

Abstract
The aim of this study was to determine whether standard treatments for Tobacco Dependence affect smoking-induced changes in intrasynaptic dopamine (DA) concentration. Forty-three otherwise healthy adult cigarette smokers (10 to 40 cigarettes per day) were treated with either practical group counseling (PGC) psychotherapy (n = 14), bupropion HCl (n = 14), or matching pill placebo (n = 15) (random assignment) for 8 weeks. Before and after treatment, each subject underwent a bolus-plus-continuous-infusion 11C-raclopride positron emission tomography (PET) scanning session, during which he or she smoked a regular cigarette. The PET scanning outcome measure of interest was percent change in smoking-induced 11C-raclopride binding potential (BPND) in the ventral caudate/nucleus accumbens (VCD/NAc), as an indirect measure of DA release. Although the entire study sample had a smaller mean smoking-induced reduction in VCD/NAc BPND after treatment (compared to before treatment), this change was highly correlated with smaller total cigarette puff volumes (and not other treatment variables). These data indicate that smoking-induced DA release is dose-dependent, and is not significantly affected by reductions in daily smoking levels or treatment type.
Keywords: tobacco dependence, 11C-raclopride, positron emission tomography, dopamine, ventral striatum, cigarette craving
1. Introduction
Recent studies using the radiotracer 11C-raclopride and positron emission tomography (PET) have demonstrated (indirectly) that cigarette smoking (Brody et al., 2004; Brody et al., 2006; Scott et al., 2007) and nicotine intake (Takahashi et al., 2008) increase intrasynaptic dopamine (DA) concentration in the ventral striatum/nucleus accumbens (VST/NAc) in human smokers (though not all studies using these and similar methods have shown this effect (Tsukada et al., 2002; Barrett et al., 2004; Montgomery et al., 2007)). In these studies, DA release has been associated with the craving-reducing (Brody et al., 2004; Brody et al., 2006) and pleasurable (Barrett et al., 2004; Brody et al., 2009) effects of smoking, as well as the severity of nicotine dependence (Scott et al., 2007). Nicotine-induced DA release has also been demonstrated in studies of non-human primates using similar methodology (Dewey et al., 1999; Tsukada et al., 2002; Marenco et al., 2004) and in rodents using microdialysis (Di Chiara and Imperato, 1988; Damsma et al., 1989; Pontieri et al., 1996; Sziraki et al., 2001). Despite these findings that point to the importance of striatal DA release in response to cigarette smoking, the effects of treatment for Tobacco Dependence (TD) on smoking-induced DA release have not been reported.
Currently, there are several effective first-line treatments for TD, including (but not limited to) the medication bupropion HCl (Zyban) and the psychotherapy ‘practical group counseling’ (PGC) (Marlatt and Gordon, 1985; Carmody, 1990; Fiore et al., 2000; Hall et al., 2002; Gold et al., 2002; Abrams et al., 2003; Holmes et al., 2004). A standard (2-month) course of treatment with bupropion HCl (administered along with brief counseling) results in abstinence rates of roughly 40% (Hurt et al., 1997), with longer term (12-month) abstinence rates of about 20 to 30% (Jorenby et al., 1999; Simon et al., 2004; Paluck et al., 2006). Abstinence rates with medication alone are typically lower than those achieved with the combination of medication plus psychotherapy (Hall et al., 2002). Similar to medication administered alone, PGC typically results in abstinence rates of about 40% after 2 months and 20% after 6 months of treatment (Fiore et al., 1994), with a recent literature review concluding that group psychotherapies have roughly twice the abstinence rates of control conditions (Stead and Lancaster, 2005).
The complete biological pathway by which bupropion HCl assists smokers in quitting remains uncertain (Horst and Preskorn, 1998; Foley et al., 2006); however, there is evidence for either diminished or unchanged smoking-induced DA release with bupropion HCl treatment. As for evidence of diminished smoking-induced DA release, the most commonly demonstrated mechanism of action for bupropion HCl is inhibition of pre-synaptic DA reuptake (Horst and Preskorn, 1998; Balfour, 2001; Stahl et al., 2004), with reports of both acute (Nomikos et al., 1989) and chronic (Ascher et al., 1995) increases in extracellular DA in the VST/NAc. In addition, a brain imaging study of subjects with depression (Argyelan et al., 2005) demonstrated that chronic bupropion HCl treatment is associated with diminished dopamine transporter availability, which would presumably increase intrasynaptic DA since the dopamine transporter is primarily responsible for clearance of intrasynaptic DA (Hoffman et al., 1998). These studies suggest that chronic bupropion HCl treatment leads to increased intrasynaptic DA, which would be expected to result in a downregulation of D2 receptors and diminished DA release in response to pharmacological stimuli (Bilder et al., 2004; Nikolaus et al., 2007a; Nikolaus et al., 2007b; Bamford et al., 2008). As for evidence of unchanged smoking-induced DA release, a recent study of rodents demonstrated that chronic bupropion HCl treatment increases extracellular DA in the nucleus accumbens shell, but does not alter the reward-facilitating effects of experimenter-administered nicotine (Paterson et al., 2007). In addition, a study of human smokers examining the effects of bupropion HCl treatment on the pleasurable effect of smoking (a symptom presumably associated with DA release (Barrett et al., 2004; Brody et al., 2009)) demonstrated no change in the pleasurable effect of a lapse cigarette (McCarthy et al., 2008). Thus, taken together, these studies suggest that a course of bupropion HCl treatment would lead to increased intrasynaptic DA and diminished smoking-induced DA release, although there is also evidence to suggest that smoking-induced DA release would be unaffected by bupropion HCl treatment.
As for practical group counseling (PGC) for TD, we are not aware of any studies examining its mechanism of action. However, it is hypothesized that talk therapy in general (as a learning experience) leads to changes in synaptic plasticity (Marlatt and Gordon, 1985; Carmody, 1990), through a retraining of implicit memory systems (Amini et al., 1996; Liggan and Kay, 1999). In addition, prior studies of other neuropsychiatric conditions indicate that changes in the brain with talk therapy typically have at least some overlap with those seen with effective medication (Roffman et al., 2005).
The goal of the study presented here was to determine if standard, first-line treatments for TD (bupropion HCl and PGC, compared to a pill placebo that matched bupropion HCl) affect smoking-induced increases in intrasynaptic DA concentration in tobacco-dependent cigarette smokers. Based on both human and animal studies, we hypothesized that these treatments would diminish smoking-induced changes in intrasynaptic DA concentration, though it was noted that there is also evidence for an absence of an effect of these treatments on smoking-induced DA release. We also sought to determine if treatment-related changes in smoking-induced DA release were associated with changes in the amount of a cigarette smoked during scanning, the number of cigarettes smoked per day, or withdrawal symptoms, hypothesizing that reduced smoking during scanning, cigarettes per day, or withdrawal symptoms would be associated with diminished smoking-induced DA release.
2. Methods
2.1 Research Participants
Two hundred and ten subjects were screened initially during a telephone interview, in which medical, psychiatric, and substance abuse histories were obtained without personal identifiers. All subjects who were qualified and wished to participate received a complete description of the study in-person, and gave written informed consent, using forms approved by the local institutional review board. Participants were then evaluated further using screening questions from the Structured Clinical Interview for DSM-IV (SCID) (First et al., 1995). Inclusion/exclusion criteria were the same as in our previous reports (Brody et al., 2004; Brody et al., 2006; Brody et al., 2009), with the central inclusion criteria being nicotine dependence, as defined by DSM-IV, smoking 10 to 40 cigarettes per day, and being treatment-seeking. Participants were excluded for any history of an Axis I psychiatric or substance abuse/dependence diagnosis other than nicotine dependence. They were also excluded for current use of medications or medical conditions that might affect brain function, or for pregnancy. The pre-treatment PET data from subjects studied here was presented in a prior report (Brody et al., 2009).
