120:8159. [Google Scholar]
- (199).Li XF, Cai ZL, Sevilla MD. J. Phys. Chem. B. 2001;105:10115. [Google Scholar]
- (200).Kumar A, Sevilla MD. J. Phys. Chem. B. 2009;113:11359. doi: 10.1021/jp903403d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (201).Sobolewski AL, Domcke W. Phys. Chem. Chem. Phys. 2004;6:2763. [Google Scholar]
- (202).Sobolewski AL, Domcke W, Hattig C. Proc. Natl. Acad. Sci. U.S.A. 2005;102:17903. doi: 10.1073/pnas.0504087102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (203).Abu-Riziq A, Grace L, Nir E, Kabelac M, Hobza P, de Vries MS. Proc. Natl. Acad. Sci. U.S.A. 2005;102:20. doi: 10.1073/pnas.0408574102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (204).Schwalb NK, Temps F. J. Am. Chem. Soc. 2007;129:9272. doi: 10.1021/ja073448+. [DOI] [PubMed] [Google Scholar]
- (205).Groenhof G, Schäfer LV, Boggio-Pasqua M, Goette M, Grubmüller H, Robb MA. J. Am. Chem. Soc. 2007;129:6812. doi: 10.1021/ja069176c. [DOI] [PubMed] [Google Scholar]
- (206).Crespo-Hernández CE, de La Harpe K, Kohler B. J. Am. Chem. Soc. 2008;130:10844. doi: 10.1021/ja802183s. [DOI] [PubMed] [Google Scholar]
- (207).de La Harpe K, Crespo-Hernández CE, Kohler B. J. Am. Chem. Soc. 2009;131:17557. doi: 10.1021/ja9076364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (208).Milligan JR, Aguilera JA, Hoang O, Ly A, Tran NQ, Ward JF. J. Am. Chem. Soc. 2004;126:1682. doi: 10.1021/ja030319u. [DOI] [PubMed] [Google Scholar]
- (209).Ly A, Tran NQ, Ward JF, Milligan JR. Biochemistry. 2004;43:9098. doi: 10.1021/bi0494830. [DOI] [PubMed] [Google Scholar]
- (210).Ly A, Tran NQ, Sullivan K, Bandong SL, Milligan JR. Org. Biomol. Chem. 2005;3:917. doi: 10.1039/b418681h. [DOI] [PubMed] [Google Scholar]
- (211).Llano J, Eriksson LA. Phys. Chem. Chem. Phys. 2004;6:4707. [Google Scholar]
- (212).Jena NR, Mishra PC, Suhai S. J. Phys. Chem. B. 2009;113:5633. doi: 10.1021/jp810468m. [DOI] [PubMed] [Google Scholar]
- (213).Sevilla MD, Paemel CV, Nichols C. J. Phys. Chem. 1972;76:3571. doi: 10.1021/j100668a012. [DOI] [PubMed] [Google Scholar]
- (214).Sevilla MD, Paemel CV, Nichols C. J. Phys. Chem. 1972;76:3577. doi: 10.1021/j100668a012. [DOI] [PubMed] [Google Scholar]
- (215).Sevilla MD. J. Phys. Chem. 1971;75:626. doi: 10.1021/j100675a005. [DOI] [PubMed] [Google Scholar]
- (216).Geimer J, Hildenbrand K, Naumov S, Beckert D. Phys. Chem. Chem. Phys. 2000;2:4199. [Google Scholar]
- (217).Krivokapic’ A, Herak JN, Sagstuen E. J. Phys. Chem. A. 2008;112:3597. doi: 10.1021/jp709855c. [DOI] [PubMed] [Google Scholar]
- (218).Nelson WH, Sagstuen E, Hole EO, Close DM. Rad. Res. 1998;149:75. [PubMed] [Google Scholar]
- (219).Shi Y, Huang C, Wang W, Kang J, Yao S, Lin N, Zheng R. Rad. Phys. Chem. 2000;58:253. [Google Scholar]
- (220).Adhikary A, Kumar A, Khanduri D, Sevilla MD. J. Am. Chem. Soc. 2008;130:10282. doi: 10.1021/ja802122s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (221).Perun S, Sobolewski AL, Domcke W. J. Phys. Chem. A. 2006;110:9031. doi: 10.1021/jp061945r. [DOI] [PubMed] [Google Scholar]
- (222).Adhikary A, Collins S, Koppen J, Becker D, Sevilla MD. Nucleic Acid Res. 2006;34:1501. doi: 10.1093/nar/gkl026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (223).Adhikary A, Malkhasian AYS, Collins S, Koppen J, Becker D, Sevilla MD. Nucleic Acids Res. 2005;33:5553. doi: 10.1093/nar/gki857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (224).Adhikary A, Collins S, Khanduri D, Sevilla MD. J. Phys. Chem. B. 2007;111:7415. doi: 10.1021/jp071107c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (225).Khanduri D, Collins S, Kumar A, Adhikary A, Sevilla MD. J. Phys. Chem. B. 2008;112:2168. doi: 10.1021/jp077429y. [DOI] [PubMed] [Google Scholar]
