

Abstract
Autism is a severe neurodevelopmental disorder characterized by a triad of complications. Autistic individuals display significant disturbances in language and reciprocal social interactions, combined with repetitive and stereotypic behaviors. Prevalence studies suggest that autism is more common than originally believed, with recent estimates citing a rate of one in 150. Although this genomic approach has yielded multiple suggestive regions, a specific risk locus has yet to be identified and widely confirmed. Because many etiologies have been suggested for this complex syndrome, we hypothesize that one of the difficulties in identifying autism genes is that multiple genetic variants may be required to significantly increase the risk of developing autism. Thus we took the alternative approach of examining 14 prominent dopamine pathway candidate genes for detailed study by genotyping 28 SNPs. Although we did observe a nominally significant association for rs2239535 (p=.008) on chromosome 20, single locus analysis did not reveal any results as significant after correction for multiple comparisons. No significant interaction was identified when Multifactor Dimensionality Reduction (MDR) was employed to test specifically for multilocus effects. Although genome-wide linkage scans in autism have provided support for linkage to various loci along the dopamine pathway, our study does not provide strong evidence of linkage or association to any specific gene or combination of genes within the pathway. These results demonstrate that common genetic variation within the tested genes located within this pathway at most play a minor to moderate role in overall autism pathogenesis.
Keywords: Autism, Dopamine, SNPs, linkage, association
Introduction
Autism is a highly heritable, genetically complex neurodevelopmental disorder that is neither distinct nor categorical. With an onset early in childhood, autism represents one extreme of a spectrum of social and communication impairment and behavioral problems, referred to as Autism Spectrum Disorders (ASD). The incidence of severe autism is estimated at one in 1,000 individuals, with males affected at a rate four times that of females (Fombonne, 1999; Williams et al., 2006). This rate of incidence increases to approximately one in 150 when the broad diagnosis is considered to include milder forms of ASD (Rice et al., 2007). Evidence from various studies indicates that ASDs have a genetically complex etiology, possibly involving epistasis and locus heterogeneity. While rare single mutations and chromosomal abnormalities are responsible for some cases (Abrahams et al., 2008), current models strongly suggest that inheritance of multiple interacting polymorphic loci contributes to a continuum of disease phenotypes in the majority of affected children (Veenstra-Vanderweele et al., 2004a; Veenstra-Vanderweele et al., 2004b).
The high heritability of autism has driven efforts to search for susceptibility loci using genome-wide linkage screens (Barrett et al., 1999; Buxbaum et al., 2001; International Molecular Genetic Study Autism Consortium (IMGSAC), 2001; Liu et al., 2001; Philippe et al., 1999; Risch et al., 1999; Shao et al., 2002; Szatmari et al., 2007; Yonan et al., 2003) and genome wide association studies (Arking et al., 2008). Although this genomic approach has yielded multiple suggestive regions, a specific risk locus has yet to be identified and widely confirmed.
Some evidence supports the involvement of neurobiological pathways, in particular the dopamine pathway (Figure 1). This connection seems compelling because the dopaminergic system has been linked to a variety of behaviors and functions: cognition, attention, motivation, and emotion (Nieoullon, 2002; Nieoullon et al., 2003). When disturbance of dopamine metabolism in autistic children was examined, affected individuals had a higher rate of urinary homovanilic acid (HVA) (Figure 1). A relationship between the severity of autism and the increased HVA levels was also observed (Garreau et al., 1980). For these reasons, the dopaminergic system is a common target for medication used in children with autism (Oswald et al., 2007). Risperidone, an antipsychotic (neuroleptic), is an antagonist of the dopamine D2 and 5HT2 receptors that has been reported to improve the restricted and repetitive stereotype (Malone et al., 2005). Other drugs such as Haldol, a dopamine antagonist, and Welbutrin, a dopamine uptake inhibitor, have proven effective in some autistic children. Thus with substantial evidence of disturbances in the dopamine pathway, we selected a set of functional candidate genes within this pathway that enabled us to examine each gene for variations in more detail. This approach entails selecting genes based on knowledge of the specific phenotypes and of the underlying neurobiology related to expected behavioral abnormalities in individuals with autism.
Figure 1. Dopamine pathway.
