

Abstract
LPS is a natural adjuvant that potentiates antigen (Ag)-specific T cell survival and Th1 differentiation by stimulating MyD88 and TRIF signaling pathways. Here, we reveal the TRIF pathway is critical for amplifying murine effector T cell accumulation into non-lymphoid tissues following immunization with Ag plus LPS. Although LPS increased the accumulation of splenic T cells in TRIF-deficient mice, markedly fewer T cells were recovered from liver and lung in comparison to WT. Most of the T cells primed in TRIF-deficient mice failed to upregulate CXCR3 and had an overall reduced capacity to produce IFN-γ, demonstrating effector T cell differentiation was linked to their migration. To investigate the role of TRIF-dependent cytokines, neutralization studies were performed in WT mice. Although TNF neutralization reduced T cell numbers, its co-neutralization with IL-10 unexpectedly restored the T cells, suggesting the balance between pro- and anti-inflammatory cytokines influences T cell survival rather than their magnitude. To investigate a role for costimulatory molecules, we tested if the T cell defect in TRIF-deficient mice could be corrected with enforced costimulation. Boosting with a CD40 agonist in addition to LPS restored the effector CD8 T cell response in livers of TRIF-deficient mice while only partially restoring CD4 T cells, suggesting that LPS primes CD8 and CD4 T cell immunity through different mechanisms. Overall, our data support targeting TRIF for vaccines aimed to direct immune responses to non-lymphoid tissues.
Keywords: T Cells, Lipopolysaccharide, Superantigens, Costimulation, Cell Activation
Introduction
Lipopolysaccharide (LPS) is a component of gram-negative bacteria with natural adjuvant activity, enhancing T cell clonal survival and effector differentiation. Thus, injecting LPS within 24 h after antigen (Ag) increases the accumulation and persistence of Ag-responsive T cells as well as their capacity to produce the Th1 cytokine interferon (IFN)-γ (1, 2). Although the innate response to LPS has been extensively studied, its link to T cell activation in vivo is not fully understood.
LPS detection is a multi-step process involving the cooperation of many germline-encoded products. In general, the serum factor LPS binding protein binds to LPS molecules and transfers them to CD14, which in turn transfers LPS to toll-like receptor (TLR) 4-MD-2 complexes on cell surfaces (3). TLR4 stimulation recruits the intracellular signaling adaptors MyD88 and TRIF, which independently propagate the signal. Both MyD88 and TRIF activate NF-kappa;B and AP-1 transcription factors and TRIF also activates IRF3 (4). These pathways result in production of pro-inflammatory cytokines such as TNF, IL-1, IL-6, and IL-12, as well as phenotypic activation of Ag presenting cells (APCs), enhancing their ability to prime T cells (5). In addition, LPS induces IL-10 which reduces levels of inflammatory cytokines (6, 7). Both MyD88 and TRIF are required for cytokine production in response to LPS while upregulation of the costimulatory molecules CD40 and CD86 only requires TRIF (8–11). Overall, the MyD88 and TRIF pathways appear to fully account for LPS responsiveness (8, 10).
LPS adjuvanticity depends upon its pro-inflammatory activity. TNF is important for T cell survival, as neutralizing this cytokine decreases T cell recovery following immunization with Ag and LPS (1). Although TNF production can be regulated by IL-10 (6), the impact of LPS-induced IL-10 on T cell survival in vivo has not been examined. Interleukin-12 appears to be specialized for promoting Th1 differentiation rather than survival of CD4 T cells, and can substitute for LPS in this respect (2). A variety of models have identified important roles for IL-12 in Th1 differentiation, however, significant Th1 responses can also be generated in its absence (12–15). Cell-associated factors such as costimulatory molecules also contribute to LPS adjuvanticity, as CD28 is required for LPS to enhance peptide-specific CD4 T cell responses (16). Interestingly, LPS increases the survival of superantigen-stimulated T cells independently of CD28 (17), indicating that redundant mechanisms can provide a costimulatory function.
Elucidating the roles of signaling adaptors is important to understand how LPS detection is linked to adjuvanticity. Previously, we reported that MyD88 is required for LPS to enhance long-term T cell survival in vivo (18). However, the acquisition of effector potential was MyD88-independent, demonstrating that distinct mechanisms are used to drive CD4 T cell survival and differentiation. In contrast to the MyD88 pathway, costimulatory agonists effectively promote CD4 T cell effector differentiation without generating robust long-term survival (18–20). These findings support the concept that pro-inflammatory cytokines are specialized for generating CD4 T cell survival while costimulation supports their differentiation. This generalization is not absolute, however, since OX40 and 4-1BB costimulatory agonists synergize to enhance CD8 T cell survival (21), and IL-18 supports effector differentiation without promoting long-term survival (22).
Previous studies have suggested that TRIF signaling has a positive impact on T cell priming. TRIF was required for LPS to generate optimal IFN-γ production and cytolytic activity among endogenous activated CD8 T cells, as well as proliferation among CD4 T cells (9). However, it was not determined if TRIF signaling specifically enhanced the survival or differentiation of T cells. Recently, TRIF was found to be required for optimal T cell clonal expansion following immunization with LPS, although long-term survival and effector differentiation were not examined (23). Even less well understood, but perhaps most importantly, is the impact of TLR signaling adaptors on responses in non-lymphoid tissues where T cells preferentially accumulate following their activation (24). Therefore, our understanding of how the LPS-TRIF pathway influences T cell activation in vivo remains incomplete.
In this study, we examined the role of TRIF in long-term T cell survival and effector function following immunization with Ag plus LPS. LPS supported T cell accumulation in TRIF-deficient lymphoid tissues even though optimal innate responsiveness required TRIF. To mimic the TRIF-deficient environment in WT mice, a pro-inflammatory (TNF) and anti-inflammatory (IL-10) cytokine were neutralized. This resulted in normal T cell survival suggesting that cytokine balance rather than magnitude determines the outcome of responses. Despite having normal lymphoid T cell accumulation, significantly fewer T cells were recovered from the liver and lungs of TRIF-deficient mice, and overall a smaller percentage of T cells were capable of producing IFN-γ. This was the case even though we initiated a powerful endogenous T cell response with the pathogen-derived toxin Staphylococcal Enterotoxin A (SEA). Using a peptide-based model revealed that CXCR3 upregulation was TRIF-dependent, offering a possible explanation for the reduced T cell accumulation in liver. Enforced CD40 costimulation restored the CD8 TRIF-deficient T cell response, suggesting the TRIF pathway supports migration to non-lymphoid tissue through the CD40 pathway. However, CD4 TRIF-deficient T cells were only partially affected suggesting they require a greater breadth of costimulation. Overall, we show the TRIF pathway preferentially supports effector T cell accumulation into non-lymphoid tissues, a result that impacts vaccine adjuvant design.
Materials and Methods
Mice
C57BL/6 mice and TRIF-deficient mice were purchased from the Jackson Laboratory (Bar Harbor, ME). TRIF-deficient and SM1 TCR transgenic mice (25) (CD4+ CD90.1+ Vβ2+ RAG−/−) were bred by our laboratory. All mice were maintained in the animal facility at the University of Connecticut Health Center under specific pathogen-free conditions and handled in accordance to National Institutes of Health federal guidelines.
Serum cytokines
Blood was taken from tail veins of mice at 1.5 h and 6 h following injection of 150 μg Salmonella typhimurium LPS (Sigma-Aldrich). Blood was kept on ice for 30 min followed by centrifugation at 13,000 rpm and 4°C for 20 min, with serum partitioning as the upper fraction. Levels of TNF, IL-10, IFN-γ, and IL-12p70 were determined by ELISA using kits from BD Biosciences.
Immunization
Reagents were diluted in PBS and injected i.p. in a total volume of 0.2 mL. SEA (Toxin Tech; Sarasota, FL) was injected at 1 μg per mouse. For adoptive transfer studies, ~5 × 105 bulk cells from lymph nodes and spleens of SM1 mice were injected i.v., corresponding to 1 × 105 SM1 T cells identified as CD4+ Vβ2+ CD90.1+. The next day, 100 μg of flagellin peptide (FL-pep; residues 427-441; Invitrogen; Carlsbad, CA) was injected. LPS (Sigma-Aldrich) was injected 18 h following SEA or FL-pep. LPS doses were determined from titration studies on individual batches to find the amount providing maximal T cell survival. For superantigen studies, 30–60 μg LPS was used while 50–180 μg LPS was used for SM1 studies. The anti-CD40 monoclonal antibody (mAb)-producing hybridoma FGK45.5 was a kind gift from Dr. A. Rolink (Basel Institute, Basel, Switzerland) (26), and the mAb was purified from hybridoma supernatants over protein G agarose (Life Technologies, Grand Island, NY). For immunization, the anti-CD40 mAb was injected at 200 μg per mouse two days prior to SEA. Anti-TNF and anti-IL-10 mAbs (clones XT-3.11 and JES5-2A5, respectively; BioExpress; West Lebanon, NH) were injected at 1 mg per mouse one hour prior to LPS. Rat IgG (Sigma-Aldrich) was injected as a control for mAbs.