2.2 Experimental Design
Forty-three otherwise healthy adult (21 to 65 years of age) cigarette smokers completed the study. Each subject underwent bolus-plus-infusion 11C-raclopride PET scanning, during which he or she smoked a regular cigarette. Within one week of the PET scanning, he or she also underwent a structural magnetic resonance imaging (MRI) (to aid in interpretation of the PET scans). Subjects were then randomized to treatment with bupropion HCl (n = 14), PGC (n = 14), or pill placebo (n = 15). After completing 8 weeks of treatment, subjects were scanned a second time with 11C-raclopride PET, with this PET session having the same protocol as the first one (including smoking a regular cigarette). Seven additional subjects participated in this study, but their data were not used for this analysis due to technical scanning issues (n = 2) or dropout from treatment (n = 5).
2.3 PET and MRI Scanning Protocols
PET and MRI scanning sessions followed the same protocols as in our previous reports (Brody et al., 2006; Brody et al., 2009) which examined smoking-induced changes in intrasynaptic DA concentration in untreated smokers. PET sessions were performed using a bolus-plus-continuous-infusion of 11C-raclopride. Subjects were instructed to smoke as per their usual habit on the morning of the PET session and to smoke a cigarette immediately prior to the testing session (for the pre-treatment session), which began at noon. Between 12:00 and 13:45 h, subjects were interviewed and completed rating scale questionnaires and other measures. Self-report rating scales were the Fagerström Test for Nicotine Dependence (FTND) (Fagerström, 1978; Heatherton et al., 1991) and the Urge to Smoke (UTS) Scale (Jarvik et al., 2000), while a study clinician (A.L.B., R.E.O., or Z.A.M.) administered the Hamilton Depression (HAM-D) (Hamilton, 1967) and Anxiety (HAM-A) (Hamilton, 1969) rating scales. Exhaled carbon monoxide (CO) levels were measured before 11C-raclopride injection using a MicroSmokerlyzer (Bedfont Scientific Ltd, Kent, UK), to provide a rough estimate of recent smoking levels. In addition to these ratings done prior to scanning, cigarette craving (UTS) was monitored after the smoke break in PET scanning as well. These rating scales were administered to ensure both low levels of psychiatric symptoms and expected reactions to smoking (such as a reduction in craving).
At 13:45 h, each subject had a 20-gauge intravenous catheter placed in the right antecubital vein, and at 14:00 h was positioned on the PET scanning bed (for acquisition of planes parallel to the orbital-meatal line). Scanning was initiated at 14:10 h with a slow bolus injection of 5 mCi 11C-raclopride in 20 ml normal saline over a 60-sec period, followed by continuous infusion of the radiotracer (5 mCi, corrected for decay) for the remainder of the testing session. This bolus-plus-continuous-infusion method was performed as described in prior studies (Carson et al., 1997; Ito et al., 1998; Brody et al., 2006; Brody et al., 2009). Brain scans were acquired continuously for the next 50-min.
At 15:00 h, each subject was removed from the scanner with the infusion still running, and had a 10-min break in an outdoor area adjacent to the PET scanning room. During the break, the subject smoked a single regular cigarette (his or her favorite brand or a standard Marlboro red package cigarette). All subjects smoked the same cigarette brand for their pre- and post- treatment PET scans, with the mean cigarette nicotine content being 1.0 (± 0.2) mg. The 3-h period of abstinence prior to the break was chosen based on the finding that craving starts to peak at this time in nicotine-dependent smokers (Schuh and Stitzer, 1995). A study investigator (A.L.B., R.E.O., or Z.A.M.) monitored subjects continuously during the break. After the break, subjects were quickly repositioned (< 1 min) in the scanner using a pre-placed mark on their forehead and a laser light from the scanner. Scanning then resumed for 30-min more.
For monitoring smoking topography during scanning, cigarettes were smoked through a Clinical Research Support System (CReSS) device (Plowshare, Baltimore, MD). While all standard recordings were obtained with this device, the primary measure of interest here was total puff volume of the cigarette (in mL), which indicated the amount of each cigarette that was smoked.
The PET scanner for this study was the GE Advance NXi PET tomograph (General Electronic Medical Systems, Milwaukee, WI, USA) with 35 slices in 3-dimensional mode. 11C-raclopride was prepared by an established procedure (Farde et al., 1986; Ehrin et al., 1987). PET scans were acquired as 5-min frames, ten before and six after the smoking break in scanning. Attenuation correction for both emission scans (before and after smoking) was performed using a 5-min transmission scan obtained with the germanium rotating rod source built into the GE scanner at the end of the scanning session.
An MRI scan of the brain was obtained with the following specifications: three-dimensional Fourier-transform (3DFT) spoiled-gradient-recalled acquisition with TR = 30 ms, TE = 7 ms, 30° angle, 2 acquisitions, 256 × 192 view matrix. The acquired volume was reconstructed as 90 contiguous, 1.5-mm thick, transaxial slices.
2.4 Tobacco Dependence Treatments
Subjects were randomly assigned (Stout et al., 1994) to treatment with either PGC (n = 14), bupropion HCl Sustained Release (SR) formula (n = 14), or matching pill placebo (n = 15). Each treatment was initiated within one week of the first PET scanning session and was continued through the second PET scanning session at week 8. The groups were counterbalanced for gender (male/female) and age (less than or greater than 40 years old), and all subjects were instructed to have a target quit date of 2 weeks after starting treatment.
Subjects randomized to PGC had twice weekly 60-min group therapy sessions for 8 weeks. PGC was based on the relapse prevention model (Marlatt and Gordon, 1985) and used standard clinical practices (Abrams et al., 2003). The therapy consisted of education about smoking addiction, withdrawal, and relapse prevention; recognizing danger situations (triggers) that could lead to relapse; developing new coping skills, such as avoiding triggers, coping with negative affective states, reducing overall stress, and distracting attention from smoking using thought-stopping techniques; developing lifestyle changes; and social support (Carmody, 1990; Fiore et al., 2000). Subjects also had exhaled CO levels monitored at each session, and were encouraged to taper off cigarettes. The manualized psychotherapy sessions were performed on a rotating basis by a study psychotherapist (S.S.).
Subjects randomized to receive bupropion HCl or matching pill placebo were treated in a double-blind manner. To do this, a research pharmacist at the Greater Los Angeles VA Healthcare System (Diep Seversen, PharmD) prepared packets of medication or placebo, which were distributed to a study physician (A.L.B. or Z.A.M.). These packets were identified by a numeric code recorded by the pharmacist. Film-coated bupropion HCl SR and placebo were obtained from the Biomedical Research Institute of New Mexico (Albuquerque, NM). Placebo ingredients (methylcellulose, hydroxypropyethycellulose, and hydroxypropylmethylcellulose) were inert and the same as those found in bupropion HCl SR tablets. Subjects were started on 1 pill per day on the day following the first PET scanning session for 3 days, with the dosage increased to 1 pill orally twice per day thereafter. For bupropion HCl SR, the dosage was 150 mg per oral once a day, then 150 mg per oral twice a day starting on the fourth treatment day. All subjects receiving pill treatment were advised of potential side effects of bupropion HCl when being given their packet of study medication/placebo, and all subjects tolerated the full dose of bupropion HCl SR for at least 4 weeks prior to the follow-up PET session. Subjects in the bupropion HCl and placebo groups met with a study physician weekly for medication management visits (15 min) for the remainder of the study. During these visits, titration of dosage, review of side effects, and monitoring of cigarette usage took place, as well as the measurement of exhaled CO. In order to isolate the effects of medication on PET findings, no counseling for cessation of tobacco use took place during these visits, and subjects in the bupropion HCl and placebo groups were instructed to take the twice daily dosing through the second PET session (including the morning of the second PET session). Given that bupropion HCl SR reaches a plasma steady state within 8 days and has a plasma mean elimination half-life of 21 h (GlaxoSmithKline, Wellbutrin SR tablets, product information, 2002), it is assumed that plasma bupropion HCl (and metabolite) levels in subjects assigned to the bupropion HCl SR group were in a steady state at the time of the follow-up PET scanning session.