- (226).Adhikary A, Khanduri D, Kumar A, Sevilla MD. J. Phys. Chem. B. 2008;112:15844. doi: 10.1021/jp808139e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (227).Becker D, Razskazovskii Y, Callaghan MU, Sevilla MD. Radiat. Res. 1996;146:361. [PubMed] [Google Scholar]
- (228).Kumar A, Sevilla MD. J. Phys. Chem. B. 2009;113:13374. doi: 10.1021/jp9058593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (229).Hammond GS. J. Am. Chem. Soc. 1955;77:334. [Google Scholar]
- (230).Aflatooni K, Gallup GA, Burrow PD. J. Phys. Chem. A. 1998;102:6205. [Google Scholar]
- (231).Rak J, Mazurkiewicz K, Kobyłecka M, Storoniak P, Harańczyk M, Dąbkowska I, Bachorz RA, Gutowski M, Radisic V, Stokes ST, Eustis SN, Wang D, Li X, Ko YJ, Bowen KH. In: Radiation Induced Molecular Phenomena in Nucleic Acids. Shukla MK, Leszczynski J, Leszczynski J, editors. Vol. 5. Springer Science + Business Media B.V.; 2008. pp. 619–667. under Challenges and Advances in Computational Chemistry and Physics. [Google Scholar]
- (232).Nelson WH, Hole EO, Sagstuen E, Close DM. Int. J. Radiat. Biol. 1988;54:963. doi: 10.1080/09553008814552361. [DOI] [PubMed] [Google Scholar]
- (233).(a) Jayatilaka N, Nelson WH. J. Phys. Chem. B. 2008;112:16908. doi: 10.1021/jp806262d. [DOI] [PubMed] [Google Scholar]; (b) Nelson WH, Sagstuen E, Hole EO, Close DM. Rad. Res. 1992;131:10. [PubMed] [Google Scholar]
- (234).Bernhard WA. Adv. Radiat. Biol. 1981;9:199. [Google Scholar]
- (235).Wang W, Sevilla MD. Rad. Res. 1994;138:9. [PubMed] [Google Scholar]
- (236).Candeias LP, Wolf P, O’Neill P, Steenken S. J. Phys. Chem. 1992;96:10302. [Google Scholar]
- (237).Harańczyk M, Gutowski M, Li X, Bowen KH. J. Phys. Chem. B. 2007;111:14073. doi: 10.1021/jp077439z. [DOI] [PubMed] [Google Scholar]
- (238).Harańczyk M, Gutowski M. Angew. Chem. Int. Ed. 2005;44:6585. doi: 10.1002/anie.200501671. [DOI] [PubMed] [Google Scholar]
- (239).Harańczyk M, Gutowski M. J. Am. Chem. Soc. 2005;127:699. doi: 10.1021/ja046622s. [DOI] [PubMed] [Google Scholar]
- (240).Chatgilialoglu C, D’Angelantonio M, Kaloudis P, Mulazzani QG, Guerra M. J. Phys. Chem. Lett. 2010;1:174. [Google Scholar]
- (241).Yamagami R, Kobayashi K, Tagawa S. J. Am. Chem. Soc. 2008;130:14772. doi: 10.1021/ja805127e. [DOI] [PubMed] [Google Scholar]
- (242).Szyperska A, Rak J, Leszczynski J, Li X, Ko YJ, Wang H, Bowen KH. J. Am. Chem. Soc. 2009;131:2663. doi: 10.1021/ja808313e. [DOI] [PubMed] [Google Scholar]
- (243).Colson A-O, Besler B, Sevilla MD. J. Phys. Chem. 1992;96:9181. [Google Scholar]
- (244).Gu J, Xie Y, Schaefer HF. J. Chem. Phys. 2007;127:155107. doi: 10.1063/1.2780148. [DOI] [PubMed] [Google Scholar]
- (245).Chen H-Y, Kao C-L, Hsu SCN. J. Am. Chem. Soc. 2009;131:15930. doi: 10.1021/ja906899p. [DOI] [PubMed] [Google Scholar]
- (246).Chen H-Y, Hsu SCN, Kao C-L. Phys. Chem. Chem. Phys. 2010;12:1253–1263. doi: 10.1039/b920603e. [DOI] [PubMed] [Google Scholar]
- (247).Sagstuen E, Hole EO, Nelson WH, Close DM. J. Phys. Chem. 1989;93:5974. [Google Scholar]
- (248).(a) Hole EO, Sagstuen E, Nelson WH, Close DM. J. Phys. Chem. 1991;95:1494. [Google Scholar]; (b) Sagstuen E, Hole EO, Nelson WH, Close DM. J. Phys. Chem. 1992;96:8269. [Google Scholar]
- (249).Barnes J, Bernhard WA, Mercer KR. Radiat. Res. 1991;126:104. [PubMed] [Google Scholar]