Cell isolation and processing
Spleens, peripheral lymph nodes (PLN; inguinal, axillary, brachial), and mesenteric lymph nodes (MLN) were crushed through nylon mesh cell strainers (Falcon/BD Biosciences; San Jose, CA), and spleens were treated with ammonium chloride to lyse RBCs. For dendritic cell analysis, spleens were digested with collagenase D (Roche; Indianapolis, IN) for ≥30 min at 37°C before passing through cell strainers, partitioned on a BSA gradient, and analyzed for expression of CD11c, CD86, and CD40. Liver and lung lymphocytes were obtained as described (24). Briefly, following perfusion livers were crushed through cell strainers, and the cells partitioned on a 35% Percoll (Sigma-Aldrich) gradient. Lungs were cut into small pieces and incubated in 1.3 mM EDTA at 37°C for 30 min, followed by collagenase (Invitrogen) for 1 h. Lung cells were then fractionated on a 44% and 67% Percoll gradient (Amersham Biosciences; Piscataway, NJ), with lymphocytes partitioning at the interface.
Cell culturing, staining, and flow cytometry
One million cells were cultured for 5 h at 37°C in 0.2 mL complete tumor medium (CTM), consisting of MEM with FBS, amino acids, salts, and antibiotics. Cells were cultured with SEA (1.0μg/mL) or FL-pep (5 μg/mL), and brefeldin A (BFA; 5 μg/mL; Calbiochem), as indicated, and stained intracellularly for cytokines.
The following mAbs were purchased from eBioscience: Allophycocyanin-conjugated CD8a, CD90.1, TNF, IL-17A, IFN-γ, rat IgG1 (isotype control), rat IgG2a; PE-conjugated CD62L, rat IgG1; FITC-conjugated rat IgG2a. The following mAbs were purchased from BD Biosciences: Allophycocyanin-conjugated CD11c; Biotinylated TCR Vβ3; Pacific Blue-conjugated CD4; PerCP-conjugated CD4 and CD90.1; PE-conjugated CD86, TCR Vβ3, IFN-γ, rat IgG2a,kappa;; FITC-conjugated CD8b.2, CD40, TCR Vβ2, IFN-γ, TNF, and rat IgG1. Anti- mouse CXCR3-Allophycocyanin was purchased from Biolegend. Surface and intracellular staining was performed as previously described (27). Briefly, cells were resuspended in staining buffer consisting of BSS, 3% FBS, and 0.1% sodium azide. Non-specific binding was blocked by a solution containing mouse serum, human IgG, and the anti-Fc mAb 2.4G2 (28), followed by incubation with fluorescently-conjugated mAbs on ice for 30 min. For intracellular staining, surface staining was performed, followed by fixation with 2% paraformaldehyde, permeabilization with 0.25% saponin, and incubation at room temperature with the anti-cytokine mAb. Flow cytometry was conducted on BD FACSCalibur and LSR II flow cytometers with data analyzed using FlowJo software (Tree Star; Ashland, OR).
Statistical analysis
Two-tailed Student’s t tests were performed with p < 0.05 representing a significant statistical difference. The type of variance was determined by F tests, with F > 0.05 corresponding to equal variance and F < 0.05 corresponding to unequal variance.
Results
LPS enhances the accumulation of effector T cells into non-lymphoid tissues through TRIF
To study the role of TRIF in LPS adjuvanticity, we utilized a model for endogenous T cell responses. The pathogen-derived agent SEA activates CD4 and CD8 T cells expressing TCR Vβ3 in vivo by interacting with conserved regions on MHC class II molecules and TCR Vβ3 chains (29). This interaction results in substantial T cell clonal expansion followed by deletion during which most Vβ3 T cells die by apoptosis (30). Injecting LPS within 24 h after SEA rescues Vβ3 T cells from deletion by promoting their long-term survival (1).
To determine if TRIF is required for LPS to rescue SEA-stimulated T cells from deletion, WT and TRIF-deficient mice were injected with SEA at time 0. One group received LPS at 18 h, and Vβ3 T cells were initially tracked in the blood. On day 5, LPS increased Vβ3 T cell levels in a partially TRIF-dependent manner, however, at those doses the effect was transient since Vβ3 T cell levels were similar between WT and TRIF-deficient mice on day 10 (data not shown). Accordingly, LPS supported the accumulation of CD4 Vβ3 T cells in spleen of WT and TRIF-deficient mice (Fig. 1A). Similar results were observed for CD8 Vβ3 T cells (Fig. 1B), however, treatment with SEA alone resulted in TRIF-deficient mice having higher CD8 Vβ3 T cell numbers suggesting the TRIF pathway may enhance peripheral T cell deletion. In the lungs and liver of WT mice, immunization with SEA plus LPS resulted in approximately 12.5-fold more CD4 Vβ3 T cells than SEA alone (Fig. 1A), and LPS increased CD8 Vβ3 T cell numbers by 22- and 12-fold in lungs and liver, respectively (Fig. 1B). Importantly, this effect was dependent on TRIF since LPS only increased Vβ3 T cells by 1- to 4-fold in the corresponding tissues of TRIF-deficient mice (Fig. 1). Therefore, lymphoid (spleen) and non-lymphoid (lung and liver) T cell responses differed strikingly in their dependence on the TRIF pathway after LPS stimulation.
Figure 1. TRIF is required for LPS to increase T cell accumulation into non-lymphoid tissues.
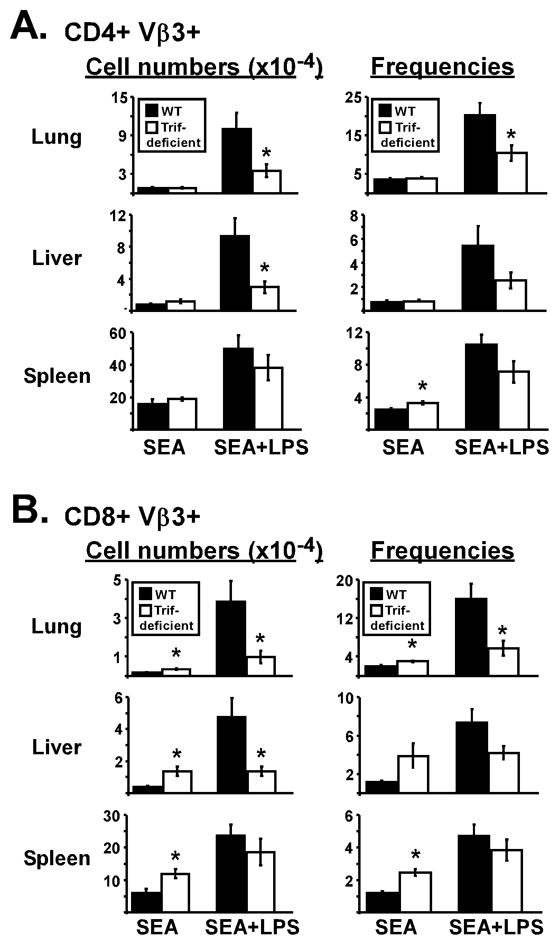
WT (closed bars) and TRIF-deficient (open bars) mice were immunized with SEA at time 0 and LPS at 18 h. (A) Left panel shows the number of CD4 Vβ3 T cells in lungs, liver, and spleen on day 10 for mice treated with SEA alone or SEA plus LPS. Right panel shows the percent of CD4 T cells expressing Vβ3 in each tissue. (B) Numbers of CD8 Vβ3 T cells (left) and the percent of CD8 T cells expressing Vβ3 (right). Data are pooled from 3 experiments with n=7–8 and represented as Mean +/- SEM. Asterisks represent significant statistical differences between similarly treated WT and TRIF-deficient mice with p = 1.5 × 10−4 – 0.038.