For all study subjects, quit status was defined at the time of the follow-up PET scanning session as a self-report of at least 7 days of continuous abstinence from smoking and an exhaled CO level of ≤ 8 ppm. To ensure that smoking a cigarette during the follow-up scan did not lead to a slip or relapse into cigarette usage in smokers who had quit smoking, all subjects were monitored for craving and withdrawal symptoms for several hours after scanning. In addition, all subjects were seen weekly by a study physician following the second PET scan for an additional 1 month of treatment.
2.5 PET Image Analysis
For analysis of PET scans, PET-to-MRI co-registration was performed using the automated image registration method (Woods et al., 1993) within MEDx 3.3 (Sensor Systems Inc., Sterling, VA). The pre-smoking (0 to 50 min after the bolus radiotracer injection) and post-smoking (60 to 90 min after injection) scans were co-registered to the MRI separately. Individual 5-min time frame data were then co-registered to MRI scans using the transformation matrix parameters from the co-registration of the summed images.
Regions of interest (ROIs) were drawn on the MRI scans and transferred onto the co-registered PET scan frames. ROI placement on each frame was verified by visual inspection by the region drawers (M.R.C. and D.S) and the P.I. (A.L.B.). For this study, the ROI value used for statistical analysis was the volume-weighted decay-corrected mean of the left and right ventral caudate (VCD) ROIs (including the nucleus accumbens [NAc]), consisting of three slices for each structure, resulting in a single value for each 5-min frame of PET scanning (Brody et al., 2006). The entire cerebellum was drawn as a reference region. These regions are similar to those drawn in prior studies by other groups (Mawlawi et al., 2001; Martinez et al., 2003). Binding potential (BPND) (Innis et al., 2007) for the VCD/NAc was calculated using the simplified reference tissue model (Lammertsma and Hume, 1996), with BPND = B′max/Kd = CVCD/NAc/Ccerebellum − 1, where C is the radioligand concentration. BPND was determined for each 5-min PET frame, and mean BPND values (corrected for radiotracer physical decay) were compared between the frames before (40 to 50 min after radiotracer injection) and after (60 to 90 min after injection) the break in scanning. These time frames are almost identical to those recommended as optimal for maximizing the signal-to-noise ratio in studies of the 11C-raclopride bolus-plus-continuous infusion method (Watabe et al., 2000; Mawlawi et al., 2001). This method assumes 40-min for the radiotracer from the bolus-plus-infusion to reach a near steady state (Ito et al., 1998; Watabe et al., 2000; Mawlawi et al., 2001). It also assumes that intrasynaptic DA concentration will remain elevated throughout the 30 min post-smoking scan, as indicated by animal microdialysis (Imperato et al., 1986; Di Chiara and Imperato, 1988; Benwell and Balfour, 1997; Janhunen and Ahtee, 2004) and imaging (Marenco et al., 2004) studies of nicotine administration.
2.6 Statistical Analyses
Means (± standard deviations) of demographic and treatment variables were determined for the entire sample and for the three treatment subgroups. To verify similarities between treatment subgroups before the initiation of treatment, Student’s t-tests and a Chi-Square test (for gender) were performed. To evaluate treatment outcomes, paired Student’s t-tests were performed for the entire sample and for the three treatment subgroups for the primary smoking outcome measures (cigarettes per day, FTND scores, and exhaled CO levels). Student’s t-tests were then performed between the active treatment subgroups (PGC- and bupropion HCl-treated) and the inactive pill placebo subgroup for the primary smoking outcome measures.
For the PET data, the variable studied was the smoking-induced percent change in 11C-raclopride binding potential (BPND), defined as 100*(BPND before smoking− BPND after smoking)/BPND before smoking, for the volume-corrected mean (of the left and right VCD/NAc) BPND value. A single BPND measure was used here, based on our prior report demonstrating nearly identical BPND changes with smoking for the left and right VCD/NAc (Brody et al., 2004).
For the central study analysis, a repeated-measures ANOVA was performed, with smoking-induced BPND percent change before and after treatment as the repeated measure and treatment group as the between-subject factor. This same analysis was also performed with pre-smoking BPND as the repeated measure, as a marker of change in baseline intrasynaptic DA from before to after treatment. Pearson Product Moment Correlation Coefficients were calculated to examine relationships between pre- to post-treatment changes in smoking-induced BPND reductions and changes in variables that might be expected to be associated with smoking-induced DA concentration change, namely total puff volume, quit status, and craving alleviation. Statistical tests were performed with SPSS version 16.0 (SPSS; Chicago, IL).
3. Results
3.1 General Study Population Characteristics
At baseline, the entire study sample consisted of adults (mean ± standard deviation - 40.3 ± 11.0 years of age), who smoked 24.2 (± 6.2) cigarettes per day, had been smoking for an average of 21.6 (± 11.2) years, and were mostly men (27 men, 16 women) (Table 1). Subjects were moderate-to-severely nicotine dependent (FTND mean score - 6.5 ± 1.9), had minimal depression (mean total HAM-D 17 score of 1.6 ± 1.7) or anxiety (mean total HAM-A score of 2.0 ± 2.4), had a mean exhaled CO of 19.9 (± 9.8) parts per million (ppm) after 1 to 2 h of abstinence prior to treatment, and had moderate-to-severe craving after 3 h abstinence (mean UTS score 4.7 ± 1.4 on a scale of 0 to 6).
Table 1.