- (250).Hissung A, Von Sonntag C. Int. J. Radiat. Biol. 1979;35:449. [PubMed] [Google Scholar]
- (251).Schuchmann H-P, Von Sonntag C. Int. J. Radiat. Biol. 1986;49:1. doi: 10.1080/09553008514552201. [DOI] [PubMed] [Google Scholar]
- (252).Mroczka NE, Mercer KR, Bernhard WA. Radiat. Res. 1997;147:560. [PubMed] [Google Scholar]
- (253).Raytchev M, Mayer E, Amann N, Wagenknecht H-A, Fiebig T. ChemPhysChem. 2004;5:706. doi: 10.1002/cphc.200301205. [DOI] [PubMed] [Google Scholar]
- (254).Netzel TL, Zhao M, Nafisi K, Headrick J, Sigman MS, Eaton BE. J. Am. Chem. Soc. 1995;117:9119. [Google Scholar]
- (255).Cai Z, Li X, Sevilla MD. J. Phys. Chem. B. 2002;106:2755. [Google Scholar]
- (256).Harańczyk M, Rak J, Gutowski M. J. Phys. Chem. A. 2005;109:11495. doi: 10.1021/jp0535590. [DOI] [PubMed] [Google Scholar]
- (257).Harańczyk M, Gutowski M, Warshel A. Phys. Chem. Chem. Phys. 2008;10:4442. doi: 10.1039/b802300j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (258).Harańczyk M, Rak J, Gutowski M, Radisic D, Stokes ST, Bowen KH. J. Phys. Chem. B. 2005;109:13383. doi: 10.1021/jp050246w. [DOI] [PubMed] [Google Scholar]
- (259).Visscher KJ, De Haas MP, Loman H, Vojnovic B, Warman JM. Int. J. Radiat. Biol. 1987;52:745. doi: 10.1080/09553008714552251. [DOI] [PubMed] [Google Scholar]
- (260).Candeias LP, Steenken S. J. Phys. Chem. 1992;96:937. [Google Scholar]
- (261).Stokes ST, Grubisic A, Li X, Ko YJ, Bowen KH. J. Chem. Phys. 2008;128:044314. doi: 10.1063/1.2823001. [DOI] [PubMed] [Google Scholar]
- (262).Li X, Bowen KH, Harańczyk M, Bachorz RA, Mazurkiewicz K, Rak J, Gutowski M. J. Chem. Phys. 2007;127:174309. doi: 10.1063/1.2795719. [DOI] [PubMed] [Google Scholar]
- (263).Mazurkiewicz K, Harańczyk M, Gutowski M, Rak J, Radisic D, Eustis SN, Wang D, Bowen KH. J. Am. Chem. 2007;129:1216. doi: 10.1021/ja066229h. [DOI] [PubMed] [Google Scholar]
- (264).Mazurkiewicz K, Harańczyk M, Storoniak P, Gutowski M, Rak J, Radisic D, Eustis SN, Wang D, Bowen KH. Chem. Phys. 2007;342:215. [Google Scholar]
- (265).Radisic D, Bowen KH, Dąbkowska I, Storoniak P, Rak J, Gutowski M. J. Am. Chem. 2005;127:6443. doi: 10.1021/ja050724g. [DOI] [PubMed] [Google Scholar]
- (266).Martin F, Burrow PD, Cai Z, Cloutier P, Hunting D, Sanche L. Phys. Rev. Lett. 2004;93:068101. doi: 10.1103/PhysRevLett.93.068101. [DOI] [PubMed] [Google Scholar]
- (267).Panajotovic R, Martin F, Cloutier PC, Hunting D, Sanche L. Radiat. Res. 2006;165:452. doi: 10.1667/rr3521.1. [DOI] [PubMed] [Google Scholar]
- (268).Li X, Sevilla MD, Sanche L. J. Am. Chem. Soc. 2003;125:13668. doi: 10.1021/ja036509m. [DOI] [PubMed] [Google Scholar]
- (269).Simons J. Acc. Chem. Res. 2006;39:772. doi: 10.1021/ar0680769. [DOI] [PubMed] [Google Scholar]
- (270).Bao X, Wang J, Gu J, Leszczynski J. Proc. Nat. Acad. Sci. USA. 2006;103:5658. doi: 10.1073/pnas.0510406103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (271).Kumar A, Sevilla MD. J. Phys. Chem. B. 2007;111:5464. doi: 10.1021/jp070800x. [DOI] [PubMed] [Google Scholar]
- (272).Kumar A, Sevilla MD. J. Am. Chem. Soc. 2008;130:2130. doi: 10.1021/ja077331x. [DOI] [PubMed] [Google Scholar]
- (273).Kumar A, Sevilla MD. ChemPhysChem. 2009;10:1426. doi: 10.1002/cphc.200900025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (274).Dąbkowska I, Rak J, Gutowski M. Eur. Phys. J. D. 2005;35:429. [Google Scholar]
- (275).Gu J, Wang J, Rak J, Leszczynski J. Angew. Chem. Int. Ed. 2007;46:3479. doi: 10.1002/anie.200604603. [DOI] [PubMed] [Google Scholar]