Effector potential was assessed by restimulating cells with SEA in vitro for 5 h and staining for intracellular IFN-γ and TNF. Immunization with SEA alone resulted in 8-10 percent of CD4 Vβ3 T cells producing IFN-γ in WT and TRIF-deficient mice, suggesting that TRIF did not impact the effector response to SEA (Fig. 2A). However, when comparing mice that were immunized with SEA plus LPS, TRIF deficiency resulted in at least 42 percent fewer CD4 Vβ3 T cells in the lungs and liver producing IFN-γ, and 30 percent fewer in the spleen (Fig. 2A). Analyzing the IFN-γ-positive cells revealed that cytokine mean fluorescence was similar among WT and TRIF-deficient mice (data not shown). To analyze the CD8 population, flow cytometry plots were gated on CD4-negative Vβ3 T cells. Similar to CD4+ cells, the percent of CD4-negative Vβ3 T cells producing IFN-γ was at least 30 percent lower in the TRIF-deficient mice treated with LPS (Fig. 2B). Stimulation with PMA plus ionomycin did not restore IFN-γ production to WT levels (data not shown), suggesting the T cells primed in TRIF-deficient mice had an intrinsic defect in Th1 differentiation. TNF production was TRIF-independent, demonstrating the effect was specific for IFN-γ. Taken together, TRIF was required for two important aspects of LPS adjuvanticity: accumulation of T cells into non-lymphoid tissues and optimal IFN-γ production by those T cells.
Figure 2. TRIF is required to LPS to increase IFN-γ production by superantigen-stimulated T cells.
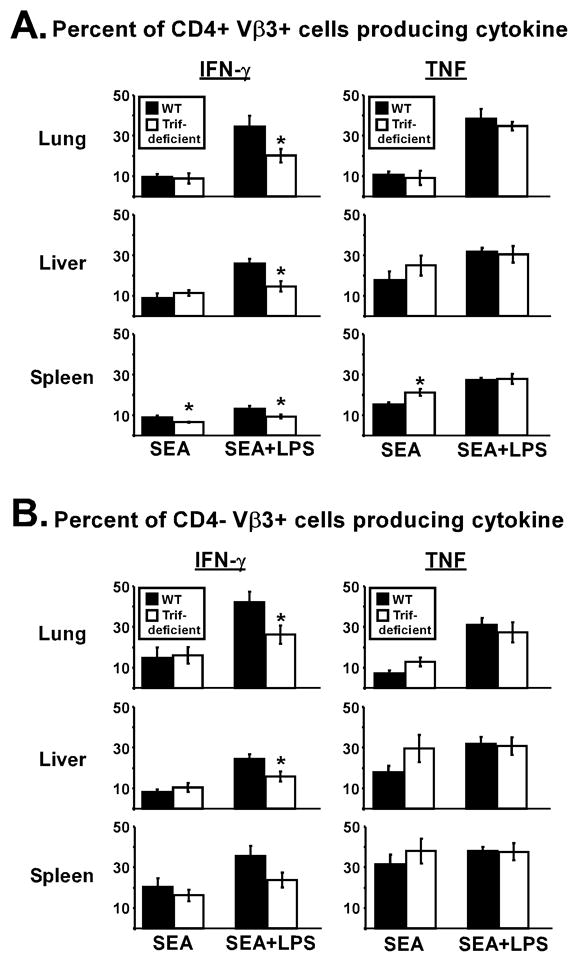
Day 10 cells from lungs, liver, and spleen were restimulated with SEA in vitro and analyzed for cytokine production. (A) Percent of CD4+ Vβ3 T cells producing IFN-γ (left) or TNF (right). (B) Percent of CD4-negative Vβ3 T cells producing IFN-γ or TNF. Cells were from the same mice used for Fig. 3. For lung samples, TNF production was measured in 2 experiments (n=4–5). Data is represented as Mean +/− SEM. Asterisks represent significant statistical differences between similarly treated WT (closed bars) and TRIF-deficient (open bars) mice with p = 0.004–0.042.
LPS-TRIF axis supports induction of peripheral effector phenotype on peptide-specific T cells
The ability of LPS to increase superantigen-specific CD4 T cells in the spleen of TRIF- deficient mice (Fig. 1A) suggested the peripheral innate response was largely intact due to MyD88-dependent signaling. Despite this result, serum levels of TNF, IL-10, and IFN-γ were reduced by more than 85 percent in TRIF-deficient mice following LPS injection, demonstrating a systemic effect on the cytokine response in vivo (Fig. 3A). Further, upregulation of CD86 and CD40 on CD11c+ cells was TRIF-dependent (Fig. 3B). We reasoned that significantly lower levels of the immunosuppressive cytokine IL-10 in TRIF-deficient mice may allow LPS to increase splenic CD4 T cell numbers when pro-survival cytokines and costimulation are suboptimal. If so, then neutralizing IL-10 in WT mice should overcome a requirement for maximal induction of survival factors like TNF (1). Alternative to this cytokine balance hypothesis, the splenic T cell response to superantigens may be dominated by memory cells that are less dependent on innate factors for survival. To eliminate this latter possibility, primary CD4 T cell responses were monitored by tracking a TCR transgenic population that was naïve prior to immunization. SM1 CD4 T cells, specific for residues 427–441 from Salmonella flagellin, referred to as FL-pep (25), were initially transferred into WT and TRIF-deficient mice to determine if they respond similarly as SEA-stimulated cells. The day after transfer, mice were immunized with FL-pep and various doses of LPS, and SM1 T cell numbers were assessed in peripheral lymph nodes (PLN) and spleen on day 10. At all doses tested, LPS enhanced the accumulation of lymphoid SM1 CD4 T cells independently of TRIF (Fig. 3C), consistent with the SEA response (Fig. 1). Further, production of IFN-γ and TNF by splenic SM1 T cells on day 10 was not significantly impacted by TRIF-deficiency although the intermediate LPS dose showed an effect (Fig. 3D).
Figure 3. LPS enhances peptide-specific CD4 T cell accumulation in lymphoid tissues independently of TRIF.
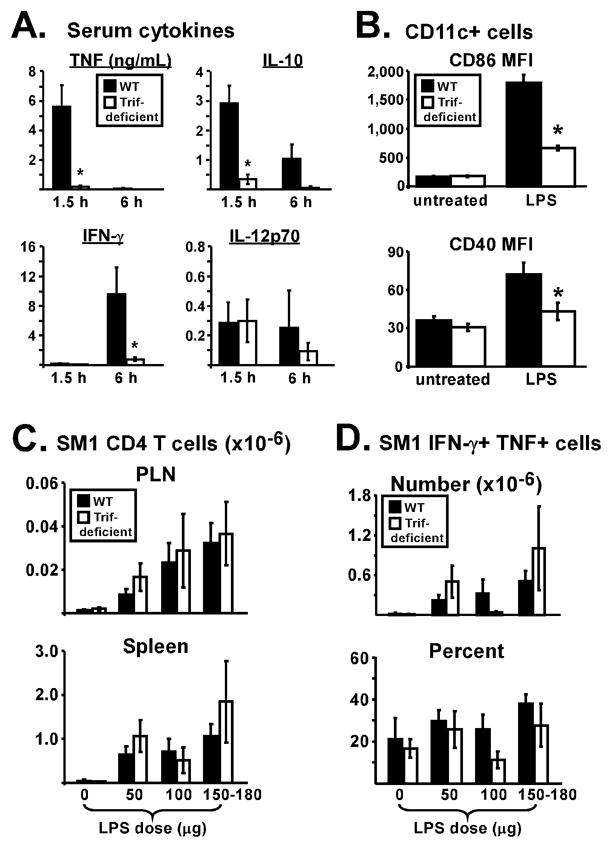
(A) Naïve WT (closed symbols) and TRIF-deficient (open symbols) mice were injected with 150 μg LPS and serum levels of TNF, IL-10, IFN-γ, and IL-12p70 were measured 1.5 h and 6 h later. Data are pooled from 3 experiments with a total of 8 mice each. (B) Mice were left untreated or injected with 150–200 μg LPS. 10–12 h later, CD11c+ cells were analyzed for expression of CD86 and CD40. Data are pooled from 3 experiments with a total of 6–7 mice per group and bar graphs display the mean fluorescence intensity (MFI) of CD86 and CD40 staining. (C) SM1 CD4 T cells were transferred, and the next day mice were immunized with FL-pep followed by varying doses of LPS (0–180 μg) 18 h later. SM1 CD4 T cell numbers were assessed in the PLN and spleen on day 10. Data are pooled from 4 experiments. For mice treated with LPS, n=7–14, while for mice not given LPS, n=5. (D) Bar graphs show the total number (top) and percent (bottom) of splenic SM1 CD4 T cells producing IFN-γ and TNF following 5 h in vitro restimulation with FL-pep. All data are shown as Mean +/− SEM with asterisks representing significant statistical differences (p < 0.05).