Subject Characteristics and Treatment Effects
| Variable | Total Group (n = 43) | Practical counseling-treated (n = 14) | Bupropion HCl-treated (n = 14) | Placebo-treated (n = 15) |
|---|---|---|---|---|
| Age | 40.3 (± 11.0) | 40.7 (± 10.0) | 41.4 (± 14.3) | 39.0 (± 10.0) |
| Percent female | 37.2% | 35.7% | 35.7% | 40.0% |
| Years smoking | 21.6 (± 11.2) | 22.1 (± 11.8) | 22.4 (± 13.1) | 20.3 (± 9.3) |
| Education (yrs) | 13.9 (± 2.3) | 14.2 (± 2.6) | 13.3 (± 1.9) | 14.2 (± 2.2) |
| HAM-D | 1.6 (± 1.7) | 1.1 (± 1.4) | 2.0 (± 2.1) | 1.5 (± 1.5) |
| HAM-A | 2.0 (± 2.4) | 1.6 (± 2.0) | 2.6 (± 3.3) | 1.8 (± 1.7) |
| FTND pre-tx | 6.5 (± 1.9) | 6.3 (± 2.0) | 6.2 (± 2.0) | 6.9 (± 1.7) |
| FTND post-tx | 3.6 (± 2.6)*** | 3.5 (± 3.0)** | 2.4 (± 2.0)*** | 4.8 (± 2.2)** |
| Cigs/day pre-tx | 24.3 (± 6.2) | 23.9 (± 6.9) | 24.3 (± 6.1) | 24.5 (± 6.0) |
| Cigs/day post-tx | 9.6 (± 7.9)*** | 8.8 (± 8.4)*** | 6.5 (± 6.9)*** | 13.4 (± 7.1)*** |
| Exhaled CO pre-tx (ppm) | 20.1 (± 7.3) | 18.1 (± 8.0) | 20.8 (± 12.8) | 20.8 (± 8.3) |
| Exhaled CO post-tx (ppm) | 12.8 (± 9.1)* | 11.2 (± 6.7)* | 9.5 (± 8.0)* | 10.4 (± 10.5)* |
| Craving change with smoking: pre-tx (UTS) | −3.9 (± 1.2) | −3.4 (± 1.6) | −3.9 (± 0.9) | −4.2 (± 1.0) |
| Craving change with smoking: post-tx (UTS) | −2.8 (± 1.4)*** | −2.5 (± 1.5)*** | −2.6 (± 1.6)*** | −3.4 (± 1.1)*** |
| Total Puff Volume pre-tx (mL) | 787.5 (± 32.7) | 766.8 (± 59.1) | 795.5 (± 72.7) | 799.5 (± 39.0) |
| Total Puff Volume post-tx (mL) | 669.5 (± 36.0)** | 716.3 (± 51.2) | 582.5 (± 70.8)** | 706.9 (± 61.6) |
Values are stated as mean (± standard deviation); Yrs = years; HAM-D = Hamilton Depression Rating Scale; HAM-A = Hamilton Anxiety Rating Scale; tx = treatment; FTND = Fagerström Test for Nicotine Dependence; cig = cigarette; CO = carbon monoxide; ppm = parts per million; UTS = Urge to Smoke Scale;
P < 0.05,
P < 0.01, and
P < 0.001, for paired Student t-tests from pre- to post- treatment
At baseline, the three treatment subgroups did not differ significantly in any of the smoking, demographic, or rating scale variables (Student’s t-tests, range of P values from 0.22 for the comparison of HAM-D scores for the two active treatment subgroups to 0.99 for comparison of pre-treatment cigarettes per day for the two active treatment subgroups) (Table 1).
From before to after treatment, 5 of the 14 subjects in each of the two active treatment subgroups (PGC- and bupropion HCl-treated) met criteria for having quit smoking, while only 1 of the 15 pill placebo-treated patients quit smoking (Chi-Square test, P = 0.05 for each active treatment subgroup compared to the placebo-treated subgroup). Of the 11 subjects total who quit smoking, 5 had been abstinent for the minimum of 7 days at the post-treatment scan, while the remaining 6 had quit earlier in treatment and remained abstinent through the post-treatment scan. The mean (± standard deviation) length of abstinence at the time of the second PET scan for those who quit smoking was 19.9 (± 9.0) days. Within the entire sample and each of the treated subgroups, the following variables decreased significantly from pre- to post- treatment: FTND scores, number of cigarettes per day, exhaled CO levels, and craving change from smoking a cigarette (Table 1). The bupropion HCl-treated group had significantly greater reductions in number of cigarettes per day and exhaled CO than the placebo-treated group (both Student’s t-tests, two-tailed, P < 0.05), while the PGC-treated subgroup had greater numerical reductions in number of cigarettes per day and exhaled CO than the placebo-treated group, but these differences did not reach statistical significance (Student’s t-tests, 2-tailed, P = 0.2 and 0.3, respectively) (Table 1). In addition, the total group and the bupropion HCl-treated subgroup smoked significantly smaller total puff volumes during PET scanning from pre- to post- treatment (both Student’s t-tests, P < .01). During the month of follow-up treatment after the second scanning session, no subjects relapsed as a result of smoking a cigarette during the post-treatment PET scanning session.
3.2 Treatment Effects on Smoking-Induced Change in DA Concentration
The entire sample had a significant mean reduction in VCD/NAc BPND with smoking for both the before (−8.6 ± 1.6%, paired Student’s t-test, 2-tailed, P < 0.0001) and after (−4.1 ± 1.7%, paired Student’s t-test, 2-tailed, P < 0.05) treatment scanning sessions, indicating that cigarette smoking resulted in a significant increase in DA concentration both before and after treatment (Table 2).
Table 2.
Treatment Effects on Ventral Caudate/Nucleus Accumbens 11C-Raclopride Binding Potential (BPND)
| Variable | Total Treated Group (n = 43) | Practical Group Counseling-treated (n = 14) | Bupropion HCl-treated (n = 14) | Pill Placebo-treated (n = 15) |
|---|---|---|---|---|
| Pre-treatment Pre-smoking |
2.48 (± 0.07) | 2.34 (± 0.14) | 2.53 (± 0.13) | 2.55 (± 0.11) |
| Pre-treatment Post-smoking |
2.26 (± 0.08) | 2.13 (± 0.13) | 2.32 (± 0.13) | 2.32 (± 0.14) |
| % change in VCD/NAc BPND pre-treatment | −8.56 (± 1.64)*** | −8.45 (± 2.92)** | −7.60 (± 3.01)** | −9.55 (± 2.76)** |
| Post-treatment Pre-smoking |
2.28 (± 0.08) | 2.11 (± 0.11) | 2.28 (± 0.16) | 2.43 (± 0.14) |
| Post-treatment Post-smoking |
2.16 (± 0.07) | 2.09 (± 0.11) | 2.12 (± 0.15) | 2.27 (± 0.13) |
| % change in VCD/NAc BPND post-treatment | −4.13 (± 0.70)* | −1.09 (± 2.06) | −5.56 (± 3.29) | −5.64 (± 3.29) |
Values are stated as mean (± standard error of the mean);
P < 0.05,
P < 0.01, and
P < 0.001, pre- to post smoking, paired Student’s t-tests; VCD/NAc = ventral caudate/nucleus accumbens; BPND = binding potential
For the central analysis of the study, there was a main effect of time (pre- to post-treatment) on smoking-induced percent change in 11C-raclopride BPND (repeated-measures ANOVA; F = 5.2; df = 1, 40; P = 0.03), but no effect of treatment subgroup (repeated-measures ANOVA; F = 0.6; df = 2, 40; n.s.), indicating decreased DA release from pre- to post- treatment during the PET procedure. For the analysis of change in baseline BPND from before to after treatment, there was a main effect of time (repeated-measures ANOVA; F = 4.9; df = 1, 40; P = 0.03), with no effect of treatment subgroup (repeated-measures ANOVA; F = 0.2; df = 2, 40; n.s.), indicating increased intrasynaptic DA from pre- to post- treatment regardless of treatment type. The correlation between percent change in smoking-induced 11C-raclopride BPND reduction and change in total puff volume was highly significant (r = −0.49, P = 0.001) (Figure 1), indicating that the amount of a cigarette smoked during scanning was closely associated with the extent of smoking-induced DA release. Other correlations between change in 11C-raclopride BPND reduction and change in number of cigarettes per day, exhaled CO levels, and craving reduction with smoking were not significant (all P values > 0.1).
Figure 1.