Following activation, T cells alter their expression of homing receptors in a manner that favors migration out of lymph nodes and into peripheral tissues. This imprinting occurs early with lasting changes observable during clonal expansion (reviewed in (31)). Since day 10 represents a time beyond the contraction phase, i.e. the end result of programming, the SM1 T cell response was next examined on day 6 when functional characteristics are expected to appear. While similar numbers of SM1 T cells were present in lymph nodes of WT and TRIF-deficient mice, there was a striking difference in their expression of CXCR3, a chemokine receptor that is upregulated by Th1 cells (Fig. 4A and refs. (32, 33)). Nearly 50 percent of SM1 T cells primed in WT mice expressed CXCR3 compared to approximately 12 percent in TRIF-deficient mice. This was associated with liver accumulation, as 2.5-fold fewer SM1 T cells were present in the liver of TRIF-deficient mice. Our findings are consistent with studies demonstrating a critical role for CXCR3 or its ligands in T cell trafficking to the liver following infection (34, 35). In WT mice, greater than 70 percent of the liver-resident SM1 T cells expressed CXCR3 versus only 35 percent in TRIF-deficient mice. TRIF-deficiency also resulted in an overall decreased ability of SM1 T cells to produce IFN-γ, similar to results observed for superantigen-stimulated T cells (Figs. 2, 4 and Table I).
Figure 4. LPS-TRIF axis supports peripheral effector phenotype on peptide-specific CD4 T cells.

SM1 CD4 T cells were transferred into WT (closed bars) and TRIF-deficient (open bars) mice, followed by immunization with FL-pep and LPS. (A) Total SM1 T cells in PLN and liver on day 6. Representative histograms show CXCR3 expression on SM1 T cells. (B) Lymphocytes were restimulated in vitro with FL-pep and brefeldin A for 5 h, followed by intracellular staining for IFN-γ and TNF. Representative dot plots are gated on liver SM1 T cells. (C) Total number of IFN-γ+ TNF+ SM1 T cells in tissues on day 6. Data are combined from 3 experiments with n=9, and bar graphs show Mean +/− SEM with asterisks representing significant statistical differences (p ≤ 0.03).
Table I.
The LPS-TRIF pathway supports effector cytokine production by peptide-specific T cells1
| Percent of SM1 T cells producing IFN-γ and TNF | |||
|---|---|---|---|
| FL-pep restimulation | Liver | Lung | Spleen |
| Wild-type | 33.1 ± 5.8 | 19.0 ± 4.4 | 25.5 ± 3.3 |
| TRIF-deficient | 12.9 ± 4.1 | 7.4 ± 1.4 | 7.2 ± 1.4 |
| PMA+iono. restimulation | |||
| Wild-type | 56.2 ± 7.2 | 33.4 ± 3.8 | 41.2 ± 3.8 |
| TRIF-deficient | 27.8 ± 7.3 | 17.1 ± 2.7 | 12.7 ± 2.1 |
SM1 T cells were transferred into wild-type and TRIF-deficient mice and immunized with FL-pep and LPS, as described in Materials and Methods. On day 6, lymphocytes were restimulated in vitro for 5 h. Shown are the percent of SM1 T cells staining double-positive for IFN-γ and TNF in response to FL-pep or PMA plus ionomycin, as indicated. Data are combined from 3 experiments with 9 total mice per group and represented as Mean ± SEM.
The balance between pro- and anti-inflammatory cytokines after LPS adjuvanticity influences CD4 T cell survival
To address our hypothesis of cytokine balance on primary CD4 T cell responses, SM1 T cells were transferred into WT mice and immunized with FL-pep at time 0 and LPS 18 h later. One hour prior to LPS, mice were injected with neutralizing mAbs for TNF, IL-10, or control rat IgG. In the spleen on day 10, LPS increased SM1 T cell numbers by 10-fold (Fig. 5, left panel). This was partially TNF-dependent since LPS only increased their numbers by 4-fold in mice treated with anti-TNF, consistent with a previous study (1). A similar trend was observed in the PLN, MLN, and liver, where TNF neutralization reduced SM1 T cell numbers by approximately 50 percent (Fig. 5). In contrast, mice treated with the IL-10 neutralizing mAb exhibited normal SM1 T cell survival after Ag and LPS. Consistent with our hypothesis, neutralizing both TNF and IL-10 also resulted in normal SM1 T cell levels, suggesting that TNF may support T cell survival by blocking IL-10-mediated suppression (Fig. 5). Since TRIF was required for inducing TNF and IL-10 (Fig. 3A), perhaps neutralizing these cytokines in WT mice resembled the environment in TRIF-deficient mice. In both cases LPS increased lymphoid T cell numbers, offering a reasonable explanation for how the MyD88 pathway can support a certain level of T cell survival in the absence of TRIF. Although TNF clearly impacted T cell numbers, providing TRIF-deficient mice with exogenous TNF in addition to LPS did not restore SM1 T cell accumulation into the liver, suggesting that cooperation among multiple TRIF-dependent factors determines extra-lymphoid T cell fate (data not shown). SM1 T cell cytokine production was not significantly affected by TNF or IL-10 neutralization (data not shown), suggesting that effector differentiation is influenced by other factors.
Figure 5. The balance of pro-inflammatory and anti-inflammatory cytokines influences T cell survival.
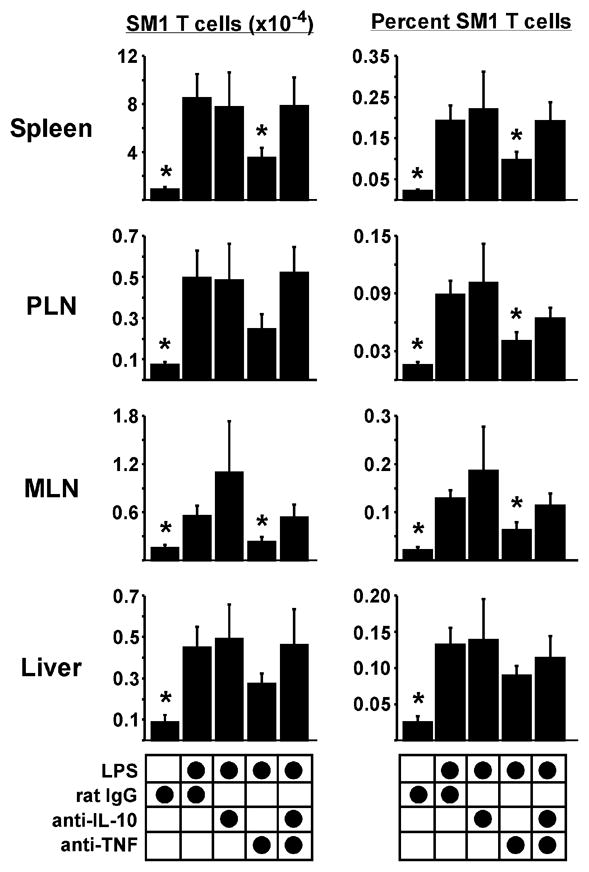
SM1 CD4 T cells were transferred into WT mice, followed by immunization with FL- pep and LPS. One hour prior to LPS, mice were injected anti-IL-10 and/or anti-TNF neutralizing mAbs, or rat IgG control. (A) SM1 T cell numbers (left) and frequencies (right) in tissues on day 10. Data are pooled from three experiments with a total of 6–9 mice per group, and are shown as Mean +/− SEM. Asterisks represent statistically significant differences of the indicated treatment compared to rat IgG plus LPS with p = 2.7 × 10−4 – 0.046.
Exogenous CD40 stimulation preferentially restores the CD8 T cell response in peripheral tissues of TRIF-deficient mice
The TRIF pathway requirement for non-lymphoid Vβ3 T cell accumulation and IFN-γ production (Figs. 1, 2) indicated it has a fundamental role in T cell priming during immunization with LPS. Since the upregulation of CD40 by LPS requires TRIF (Ref (9) and Fig. 3B), perhaps CD40 stimulation was suboptimal in the TRIF-deficient mice. If so, this defect could be restored by an anti-CD40 agonist mAb which enhances the clonal expansion of SEA-stimulated T cells but does not prevent their ultimate deletion (36). Interestingly, combining the CD40 agonist with LPS results in a synergistic enhancement of T cell survival (20, 37). To determine if enforced CD40 stimulation restores T cell responses in the lung and liver, TRIF-deficient mice were immunized with anti-CD40, SEA, and LPS as described in Materials and Methods. Control groups received rat IgG in place of anti-CD40, or did not receive LPS. Although the LPS dose was 40 percent lower than normal to accommodate CD40 stimulation (20), LPS potentiated T cell survival on day 10 in WT mice, resulting in 6-fold more Vβ3 T cells than anti-CD40 by itself (Table II). The combination of anti-CD40 plus LPS resulted in higher Vβ3 T cell levels, ranging 15- to 18-fold greater than anti-CD40 by itself. TRIF-deficient mice demonstrated an impaired response to rat IgG plus LPS, producing only twice as many Vβ3 T cells as anti-CD40 treatment (Table II). However, similar to WT, the anti-CD40 and LPS combination increased T cell accumulation in TRIF-deficient mice, resulting in the restoration of CD8 Vβ3 T cell numbers to nearly normal levels (Table II). A similar trend was observed for CD4 Vβ3 T cells, where the combination resulted in a 2.7-fold increase over rat IgG plus LPS. However, the CD4 population did not reach WT levels in TRIF-deficient mice (Table II). Therefore, enforced CD40 costimulation preferentially restored CD8 over CD4 T cell accumulation in the lungs and liver of TRIF-deficient mice.