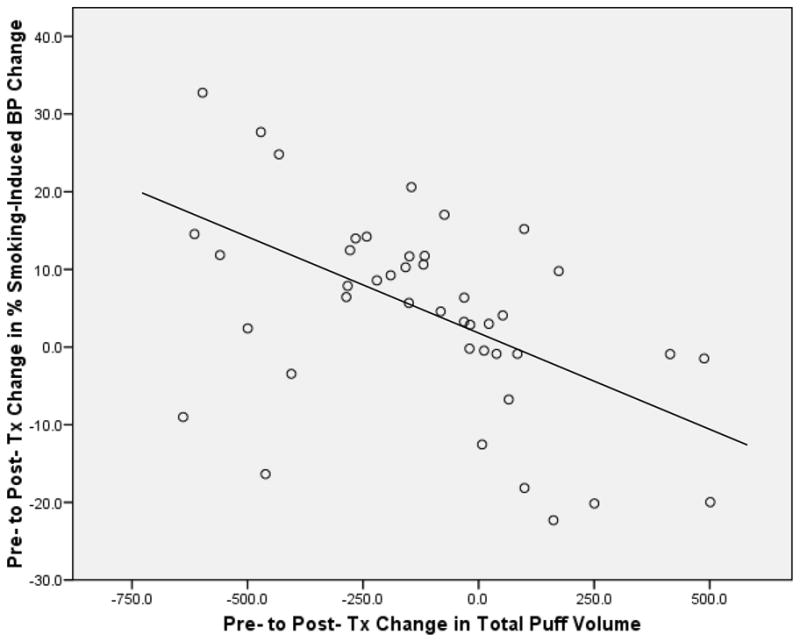
Scatterplot showing the significant correlation (r = −0.49, P = 0.001) between pre- to post- treatment changes in total puff volume (x axis) and percent change in smoking-induced binding potential (BP) in the ventral striatum (y axis).
4. Discussion
The central finding of this study was a reduction in smoking-induced change in 11C-raclopride binding potential (BPND) from pre- to post- treatment that was correlated with the total puff volume of a cigarette smoked during scanning. There were no significant associations between the PET measure and treatment type, quit status, or withdrawal symptoms. Because percent change in 11C-raclopride BPND is an indirect measure of change in intrasynaptic DA concentration (and therefore DA release), the association between this measure and total puff volume indicates that smoking-induced DA release is dose-dependent, regardless of treatment effects or smoking status, at least early in abstinence as was studied here.
The timing of the length of abstinence may be particularly important in interpreting study results, given the known time course of normalization of nicotinic acetylcholine receptor (nAChR) densities in brain DA pathways. In this study, 5 of the 11 quitters had 1 cigarette-free week at the time of the second scan, and the quitter group as a whole had a mean 19.9 days abstinence. On a cellular level, the interaction of inhaled nicotine from smoking with nAChRs on cells of the midbrain ventral tegmental area is thought to lead to DA release in the VST/NAc (Nisell et al., 1994). Studies show that nAChRs are upregulated in cigarette smokers (Benwell et al., 1988; Breese et al., 1997; Mamede et al., 2007) and remain elevated in the first 10 days of abstinence (Mamede et al., 2007), but normalize to levels of non-smokers in 3 weeks (Mamede et al., 2007) to 2 months (Breese et al., 1997). Given that our study indicated that smoking-induced DA release was dose-dependent (regardless of smoking status in a group of smokers with a mean of slightly less than 3 weeks abstinence), this study did not rule out the possibility that a longer period of abstinence might result in alterations in the relationship between amount of a cigarette smoked and smoking-induced DA release. Alternatively, study results may indicate that smoking-induced DA release indeed remains dose-dependent regardless of smoking status or treatment effects. Studies of smokers with longer durations of abstinence would help resolve this issue.
Other PET findings of interest included the post-treatment smoking-induced decrease in 11C-raclopride BPND, which supports earlier reports in untreated smokers (Brody et al., 2004; Brody et al., 2006; Scott et al., 2007), and the somewhat surprising absence of an association between 11C-raclopride BPND and craving, as was observed in prior work (Brody et al., 2004; Brody et al., 2006). This latter effect may have been due to sample size/power issues, or may indicate that treatment alters the relationship between craving and DA concentration.
Results of the clinical treatment portion of the study demonstrated the expected decreases in smoking measures (FTND scores, reported numbers of cigarettes per day, and exhaled CO levels) for all treatments, with more robust findings in the actively-treated than the placebo-treated groups (Table 1). These findings were consistent with prior research examining the effects of bupropion HCl treatment on smoking measures (Fiore et al., 1994; Hurt et al., 1997). In addition, the entire sample (3 subgroups combined) had lower total puff volumes of the cigarette smoked during the follow-up PET session, and this effect was strongest in the bupropion-treated group (Table 1). These outcomes may have been due to treated smokers intentionally inhaling less deeply due a desire to avoid smoking during their attempt to remain abstinent or may have been due to a pharmacological effect of chronic bupropion HCl treatment resulting in increased intrasynaptic DA (Horst and Preskorn, 1998; Holm and Spencer, 2000; Balfour, 2001; Stahl et al., 2004) with less of a need to inhale a cigarette as deeply to achieve the same intrasynaptic DA level. This last point is supported by the overall group having lower baseline 11C-raclopride BPND from pre- to post- treatment, which may indicate increased baseline intrasynaptic DA (though differences in radiotracer metabolism or small variability in radiotracer dosing may have contributed to this finding).
Strengths of this study included: a homogeneous subject sample in regard to demographic variables, cigarette usage, and absence of co-morbidity; a statistical method that minimized type I error by using a single ROI BPND value; and the isolation of treatment effects by providing each treatment type alone. Weaknesses were primarily ones inherent to PET scanning with 11C-raclopride. While this radiotracer is commonly used for the evaluation of DA release (including work by our group (Brody et al., 2004; Brody et al., 2006; Brody et al., 2009) and others (e.g- (Volkow et al., 1994; Carson et al., 1997; Schlaepfer et al., 1997; Ginovart et al., 2002; Marenco et al., 2004; Barrett et al., 2004; Scott et al., 2007)), and consistently demonstrates DA release when expected, the short half-life and variable pharmacokinetics of the radiotracer make its use logistically challenging for human studies, and resulted in considerable variability within our data set (Table 2). Other limitations of the study included the modest sample size (which may have diminished the ability to detect group differences) and the absence of plasma nicotine levels. The duration of treatment (8 weeks), which was chosen in order to have smokers who were non-responsive to medication/placebo treated for the shortest period of time while including a typical 12-session course of PGC, was also a potentially confounding factor. This relatively short duration of treatment may have limited the ability to elicit an effect on smoking-induced DA release, especially for the PGC-treated group. Despite these limitations, this study did demonstrate that cigarette smoking resulted in VCD/NAc BPND change in the hypothesized direction (both before and after treatment), and these results add confidence to the findings here.