Table II.
Total Vβ3 T cell numbers in peripheral tissues on day 10 (×10−3)1
| Wild-type | TRIF-deficient | |||||
|---|---|---|---|---|---|---|
| CD8 Vβ3 | anti-CD40 | rat IgG +LPS | Both | anti-CD40 | rat IgG +LPS | Both |
| Lung | 2.7 ± 0.5 | 16.2 ± 3.5 | 52.9 ± 14.6 | 3.2 ± 0.7 | 9.9 ± 4.2 | 42.4 ± 22.6 |
| Liver | 7.1 ± 2.1 | 44.7 ± 8.2 | 71.5 ± 17.1 | 9.5 ± 2.7 | 11.1 ± 4.7 | 57.5 ± 23.7 |
| Avg fold increase | 1.0 | 6.1 | 14.7 | 1.0 | 2.1 | 9.6 |
| CD4 Vβ3 | ||||||
| Lung | 6.7 ± 0.8 | 36.3 ± 6.2 | 133.0 ± 17.4 | 6.7 ± 0.9 | 17.2 ± 5.3 | 38.4 ± 12.4 |
| Liver | 9.8 ± 1.4 | 69.6 ± 12.4 | 159.7 ± 23.1 | 8.7 ± 3.1 | 13.0 ± 5.5 | 44.3 ± 14.5 |
| Avg fold increase | 1.0 | 6.3 | 18.1 | 1.0 | 2.0 | 5.4 |
Wild-type and TRIF-deficient mice were immunized with SEA along with anti-CD40, LPS, or both as described in Materials and Methods. On day 10, the total number of CD8 (top) and CD4 (bottom) T cells expressing Vβ3 was determined in the lungs and liver. Data are combined from 4 experiments with 6-7 total mice per group and represented as Mean +/− SEM. Average fold increase represents the number of Vβ3 T cells from a specific treatment divided by the number obtained from anti-CD40 alone treatment.
Liver T cell function was assessed by intracellular cytokine staining. In WT mice, rat IgG plus LPS produced a better IFN-γ response than anti-CD40, and treatment with the combination did not further increase IFN-γ production over LPS alone (Fig. 6). LPS also significantly increased the percent of CD4 Vβ3 T cells producing IL-17A, a cytokine detected in various tissues including the intestine, liver, and lung (38-40). In TRIF-deficient mice, unlike WT mice, there was no significant difference between the anti-CD40 and rat IgG plus LPS groups (Fig. 6), consistent with an impaired Th1 response (Fig. 2). This was not due to conversion to a Th17 phenotype since the number of IL-17A-producers was even lower than IFN-γ-producers. Importantly, treatment with the combination significantly increased IFN-γ and IL-17A capacity in TRIF-deficient mice (Fig. 6, right panel, compare “anti-CD40” to “both”). The most dramatic effect was observed for CD8 Vβ3 T cells, where IFN-γ production was restored to WT levels (Fig. 6A).
Figure 6. Exogenous CD40 stimulation in combination with LPS enhances superantigen-specific cytokine production independently of TRIF.

WT (left panels) and TRIF-deficient (right panels) mice were immunized with SEA and either anti-CD40, rat IgG plus LPS, or anti-CD40 plus LPS (both), as described in Materials and Methods. On day 10, liver lymphocytes were restimulated with SEA in vitro and analyzed for production of IFN-γ and IL-17A by intracellular cytokine staining. Scatter plots show the percent of CD8 Vβ3 T cells (A) or CD4 Vβ3 T cells (B), producing cytokine from individual mice. Cells were from the same mice used for Table I and horizontal bars represent the Mean for each group. Two-tailed Student’s t tests were used to evaluate statistical significance between anti-CD40 treatment and the other groups, with p values listed. (C) Representative flow cytometry plots from each treatment group gated on CD4 Vβ3 T cells.
This preferential effect on the CD8 response was best observed when analyzing total effector cell numbers (Fig. 7). Thus, TRIF-deficient mice immunized with anti-CD40 plus LPS generated normal levels of IFN-γ-producing CD8 Vβ3 T cells (Fig. 7A), but not CD4 Vβ3 T cells (Fig. 7B). Therefore, studying the role of TRIF in T cell responses generated with anti-CD40 and LPS revealed a mechanistic dichotomy among CD4 and CD8 T cell subsets in their differentiation and accumulation in liver.
Figure 7. Exogenous CD40 stimulation in combination with LPS preferentially restored CD8 effector T cell accumulation in TRIF-deficient mice.
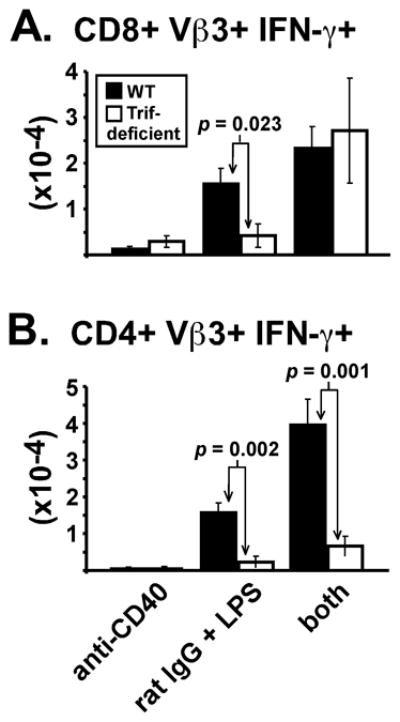
Bar graphs show the total number of liver CD8 Vβ3 T cells (A) and CD4 Vβ3 T cells (B) producing IFN-γ on day 10. Data are from the same experiments used for Table I and Fig. 6. Two-tailed Student’s t tests were used to evaluate statistical significance between similarly immunized WT (closed bars) and TRIF-deficient (open bars) mice, with p values listed.
Discussion
T cells migrate into non-lymphoid tissues following infection (24), and microbial components are often used to mimic this response (41) and generate protective immunity. This study identifies the TRIF pathway as critical for LPS to increase effector T cell accumulation into non-lymphoid tissues such as liver and lung. Our data complement the work from other groups demonstrating a role for TRIF in T cell responses generated by LPS (9, 23). Those studies did not distinguish between T cell survival and effector differentiation (9), analyze effector function (23), or analyze non-lymphoid tissues where the most dramatic effect of LPS in TRIF-deficient mice was observed (Fig. 1). Consistent with Mata-Haro, et al. (23), we found that early CD8 T cell expansion in response to Ag plus LPS was TRIF-dependent (data not shown); however, LPS increased splenic T cell numbers in TRIF-deficient mice on day 10 (Figs. 1, 3). This suggests that MyD88 can support some level of T cell survival in the absence of TRIF. In conjunction with previous work demonstrating its specific effect on long-term T cell survival (18), our data suggests the role of MyD88 in LPS adjuvanticity is to sustain the response elicited by TRIF stimulation. The dual requirement for TRIF in non-lymphoid T cell accumulation and effector function (Figs. 1, 2, 4) suggests these processes are part of the same differentiation program, similar to infection models (24, 42). Accordingly, a higher frequency of T cells isolated from non-lymphoid tissues produced IFN-γ upon recall than splenic T cells, especially within the CD4 T cell population (Fig. 2).
The mechanism by which TRIF stimulation impacts extra-lymphoid T cell responses may involve proliferation, peripheral survival, or tissue homing capability. Strikingly, TRIF was required for the upregulation of CXCR3 by peptide-specific CD4 T cells in lymph nodes and spleen, correlating with reduced T cell numbers in liver (Fig. 4A, and data not shown). CXCR3 expression by Th1 cells is associated with their accumulation at sites of inflammation (43), and blocking CXCR3 ligands in vivo reduces T cell recruitment to liver following infection with Toxoplasma gondii and murine cytomegalovirus (34, 35). Since CXCR3 is induced through T-bet (44), it can be a marker for Th1 activation and represents a possible mechanism for how TRIF stimulation by LPS supports peripheral T cell immunity. The involvement of CXCR3 appears to be tissue-dependent, however, since lung-resident SM1 T cells did not express the receptor and similar SM1 T cell numbers were recovered from the lungs of WT and TRIF-deficient mice on day 6 (data not shown). This is consistent with a recent report showing that CCR5, not CXCR3, is important for T cell migration to the lung following infection (45). In addition to the impaired CXCR3 upregulation, 40 percent more splenic SM1 T cells expressed the lymphoid-associated marker CD62L when they were primed in TRIF-deficient mice, further showing the T cells had undergone suboptimal activation (data not shown). Providing TNF to TRIF-deficient mice in addition to LPS restored splenic CD62L expression to WT levels, suggesting that TNF may contribute to the migration phenotype (data not shown).