Acknowledgments
Supported by the Tobacco-Related Disease Research Program (A.L.B. [11RT-0024 and 16RT-0098], the National Institute on Drug Abuse (A.L.B. [R01 DA15059 and DA20872]), a Veterans Affairs Type I Merit Review Award (A.L.B.), and the Office of National Drug Control Policy (E.D.L [DABT63-00-C-1003]). The authors thank Josephine Ribe and Michael Clark for technical support in performing positron emission tomography and magnetic resonance imaging scans, respectively.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- Abrams DB, Niaura RS, Brown RA, Emmons KM, Goldstein MG, Monti PM. The Tobacco Dependence Treatment Handbook: A Guide to Best Practices. The Guilford Press; 2003. [Google Scholar]
- Amini F, Lewis T, Lannon R, Louie A, Baumbacher G, McGuinness T, Schiff EZ. Affect, attachment, memory: contributions toward psychobiologic integration. Psychiatry. 1996;59:213–239. [PubMed] [Google Scholar]
- Argyelan M, Szabo Z, Kanyo B, Tanacs A, Kovacs Z, Janka Z, Pavics L. Dopamine transporter availability in medication free and in bupropion treated depression: a 99mTc-TRODAT-1 SPECT study. Journal of Affective Disorders. 2005;89:115–123. doi: 10.1016/j.jad.2005.08.016. [DOI] [PubMed] [Google Scholar]
- Ascher JA, Cole JO, Colin JN, Feighner JP, Ferris RM, Fibiger HC, Golden RN, Martin P, Potter WZ, Richelson E, Sulser F. Bupropion: a review of its mechanism of antidepressant activity. Journal of Clinical Psychiatry. 1995;56:395–401. [PubMed] [Google Scholar]
- Balfour DJ. The pharmacology underlying pharmacotherapy for tobacco dependence: A focus on bupropion. International Journal of Clinical Practice. 2001;55:53–57. [PubMed] [Google Scholar]
- Bamford NS, Zhang H, Joyce JA, Scarlis CA, Hanan W, Wu NP, Andre VM, Cohen R, Cepeda C, Levine MS, Harleton E, Sulzer D. Repeated exposure to methamphetamine causes long-lasting presynaptic corticostriatal depression that is renormalized with drug readministration. Neuron. 2008;58:89–103. doi: 10.1016/j.neuron.2008.01.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett SP, Boileau I, Okker J, Pihl RO, Dagher A. The hedonic response to cigarette smoking is proportional to dopamine release in the human striatum as measured by positron emission tomography and [(11)C]raclopride. Synapse. 2004;54:65–71. doi: 10.1002/syn.20066. [DOI] [PubMed] [Google Scholar]
- Benwell ME, Balfour DJ. Regional variation in the effects of nicotine on catecholamine overflow in rat brain. European Journal of Pharmacology. 1997;325:13–20. doi: 10.1016/s0014-2999(97)00101-5. [DOI] [PubMed] [Google Scholar]
- Benwell ME, Balfour DJK, Anderson JM. Evidence that tobacco smoking increases the density of (−)-[3H]nicotine binding sites in human brain. Journal of Neurochemistry. 1988;50:1243–1247. doi: 10.1111/j.1471-4159.1988.tb10600.x. [DOI] [PubMed] [Google Scholar]
- Bilder RM, Volavka J, Lachman HM, Grace AA. The catechol-o-methyltransferase polymorphism: relations to the tonic-phasic dopamine hypothesis and neuropsychiatric phenotypes. Neuropsychopharmacology. 2004;29:1943–1961. doi: 10.1038/sj.npp.1300542. [DOI] [PubMed] [Google Scholar]
- Breese CR, Marks MJ, Logel J, Adams CE, Sullivan B, Collins AC, Leonard S. Effect of smoking history on [3H]nicotine binding in human postmortem brain. Journal of Pharmacology and Experimental Therapeutics. 1997;282:7–13. [PubMed] [Google Scholar]
- Brody AL, Mandelkern MA, Olmstead RE, Allen-Martinez Z, Scheibal D, Abrams AL, Costello MR, Farahi J, Saxena S, Monterosso J, London ED. Ventral striatal dopamine release in response to smoking a regular vs a denicotinized cigarette. Neuropsychopharmacology. 2009;34:282–289. doi: 10.1038/npp.2008.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brody AL, Mandelkern MA, Olmstead RE, Scheibal D, Hahn E, Shiraga S, Zamora-Paja E, Farahi J, Saxena S, London ED, McCracken JT. Gene variants of brain dopamine pathways and smoking-induced dopamine release in the ventral caudate/nucleus accumbens. Archives of General Psychiatry. 2006;63:808–816. doi: 10.1001/archpsyc.63.7.808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brody AL, Olmstead RE, London ED, Farahi J, Meyer JH, Grossman P, Lee GS, Huang J, Hahn EL, Mandelkern MA. Smoking-induced ventral striatum dopamine release. American Journal of Psychiatry. 2004;161:1211–1218. doi: 10.1176/appi.ajp.161.7.1211. [DOI] [PubMed] [Google Scholar]
- Carmody TP. Preventing relapse in the treatment of nicotine addiction: current issues and future directions. Journal of Psychoactive Drugs. 1990;22:211–238. doi: 10.1080/02791072.1990.10472545. [DOI] [PubMed] [Google Scholar]
- Carson RE, Breier A, de Bartolomeis A, Saunders RC, Su TP, Schmall B, Der MG, Pickar D, Eckelman WC. Quantification of amphetamine-induced changes in [11C]raclopride binding with continuous infusion. Journal of Cerebral Blood Flow and Metabolism. 1997;17:437–447. doi: 10.1097/00004647-199704000-00009. [DOI] [PubMed] [Google Scholar]
- Damsma G, Day J, Fibiger HC. Lack of tolerance to nicotine-induced dopamine release in the nucleus accumbens. European Journal of Pharmacology. 1989;168:363–368. doi: 10.1016/0014-2999(89)90798-x. [DOI] [PubMed] [Google Scholar]
- Dewey SL, Brodie JD, Gerasimov M, Horan B, Gardner EL, Ashby CRJ. A pharmacologic strategy for the treatment of nicotine addiction. Synapse. 1999;31:76–86. doi: 10.1002/(SICI)1098-2396(199901)31:1<76::AID-SYN10>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- Di Chiara G, Imperato A. Drugs abused by humans preferentially increase synaptic dopamine concentrations in the mesolimbic system of freely moving rats. Proceedings of the National Academy of Sciences of the United States of America. 1988;85:5274–5278. doi: 10.1073/pnas.85.14.5274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrin E, Gawell L, Hogberg T, Depaulis T, Strom P. Synthesis of [Methoxy-H-3]- and [Methoxy-C-11]-Labeled Raclopride - Specific Dopamine-D2 Receptor Ligands. Journal of Labelled Compounds & Radiopharmaceuticals. 1987;24:931–940. [Google Scholar]
- Fagerström KO. Measuring the degree of physical dependence to tobacco smoking with reference to individualization of treatment. Addictive Behaviors. 1978;3:235–241. doi: 10.1016/0306-4603(78)90024-2. [DOI] [PubMed] [Google Scholar]
- Farde L, Hall H, Ehrin E, Sedvall G. Quantitative analysis of D2 dopamine receptor binding in the living human brain by PET. Science. 1986;231:258–261. doi: 10.1126/science.2867601. [DOI] [PubMed] [Google Scholar]
- Fiore MC, Bailey WC, Cohen SJ, Dorfman SF, Goldstein MG, Gritz ER, Heyman RB, Jaen CR, Kottke TE, Lando HA, Mecklenburg RE, Mullen PD, Nett LM, Robinson L, Stitzer ML, Tommasello AC, Villejo L, Wewers ME. Clinical Practice Guideline US Department of Health and Human Services. Public Health Service; Rockville, MD: 2000. Treating Tobacco Use and Dependence. [Google Scholar]