Elucidation of the cell types contributing to TRIF-dependent responses should provide useful vaccine targets. Dendritic cells are likely candidates since they are required to elicit CD8 T cell responses (46), and for optimal Th1 differentiation driven by LPS (18), correlating with the TRIF phenotype (Fig. 2). Macrophages also contribute to LPS responsiveness in vivo (47), however, their specific role in the adjuvant effect is unknown.
LPS adjuvanticity depends upon the induction of inflammatory cytokines (1). Since TNF production was TRIF-dependent (Fig. 3A), we initially predicted a global effect on the T cell response. However, the ability of LPS to increase T cells in the lymphoid tissues of TRIF-deficient mice (Fig. 1, 3C) prompted us to examine this further. Based on our data examining TNF and IL-10 neutralization (Fig. 5), we propose that the balance, in contrast to the magnitude, of pro- and anti-inflammatory cytokines determines the level of T cell accumulation. Thus, TRIF-deficient mice, which produce less IL-10 as well as less TNF upon LPS stimulation (Fig. 3A), were able to sustain lymphoid T cell survival. Since adjuvants induce both pro- and anti-inflammatory cytokines, it is important to consider the balance of these factors when designing vaccines. Another important consideration is the adjuvant receptor. TRIF is known to function downstream of TLR3 and TLR4, and TLR4 is thought to mediate the LPS adjuvant effect. In the future it will be important to determine the receptor(s) and signaling pathways involved in the TRIF phenotype. It has been shown that TRIF stimulates apoptosis through caspase-8 (48, 49), providing a possible explanation for why more endogenous CD8 T cells were observed in tissues of TRIF-deficient mice treated with superantigen alone (Fig. 1). The ability of TRIF to induce apoptosis adds an underappreciated level of complexity to its regulation of adaptive immunity.
LPS is a unique adjuvant in that TLR4 stimulation results in activation of both MyD88 and TRIF signaling pathways. Previously, we reported that MyD88 is required for LPS to drive T cell survival but not effector differentiation (18), while here we show that TRIF drives effector T cell differentiation mostly in non-lymphoid tissue sites (Fig. 2), suggesting the signaling adaptors have non-redundant functions. Phenotypic differences may be explained by the ability of TLR4 to induce type I IFN through TRIF but not MyD88 (4). Expression of the type I IFN-receptor by T cells is important for their response to lymphocytic choriomeningitis virus and Listeria monocytogenes (50–53), although these pathogens do not synthesize LPS. In addition to directly stimulating T cells, type I IFNs activate APCs and can thus indirectly contribute to T cell priming. The ability of exogenous CD40 stimulation to fully restore the CD8 T cell response in TRIF-deficient mice suggested an important role for costimulation (Fig. 7A). Although exogenous CD40 stimulation also enhanced the CD4 response in TRIF-deficient mice, total effector cell numbers remained less than WT (Table I, Fig. 7B), demonstrating that CD40 signaling does not fully explain the contribution of TRIF in LPS adjuvanticity. More studies are needed to address the differential role of TRIF in amplifying CD8 versus CD4 T cell responses.
In addition to Th1 cell accumulation, LPS supported a Th17 response in the liver (Fig. 6). Since the total number of IFN-γ producers was about 3-fold higher than IL-17-producers (data not shown), the overall T cell response was Th1-biased. A small percentage of CD4 Vβ3 T cells produced both IFN-γ and IL-17 upon recall (Fig. 6C), suggesting that some Th1 and Th17 cells can share a common developmental pathway. Although whole cell pertussis vaccines have been shown to generate TLR4-dependent Th17 responses (54), we provide the first evidence that superantigen in combination with LPS supports Th17 responses in vivo. It will be important to determine if this effect is specific for superantigens or generalizeable to peptide-specific T cells.
Mata-Haro, et al. has demonstrated the LPS derivative monophosphoryl lipid A enhances Ag-specific T cell clonal expansion by preferentially stimulating the TRIF pathway (23). This finding is directly applicable to humans since monophosphoryl lipid A is generally non-toxic and associated with only mild side effects (55–57). Our data show that stimulating TRIF through LPS generates robust systemic T cell responses (Figs. 1, 2), providing further impetus for targeting this pathway in vaccine design. The quality of T cell responses must not be overlooked, as LPS is pleiotropic and can support Th1, Th2, Th17, and regulatory T cell function, depending on the experimental model (Refs. (2, 58–62), and Fig. 6). Perhaps partial TLR4 agonists affect the balance between functional T cell subsets differently than LPS, resulting in qualitatively distinct responses. If so, then TLR4 may potentially be a therapeutic target for a variety of immunological conditions.
Footnotes
This work was supported by National Institutes of Health Grants R01-AI42858 and R01-AI52108 (to A.T.V), and J.P.M. was also supported by T32-AI07080.
Abbreviations: BSS: balanced salt solution; FL-pep: flagellin peptide; mAb: monoclonal antibody; PLN: peripheral lymph nodes; MLN: mesenteric lymph nodes; SEA: Staphylococcal Enterotoxin A; SEM: standard error of the mean.
Disclosures
The authors declare no conflicting financial interests.
References
- 1.Vella AT, McCormack JE, Linsley PS, Kappler JW, Marrack P. Lipopolysaccharide interferes with the induction of peripheral T cell death. Immunity. 1995;2:261–270. doi: 10.1016/1074-7613(95)90050-0. [DOI] [PubMed] [Google Scholar]
- 2.Pape KA, Khoruts A, Mondino A, Jenkins MK. Inflammatory cytokines enhance the in vivo clonal expansion and differentiation of antigen-activated CD4+ T cells. J Immunol. 1997;159:591–598. [PubMed] [Google Scholar]
- 3.Palsson-McDermott EM, O'Neill LA. Signal transduction by the lipopolysaccharide receptor, Toll-like receptor-4. Immunology. 2004;113:153–162. doi: 10.1111/j.1365-2567.2004.01976.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kawai T, Akira S. TLR signaling. Cell Death Differ. 2006;13:816–825. doi: 10.1038/sj.cdd.4401850. [DOI] [PubMed] [Google Scholar]
- 5.Iwasaki A, Medzhitov R. Toll-like receptor control of the adaptive immune responses. Nat Immunol. 2004;5:987–995. doi: 10.1038/ni1112. [DOI] [PubMed] [Google Scholar]
- 6.Fiorentino DF, Zlotnik A, Mosmann TR, Howard M, O'Garra A. IL-10 inhibits cytokine production by activated macrophages. J Immunol. 1991;147:3815–3822. [PubMed] [Google Scholar]
- 7.Durez P, Abramowicz D, Gerard C, Van Mechelen M, Amraoui Z, Dubois C, Leo O, Velu T, Goldman M. In vivo induction of interleukin 10 by anti-CD3 monoclonal antibody or bacterial lipopolysaccharide: differential modulation by cyclosporin A. J Exp Med. 1993;177:551–555. doi: 10.1084/jem.177.2.551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hoebe K, Du X, Georgel P, Janssen E, Tabeta K, Kim SO, Goode J, Lin P, Mann N, Mudd S, Crozat K, Sovath S, Han J, Beutler B. Identification of Lps2 as a key transducer of MyD88-independent TIR signalling. Nature. 2003;424:743–748. doi: 10.1038/nature01889. [DOI] [PubMed] [Google Scholar]