- Fiore MC, Kenford SL, Jorenby DE, Wetter DW, Smith SS, Baker TB. Two studies of the clinical effectiveness of the nicotine patch with different counseling treatments. Chest. 1994;105:524–533. doi: 10.1378/chest.105.2.524. [DOI] [PubMed] [Google Scholar]
- First MB, Spitzer RL, Gibbon M, Williams JBW. Structured Clinical Interview for DSM-IV Axis I Disorders. 1995 doi: 10.1001/archpsyc.1992.01820080032005. Patient Edition (SCID-I/P, version 2.0) [DOI] [PubMed] [Google Scholar]
- Foley KF, DeSanty KP, Kast RE. Bupropion: pharmacology and therapeutic applications. Expert Reviews of Neurotherapy. 2006;6:1249–1265. doi: 10.1586/14737175.6.9.1249. [DOI] [PubMed] [Google Scholar]
- Ginovart N, Hassoun W, Le Cavorsin M, Veyre L, Le Bars D, Leviel V. Effects of amphetamine and evoked dopamine release on [C-11]raclopride binding in anesthetized cats. Neuropsychopharmacology. 2002;27:72–84. doi: 10.1016/S0893-133X(02)00285-3. [DOI] [PubMed] [Google Scholar]
- Gold PB, Rubey RN, Harvey RT. Naturalistic, self-assignment comparative trial of bupropion SR, a nicotine patch, or both for smoking cessation treatment in primary care. American Journal of Addiction. 2002;11:315–331. doi: 10.1080/1055049029008811. [DOI] [PubMed] [Google Scholar]
- Hall SM, Humfleet GL, Reus VI, Munoz RF, Hartz DT, Maude-Griffin R. Psychological intervention and antidepressant treatment in smoking cessation. Archives of General Psychiatry. 2002;59:930–936. doi: 10.1001/archpsyc.59.10.930. [DOI] [PubMed] [Google Scholar]
- Hamilton M. Development of a rating scale for primary depressive illness. British Journal of Social Psychology. 1967;6:278–296. doi: 10.1111/j.2044-8260.1967.tb00530.x. [DOI] [PubMed] [Google Scholar]
- Hamilton M. Diagnosis and rating of anxiety. British Journal of Psychiatry. 1969;3:76–79. [Google Scholar]
- Heatherton TF, Kozlowski LT, Frecker RC, Fagerström KO. The Fagerström Test for Nicotine Dependence: a revision of the Fagerström Tolerance Questionnaire. British Journal of Addiction. 1991;86:1119–1127. doi: 10.1111/j.1360-0443.1991.tb01879.x. [DOI] [PubMed] [Google Scholar]
- Hoffman BJ, Hansson SR, Mezey E, Palkovits M. Localization and dynamic regulation of biogenic amine transporters in the mammalian central nervous system. Frontiers in Neuroendocrinology. 1998;19:187–231. doi: 10.1006/frne.1998.0168. [DOI] [PubMed] [Google Scholar]
- Holm KJ, Spencer CM. Bupropion: A review of its use in the management of smoking cessation. Drugs. 2000;59:1007–1024. doi: 10.2165/00003495-200059040-00019. [DOI] [PubMed] [Google Scholar]
- Holmes S, Zwar N, Jimenez-Ruiz CA, Ryan PJ, Browning D, Bergmann L, Johnston JA. Bupropion as an aid to smoking cessation: a review of real-life effectiveness. International Journal of Clinical Practice. 2004;58:285–291. doi: 10.1111/j.1368-5031.2004.00153.x. [DOI] [PubMed] [Google Scholar]
- Horst WD, Preskorn SH. Mechanisms of action and clinical characteristics of three atypical antidepressants: venlafaxine, nefazodone, bupropion. Journal of Affective Disorders. 1998;51:237–254. doi: 10.1016/s0165-0327(98)00222-5. [DOI] [PubMed] [Google Scholar]
- Hurt RD, Sachs DP, Glover ED, Offord KP, Johnston JA, Dale LC, Khayrallah MA, Schroeder DR, Glover PN, Sullivan CR, Croghan IT, Sullivan PM. A comparison of sustained-release bupropion and placebo for smoking cessation. New England Journal of Medicine. 1997;337:1195–1202. doi: 10.1056/NEJM199710233371703. [DOI] [PubMed] [Google Scholar]
- Imperato A, Mulas A, Di Chiara G. Nicotine preferentially stimulates dopamine release in the limbic system of freely moving rats. European Journal of Pharmacology. 1986;132:337–338. doi: 10.1016/0014-2999(86)90629-1. [DOI] [PubMed] [Google Scholar]
- Innis RB, Cunningham VJ, Delforge J, Fujita M, Gjedde A, Gunn RN, Holden J, Houle S, Huang SC, Ichise M, Iida H, Ito H, Kimura Y, Koeppe RA, Knudsen GM, Knuuti J, Lammertsma AA, Laruelle M, Logan J, Maguire RP, Mintun MA, Morris ED, Parsey R, Price JC, Slifstein M, Sossi V, Suhara T, Votaw JR, Wong DF, Carson RE. Consensus nomenclature for in vivo imaging of reversibly binding radioligands. Journal of Cerebral Blood Flow and Metabolism. 2007;27:1533–1539. doi: 10.1038/sj.jcbfm.9600493. [DOI] [PubMed] [Google Scholar]
- Ito H, Hietala J, Blomqvist G, Halldin C, Farde L. Comparison of the transient equilibrium and continuous infusion method for quantitative PET analysis of [11C]raclopride binding. Journal of Cerebral Blood Flow and Metabolism. 1998;18:941–950. doi: 10.1097/00004647-199809000-00003. [DOI] [PubMed] [Google Scholar]
- Janhunen S, Ahtee L. Comparison of the effects of nicotine and epibatidine on the striatal extracellular dopamine. European Journal of Pharmacology. 2004;494:167–177. doi: 10.1016/j.ejphar.2004.05.015. [DOI] [PubMed] [Google Scholar]
- Jarvik ME, Madsen DC, Olmstead RE, Iwamoto-Schaap PN, Elins JL, Benowitz NL. Nicotine blood levels and subjective craving for cigarettes. Pharmacology Biochemistry and Behavior. 2000;66:553–558. doi: 10.1016/s0091-3057(00)00261-6. [DOI] [PubMed] [Google Scholar]
- Jorenby DE, Leischow SJ, Nides MA, Rennard SI, Johnston JA, Hughes AR, Smith SS, Muramoto ML, Daughton DM, Doan K, Fiore MC, Baker TB. A controlled trial of sustained-release bupropion, a nicotine patch, or both for smoking cessation. New England Journal of Medicine. 1999;340:685–691. doi: 10.1056/NEJM199903043400903. [DOI] [PubMed] [Google Scholar]
- Lammertsma AA, Hume SP. Simplified reference tissue model for PET receptor studies. Neuroimage. 1996;4:153–158. doi: 10.1006/nimg.1996.0066. [DOI] [PubMed] [Google Scholar]
- Liggan DY, Kay J. Some neurobiological aspects of psychotherapy: A review. Journal of Psychotherapy Practice Research. 1999;8:103–114. [PMC free article] [PubMed] [Google Scholar]
- Mamede M, Ishizu K, Ueda M, Mukai T, Iida Y, Kawashima H, Fukuyama H, Togashi K, Saji H. Temporal change in human nicotinic acetylcholine receptor after smoking cessation: 5IA SPECT study. Journal of Nuclear Medicine. 2007;48:1829–1835. doi: 10.2967/jnumed.107.043471. [DOI] [PubMed] [Google Scholar]
- Marenco S, Carson RE, Berman KF, Herscovitch P, Weinberger DR. Nicotine-induced dopamine release in primates measured with [C-11]raclopride PET. Neuropsychopharmacology. 2004;29:259–268. doi: 10.1038/sj.npp.1300287. [DOI] [PubMed] [Google Scholar]
- Marlatt GA, Gordon JRE. Relapse Prevention: maintenance strategies in the treatment of addictive behaviors. Guilford; New York: 1985. [Google Scholar]