- 9.Hoebe K, Janssen EM, Kim SO, Alexopoulou L, Flavell RA, Han J, Beutler B. Upregulation of costimulatory molecules induced by lipopolysaccharide and double-stranded RNA occurs by Trif-dependent and Trif-independent pathways. Nat Immunol. 2003;4:1223–1229. doi: 10.1038/ni1010. [DOI] [PubMed] [Google Scholar]
- 10.Yamamoto M, Sato S, Hemmi H, Hoshino K, Kaisho T, Sanjo H, Takeuchi O, Sugiyama M, Okabe M, Takeda K, Akira S. Role of adaptor TRIF in the MyD88-independent toll-like receptor signaling pathway. Science. 2003;301:640–643. doi: 10.1126/science.1087262. [DOI] [PubMed] [Google Scholar]
- 11.Boonstra A, Rajsbaum R, Holman M, Marques R, Asselin-Paturel C, Pereira JP, Bates EE, Akira S, Vieira P, Liu YJ, Trinchieri G, O'Garra A. Macrophages and myeloid dendritic cells, but not plasmacytoid dendritic cells, produce IL-10 in response to MyD88- and TRIF-dependent TLR signals, and TLR-independent signals. J Immunol. 2006;177:7551–7558. doi: 10.4049/jimmunol.177.11.7551. [DOI] [PubMed] [Google Scholar]
- 12.Magram J, Connaughton SE, Warrier RR, Carvajal DM, Wu CY, Ferrante J, Stewart C, Sarmiento U, Faherty DA, Gately MK. IL-12-deficient mice are defective in IFN gamma production and type 1 cytokine responses. Immunity. 1996;4:471–481. doi: 10.1016/s1074-7613(00)80413-6. [DOI] [PubMed] [Google Scholar]
- 13.Schijns VE, Haagmans BL, Wierda CM, Kruithof B, Heijnen IA, Alber G, Horzinek MC. Mice lacking IL-12 develop polarized Th1 cells during viral infection. J Immunol. 1998;160:3958–3964. [PubMed] [Google Scholar]
- 14.Jankovic D, Kullberg MC, Hieny S, Caspar P, Collazo CM, Sher A. In the absence of IL-12, CD4(+) T cell responses to intracellular pathogens fail to default to a Th2 pattern and are host protective in an IL-10(−/−) setting. Immunity. 2002;16:429–439. doi: 10.1016/s1074-7613(02)00278-9. [DOI] [PubMed] [Google Scholar]
- 15.Trinchieri G. Interleukin-12 and the regulation of innate resistance and adaptive immunity. Nat Rev Immunol. 2003;3:133–146. doi: 10.1038/nri1001. [DOI] [PubMed] [Google Scholar]
- 16.Khoruts A, Mondino A, Pape KA, Reiner SL, Jenkins MK. A natural immunological adjuvant enhances T cell clonal expansion through a CD28-dependent, interleukin (IL)-2-independent mechanism. J Exp Med. 1998;187:225–236. doi: 10.1084/jem.187.2.225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vella AT, Mitchell T, Groth B, Linsley PS, Green JM, Thompson CB, Kappler JW, Marrack P. CD28 engagement and proinflammatory cytokines contribute to T cell expansion and long-term survival in vivo. J Immunol. 1997;158:4714–4720. [PubMed] [Google Scholar]
- 18.McAleer JP, Zammit DJ, Lefrancois L, Rossi RJ, Vella AT. The lipopolysaccharide adjuvant effect on T cells relies on nonoverlapping contributions from the MyD88 pathway and CD11c+ cells. J Immunol. 2007;179:6524–6535. doi: 10.4049/jimmunol.179.10.6524. [DOI] [PubMed] [Google Scholar]
- 19.Maxwell JR, Weinberg A, Prell RA, Vella AT. Danger and OX40 receptor signaling synergize to enhance memory T cell survival by inhibiting peripheral deletion. J Immunol. 2000;164:107–112. doi: 10.4049/jimmunol.164.1.107. [DOI] [PubMed] [Google Scholar]
- 20.Maxwell JR, Ruby C, Kerkvliet NI, Vella AT. Contrasting the roles of costimulation and the natural adjuvant lipopolysaccharide during the induction of T cell immunity. J Immunol. 2002;168:4372–4381. doi: 10.4049/jimmunol.168.9.4372. [DOI] [PubMed] [Google Scholar]
- 21.Lee SJ, Myers L, Muralimohan G, Dai J, Qiao Y, Li Z, Mittler RS, Vella AT. 4-1BB and OX40 dual costimulation synergistically stimulate primary specific CD8 T cells for robust effector function. J Immunol. 2004;173:3002–3012. doi: 10.4049/jimmunol.173.5.3002. [DOI] [PubMed] [Google Scholar]
- 22.Maxwell JR, Yadav R, Rossi RJ, Ruby CE, Weinberg AD, Aguila HL, Vella AT. IL-18 bridges innate and adaptive immunity through IFN-gamma and the CD134 pathway. J Immunol. 2006;177:234–245. doi: 10.4049/jimmunol.177.1.234. [DOI] [PubMed] [Google Scholar]
- 23.Mata-Haro V, Cekic C, Martin M, Chilton PM, Casella CR, Mitchell TC. The vaccine adjuvant monophosphoryl lipid A as a TRIF-biased agonist of TLR4. Science. 2007;316:1628–1632. doi: 10.1126/science.1138963. [DOI] [PubMed] [Google Scholar]
- 24.Masopust D, Vezys V, Marzo AL, Lefrancois L. Preferential localization of effector memory cells in nonlymphoid tissue. Science. 2001;291:2413–2417. doi: 10.1126/science.1058867. [DOI] [PubMed] [Google Scholar]
- 25.McSorley SJ, Asch S, Costalonga M, Reinhardt RL, Jenkins MK. Tracking salmonella-specific CD4 T cells in vivo reveals a local mucosal response to a disseminated infection. Immunity. 2002;16:365–377. doi: 10.1016/s1074-7613(02)00289-3. [DOI] [PubMed] [Google Scholar]
- 26.Rolink A, Melchers F, Andersson J. The SCID but not the RAG-2 gene product is required for S mu-S epsilon heavy chain class switching. Immunity. 1996;5:319–330. doi: 10.1016/s1074-7613(00)80258-7. [DOI] [PubMed] [Google Scholar]
- 27.Maxwell JR, Rossi RJ, McSorley SJ, Vella AT. T cell clonal conditioning: a phase occurring early after antigen presentation but before clonal expansion is impacted by Toll-like receptor stimulation. J Immunol. 2004;172:248–259. doi: 10.4049/jimmunol.172.1.248. [DOI] [PubMed] [Google Scholar]
- 28.Unkeless JC. Characterization of a monoclonal antibody directed against mouse macrophage and lymphocyte Fc receptors. J Exp Med. 1979;150:580–596. doi: 10.1084/jem.150.3.580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Marrack P, Kappler J. The staphylococcal enterotoxins and their relatives. Science. 1990;248:705–711. doi: 10.1126/science.2185544. [DOI] [PubMed] [Google Scholar]
- 30.McCormack JE, Callahan JE, Kappler J, Marrack PC. Profound deletion of mature T cells in vivo by chronic exposure to exogenous superantigen. J Immunol. 1993;150:3785–3792. [PubMed] [Google Scholar]
- 31.Masopust D, Kaech SM, Wherry EJ, Ahmed R. The role of programming in memory T-cell development. Curr Opin Immunol. 2004;16:217–225. doi: 10.1016/j.coi.2004.02.005. [DOI] [PubMed] [Google Scholar]
- 32.Bonecchi R, Bianchi G, Bordignon PP, D'Ambrosio D, Lang R, Borsatti A, Sozzani S, Allavena P, Gray PA, Mantovani A, Sinigaglia F. Differential expression of chemokine receptors and chemotactic responsiveness of type 1 T helper cells (Th1s) and Th2s. J Exp Med. 1998;187:129–134. doi: 10.1084/jem.187.1.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sallusto F, Lenig D, Mackay CR, Lanzavecchia A. Flexible programs of chemokine receptor expression on human polarized T helper 1 and 2 lymphocytes. J Exp Med. 1998;187:875–883. doi: 10.1084/jem.187.6.875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Khan IA, MacLean JA, Lee FS, Casciotti L, DeHaan E, Schwartzman JD, Luster AD. IP-10 is critical for effector T cell trafficking and host survival in Toxoplasma gondii infection. Immunity. 2000;12:483–494. doi: 10.1016/s1074-7613(00)80200-9. [DOI] [PubMed] [Google Scholar]
- 35.Hokeness KL, Deweerd ES, Munks MW, Lewis CA, Gladue RP, Salazar-Mather TP. CXCR3-dependent recruitment of antigen-specific T lymphocytes to the liver during murine cytomegalovirus infection. J Virol. 2007;81:1241–1250. doi: 10.1128/JVI.01937-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Maxwell JR, Campbell JD, Kim CH, Vella AT. CD40 activation boosts T cell immunity in vivo by enhancing T cell clonal expansion and delaying peripheral T cell deletion. J Immunol. 1999;162:2024–2034. [PubMed] [Google Scholar]
- 37.Ahonen CL, Doxsee CL, McGurran SM, Riter TR, Wade WF, Barth RJ, Vasilakos JP, Noelle RJ, Kedl RM. Combined TLR and CD40 triggering induces potent CD8+ T cell expansion with variable dependence on type I IFN. J Exp Med. 2004;199:775–784. doi: 10.1084/jem.20031591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ivanov, McKenzie BS, Zhou L, Tadokoro CE, Lepelley A, Lafaille JJ, Cua DJ, Littman DR. The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell. 2006;126:1121–1133. doi: 10.1016/j.cell.2006.07.035. [DOI] [PubMed] [Google Scholar]
- 39.Heninger E, Hogan LH, Karman J, Macvilay S, Hill B, Woods JP, Sandor M. Characterization of the Histoplasma capsulatum-induced granuloma. J Immunol. 2006;177:3303–3313. doi: 10.4049/jimmunol.177.5.3303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Khader SA, Bell GK, Pearl JE, Fountain JJ, Rangel-Moreno J, Cilley GE, Shen F, Eaton SM, Gaffen SL, Swain SL, Locksley RM, Haynes L, Randall TD, Cooper AM. IL-23 and IL-17 in the establishment of protective pulmonary CD4+ T cell responses after vaccination and during Mycobacterium tuberculosis challenge. Nat Immunol. 2007;8:369–377. doi: 10.1038/ni1449. [DOI] [PubMed] [Google Scholar]
- 41.Reinhardt RL, Khoruts A, Merica R, Zell T, Jenkins MK. Visualizing the generation of memory CD4 T cells in the whole body. Nature. 2001;410:101–105. doi: 10.1038/35065111. [DOI] [PubMed] [Google Scholar]
- 42.von Andrian UH, Mackay CR. T-cell function and migration. Two sides of the same coin. N Engl J Med. 2000;343:1020–1034. doi: 10.1056/NEJM200010053431407. [DOI] [PubMed] [Google Scholar]
- 43.Qin S, Rottman JB, Myers P, Kassam N, Weinblatt M, Loetscher M, Koch AE, Moser B, Mackay CR. The chemokine receptors CXCR3 and CCR5 mark subsets of T cells associated with certain inflammatory reactions. J Clin Invest. 1998;101:746–754. doi: 10.1172/JCI1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lord GM, Rao RM, Choe H, Sullivan BM, Lichtman AH, Luscinskas FW, Glimcher LH. T-bet is required for optimal proinflammatory CD4+ T-cell trafficking. Blood. 2005;106:3432–3439. doi: 10.1182/blood-2005-04-1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kohlmeier JE, Miller SC, Smith J, Lu B, Gerard C, Cookenham T, Roberts AD, Woodland DL. The chemokine receptor CCR5 plays a key role in the early memory CD8+ T cell response to respiratory virus infections. Immunity. 2008;29:101–113. doi: 10.1016/j.immuni.2008.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jung S, Unutmaz D, Wong P, Sano G, De los Santos K, Sparwasser T, Wu S, Vuthoori S, Ko K, Zavala F, Pamer EG, Littman DR, Lang RA. In vivo depletion of CD11c(+) dendritic cells abrogates priming of CD8(+) T cells by exogenous cell-associated antigens. Immunity. 2002;17:211–220. doi: 10.1016/s1074-7613(02)00365-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yang Y, Liu B, Dai J, Srivastava PK, Zammit DJ, Lefrancois L, Li Z. Heat shock protein gp96 is a master chaperone for toll-like receptors and is important in the innate function of macrophages. Immunity. 2007;26:215–226. doi: 10.1016/j.immuni.2006.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Han KJ, Su X, Xu LG, Bin LH, Zhang J, Shu HB. Mechanisms of the TRIF-induced interferon-stimulated response element and NF-kappaB activation and apoptosis pathways. J Biol Chem. 2004;279:15652–15661. doi: 10.1074/jbc.M311629200. [DOI] [PubMed] [Google Scholar]
- 49.Kaiser WJ, Offermann MK. Apoptosis induced by the toll-like receptor adaptor TRIF is dependent on its receptor interacting protein homotypic interaction motif. J Immunol. 2005;174:4942–4952. doi: 10.4049/jimmunol.174.8.4942. [DOI] [PubMed] [Google Scholar]
- 50.Kolumam GA, Thomas S, Thompson LJ, Sprent J, Murali-Krishna K. Type I interferons act directly on CD8 T cells to allow clonal expansion and memory formation in response to viral infection. J Exp Med. 2005;202:637–650. doi: 10.1084/jem.20050821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Aichele P, Unsoeld H, Koschella M, Schweier O, Kalinke U, Vucikuja S. CD8 T cells specific for lymphocytic choriomeningitis virus require type I IFN receptor for clonal expansion. J Immunol. 2006;176:4525–4529. doi: 10.4049/jimmunol.176.8.4525. [DOI] [PubMed] [Google Scholar]
- 52.Havenar-Daughton C, Kolumam GA, Murali-Krishna K. Cutting Edge: The direct action of type I IFN on CD4 T cells is critical for sustaining clonal expansion in response to a viral but not a bacterial infection. J Immunol. 2006;176:3315–3319. doi: 10.4049/jimmunol.176.6.3315. [DOI] [PubMed] [Google Scholar]
- 53.Way SS, Havenar-Daughton C, Kolumam GA, Orgun NN, Murali-Krishna K. IL-12 and type-I IFN synergize for IFN-gamma production by CD4 T cells, whereas neither are required for IFN-gamma production by CD8 T cells after Listeria monocytogenes infection. J Immunol. 2007;178:4498–4505. doi: 10.4049/jimmunol.178.7.4498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Higgins SC, Jarnicki AG, Lavelle EC, Mills KH. TLR4 mediates vaccine-induced protective cellular immunity to Bordetella pertussis: role of IL-17-producing T cells. J Immunol. 2006;177:7980–7989. doi: 10.4049/jimmunol.177.11.7980. [DOI] [PubMed] [Google Scholar]
- 55.Fries LF, Gordon DM, Richards RL, Egan JE, Hollingdale MR, Gross M, Silverman C, Alving CR. Liposomal malaria vaccine in humans: a safe and potent adjuvant strategy. Proc Natl Acad Sci U S A. 1992;89:358–362. doi: 10.1073/pnas.89.1.358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Stoute JA, Slaoui M, Heppner DG, Momin P, Kester KE, Desmons P, Wellde BT, Garcon N, Krzych U, Marchand M. A preliminary evaluation of a recombinant circumsporozoite protein vaccine against Plasmodium falciparum malaria. RTS,S Malaria Vaccine Evaluation Group. N Engl J Med. 1997;336:86–91. doi: 10.1056/NEJM199701093360202. [DOI] [PubMed] [Google Scholar]
- 57.Thoelen S, Van Damme P, Mathei C, Leroux-Roels G, Desombere I, Safary A, Vandepapeliere P, Slaoui M, Meheus A. Safety and immunogenicity of a hepatitis B vaccine formulated with a novel adjuvant system. Vaccine. 1998;16:708–714. doi: 10.1016/s0264-410x(97)00254-5. [DOI] [PubMed] [Google Scholar]
- 58.Eisenbarth SC, Piggott DA, Huleatt JW, Visintin I, Herrick CA, Bottomly K. Lipopolysaccharide-enhanced, toll-like receptor 4-dependent T helper cell type 2 responses to inhaled antigen. J Exp Med. 2002;196:1645–1651. doi: 10.1084/jem.20021340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Caramalho I, Lopes-Carvalho T, Ostler D, Zelenay S, Haury M, Demengeot J. Regulatory T cells selectively express toll-like receptors and are activated by lipopolysaccharide. J Exp Med. 2003;197:403–411. doi: 10.1084/jem.20021633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lewkowicz P, Lewkowicz N, Sasiak A, Tchorzewski H. Lipopolysaccharide-activated CD4+CD25+ T regulatory cells inhibit neutrophil function and promote their apoptosis and death. J Immunol. 2006;177:7155–7163. doi: 10.4049/jimmunol.177.10.7155. [DOI] [PubMed] [Google Scholar]
- 61.den Haan JM, Kraal G, Bevan MJ. Cutting edge: Lipopolysaccharide induces IL-10-producing regulatory CD4+ T cells that suppress the CD8+ T cell response. J Immunol. 2007;178:5429–5433. doi: 10.4049/jimmunol.178.9.5429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Murakami D, Yamada H, Yajima T, Masuda A, Komune S, Yoshikai Y. Lipopolysaccharide inhalation exacerbates allergic airway inflammation by activating mast cells and promoting Th2 responses. Clin Exp Allergy. 2007;37:339–347. doi: 10.1111/j.1365-2222.2006.02633.x. [DOI] [PubMed] [Google Scholar]