- Martinez D, Slifstein M, Broft A, Mawlawi O, Hwang DR, Huang Y, Cooper T, Kegeles L, Zarahn E, Abi-Dargham A, Haber SN, Laruelle M. Imaging Human Mesolimbic Dopamine Transmission With Positron Emission Tomography. Part II: Amphetamine-Induced Dopamine Release in the Functional Subdivisions of the Striatum. Journal of Cerebral Blood Flow and Metabolism. 2003;23:285–300. doi: 10.1097/01.WCB.0000048520.34839.1A. [DOI] [PubMed] [Google Scholar]
- Mawlawi O, Martinez D, Slifstein M, Broft A, Chatterjee R, Hwang DR, Huang Y, Simpson N, Ngo K, Van Heertum R, Laruelle M. Imaging human mesolimbic dopamine transmission with positron emission tomography: I. Accuracy and precision of D(2) receptor parameter measurements in ventral striatum. Journal of Cerebral Blood Flow and Metabolism. 2001;21:1034–1057. doi: 10.1097/00004647-200109000-00002. [DOI] [PubMed] [Google Scholar]
- McCarthy DE, Piasecki TM, Lawrence DL, Jorenby DE, Shiffman S, Baker TB. Psychological mediators of bupropion sustained-release treatment for smoking cessation. Addiction. 2008;103:1521–1533. doi: 10.1111/j.1360-0443.2008.02275.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montgomery AJ, Lingford-Hughes AR, Egerton A, Nutt DJ, Grasby PM. The effect of nicotine on striatal dopamine release in man: A [11C]raclopride PET study. Synapse. 2007;61:637–645. doi: 10.1002/syn.20419. [DOI] [PubMed] [Google Scholar]
- Nikolaus S, Antke C, Kley K, Poeppel TD, Hautzel H, Schmidt D, Muller HW. Investigating the dopaminergic synapse in vivo. I. Molecular imaging studies in humans. Reviews in the Neurosciences. 2007a;18:439–472. doi: 10.1515/revneuro.2007.18.6.439. [DOI] [PubMed] [Google Scholar]
- Nikolaus S, Larisch R, Beu M, Antke C, Kley K, Forutan F, Wirrwar A, Muller HW. Investigating the dopaminergic synapse in vivo. II. Molecular imaging studies in small laboratory animals. Reviews in the Neurosciences. 2007b;18:473–504. doi: 10.1515/revneuro.2007.18.6.473. [DOI] [PubMed] [Google Scholar]
- Nisell M, Nomikos GG, Svensson TH. Systemic nicotine-induced dopamine release in the rat nucleus accumbens is regulated by nicotinic receptors in the ventral tegmental area. Synapse. 1994;16:36–44. doi: 10.1002/syn.890160105. [DOI] [PubMed] [Google Scholar]
- Nomikos GG, Damsma G, Wenkstern D, Fibiger HC. Acute effects of bupropion on extracellular dopamine concentrations in rat striatum and nucleus accumbens studied by in vivo microdialysis. Neuropsychopharmacology. 1989;2:273–279. doi: 10.1016/0893-133x(89)90031-6. [DOI] [PubMed] [Google Scholar]
- Paluck EC, McCormack JP, Ensom MH, Levine M, Soon JA, Fielding DW. Outcomes of bupropion therapy for smoking cessation during routine clinical use. Annals of Pharmacotherapy. 2006;40:185–190. doi: 10.1345/aph.1G324. [DOI] [PubMed] [Google Scholar]
- Paterson NE, Balfour DJ, Markou A. Chronic bupropion attenuated the anhedonic component of nicotine withdrawal in rats via inhibition of dopamine reuptake in the nucleus accumbens shell. European Journal of Neuroscience. 2007;25:3099–3108. doi: 10.1111/j.1460-9568.2007.05546.x. [DOI] [PubMed] [Google Scholar]
- Pontieri FE, Tanda G, Orzi F, Di Chiara G. Effects of nicotine on the nucleus accumbens and similarity to those of addictive drugs. Nature. 1996;382:255–257. doi: 10.1038/382255a0. [DOI] [PubMed] [Google Scholar]
- Roffman JL, Marci CD, Glick DM, Dougherty DD, Rauch SL. Neuroimaging and the functional neuroanatomy of psychotherapy. Psychological Medicine. 2005;35:1385–1398. doi: 10.1017/S0033291705005064. [DOI] [PubMed] [Google Scholar]
- Schlaepfer TE, Pearlson GD, Wong DF, Marenco S, Dannals RF. PET study of competition between intravenous cocaine and [11C]raclopride at dopamine receptors in human subjects. American Journal of Psychiatry. 1997;154:1209–1213. doi: 10.1176/ajp.154.9.1209. [DOI] [PubMed] [Google Scholar]
- Schuh KJ, Stitzer ML. Desire to smoke during spaced smoking intervals. Psychopharmacology (Berl) 1995;120:289–295. doi: 10.1007/BF02311176. [DOI] [PubMed] [Google Scholar]
- Scott DJ, Domino EF, Heitzeg MM, Koeppe RA, Ni L, Guthrie S, Zubieta JK. Smoking modulation of mu-opioid and dopamine D2 receptor-mediated neurotransmission in humans. Neuropsychopharmacology. 2007;32:450–457. doi: 10.1038/sj.npp.1301238. [DOI] [PubMed] [Google Scholar]
- Simon JA, Duncan C, Carmody TP, Hudes ES. Bupropion for smoking cessation: a randomized trial. Archives of Internal Medicine. 2004;164:1797–1803. doi: 10.1001/archinte.164.16.1797. [DOI] [PubMed] [Google Scholar]
- Stahl SM, Pradko JF, Haight BR, Modell JG, Rockett CB, Learned-Coughlin S. A Review of the Neuropharmacology of Bupropion, a Dual Norepinephrine and Dopamine Reuptake Inhibitor. Primary Care Companion Journal of Clinical Psychiatry. 2004;6:159–166. doi: 10.4088/pcc.v06n0403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stead LF, Lancaster T. Group behaviour therapy programmes for smoking cessation. Cochrane Database Syst Rev. 2005:CD001007. doi: 10.1002/14651858.CD001007.pub2. [DOI] [PubMed] [Google Scholar]
- Stout RL, Wirtz PW, Carbonari JP, Del Boca FK. Ensuring balanced distribution of prognostic factors in treatment outcome research. Journal of Studies on Alcohol Suppl. 1994;12:70–75. doi: 10.15288/jsas.1994.s12.70. [DOI] [PubMed] [Google Scholar]
- Sziraki I, Lipovac MN, Hashim A, Sershen H, Allen D, Cooper T, Czobor P, Lajtha A. Differences in nicotine-induced dopamine release and nicotine pharmacokinetics between Lewis and Fischer 344 rats. Neurochemistry Research. 2001;26:609–617. doi: 10.1023/a:1010979018217. [DOI] [PubMed] [Google Scholar]
- Takahashi H, Fujimura Y, Hayashi M, Takano H, Kato M, Okubo Y, Kanno I, Ito H, Suhara T. Enhanced dopamine release by nicotine in cigarette smokers: a double-blind, randomized, placebo-controlled pilot study. International Journal of Neuropsychopharmacology. 2008;11:413–417. doi: 10.1017/S1461145707008103. [DOI] [PubMed] [Google Scholar]
- Tsukada H, Miyasato K, Kakiuchi T, Nishiyama S, Harada N, Domino EF. Comparative effects of methamphetamine and nicotine on the striatal [C-11]raclopride binding in unanesthetized monkeys. Synapse. 2002;45:207–212. doi: 10.1002/syn.10102. [DOI] [PubMed] [Google Scholar]
- Volkow ND, Wang GJ, Fowler JS, Logan J, Schlyer D, Hitzemann R, Lieberman J, Angrist B, Pappas N, MacGregor R, Burr G, Cooper T, Wolf AP. Imaging endogenous dopamine competition with [11C]raclopride in the human brain. Synapse. 1994;16:255–262. doi: 10.1002/syn.890160402. [DOI] [PubMed] [Google Scholar]
- Watabe H, Endres CJ, Breier A, Schmall B, Eckelman WC, Carson RE. Measurement of dopamine release with continuous infusion of [11C]raclopride: optimization and signal-to-noise considerations. Journal of Nuclear Medicine. 2000;41:522–530. [PubMed] [Google Scholar]
- Woods RP, Mazziotta JC, Cherry SR. MRI-PET registration with automated algorithm. Journal of Computer Assisted Tomography. 1993;17:536–546. doi: 10.1097/00004728-199307000-00004. [DOI] [PubMed] [Google Scholar]