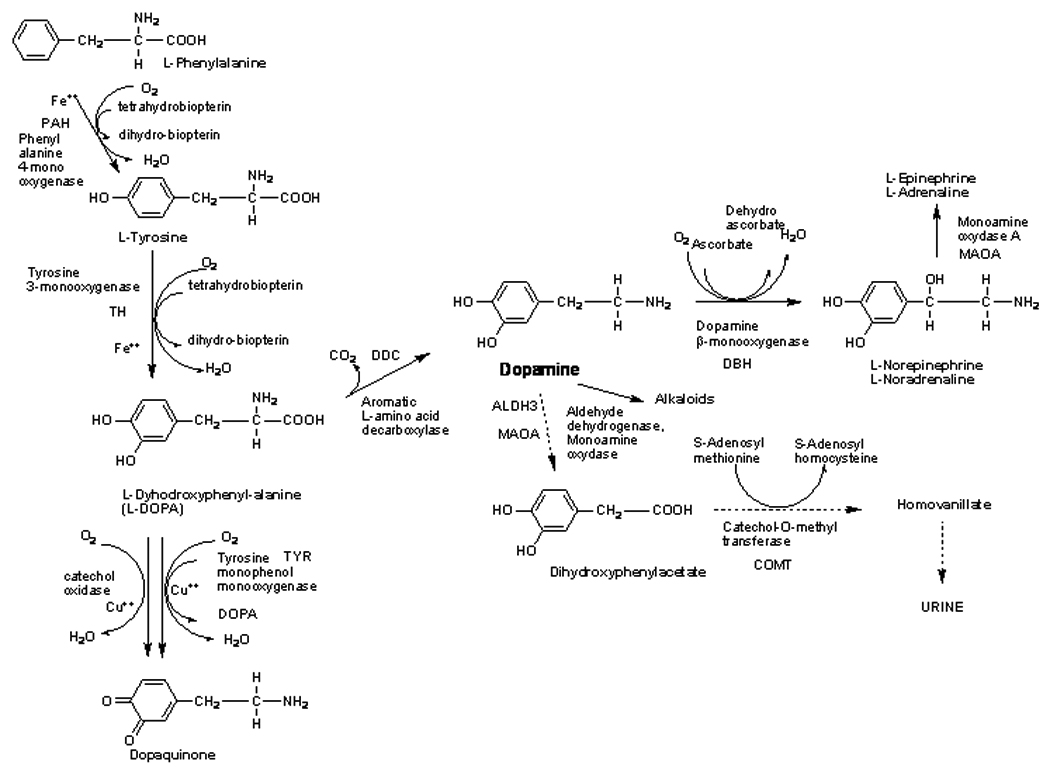
Representation of the dopamine pathway with the metabolic genes indicated. The other genes genotyped in this study are SLC6A3, the dopamine transporter. PTS (6-pyruvoyltetrahydropterin synthase) an enzyme part of the tetrahydrobiopterin pathway, and YWHAB (Tyrosine-3-monooxygenase activation protein).
Materials and Methods
Dataset
Our analysis was conducted on a dataset consisting of 403 non-Hispanic Caucasian American families collected in the Southeast United States by the Center for Human Genetics Research at Vanderbilt University and the Institute for Human Genomics at the University of Miami (Table 1). Probands for the study consisted of individuals between the ages of 3 and 21 years who were clinically diagnosed with autism using Diagnostic and Statistical Manual (DSM)-IV criteria. The clinical diagnosis of autism was confirmed based on clinical evaluation using DSM-IV diagnostic criteria supported by the Autism Diagnostic Interview-Revised (ADI-R) and medical records. Exclusion criteria for participation in the larger genetics study included developmental level below 18 months, severe sensory problems (e.g., visual impairment or hearing loss), significant motor impairments (e.g., failure to sit by 12 months or walk by 24 months), or identified metabolic, genetic, or progressive neurological disorders Parents/caregivers were informed of the purposes, risks, and benefits of participating in this project and provided informed consent.
Table 1.
Dataset Distributions
| Total | Multiplex | Trios | Unaffected sibs |
|
|---|---|---|---|---|
| All | 403 | 151 | 252 | 231 |
| Male Only Subset | 303 | 89 | 214 | 185 |
Molecular Analysis
Genomic DNA was extracted from blood using standard protocols and a commercial system (Puregene; Gentra Systems, Minneapolis, MN). All single nucleotide polymorphisms (SNPs) were identified using the Ensembl (www.ensembl.org), dbSNP (www.ncbi.nlm.nih.gov/projects/SNP), and AppliedBiosystems (http://www.appliedbiosystems.com/) databases. Multiple SNPs spanning each gene were chosen using a hierarchy of nonsynonymous coding change, minor allele frequency > 0.10, location within the gene, with the goal of capturing as much of the common variation as possible. We calculated the linkage disequilibrium between SNPs to assess the coverage of the genes with the goal of capturing most of the common variations (supplementary material). A total of 28 SNPs were genotyped for nine genes (Table 2). The genes were selected based on the metabolic proximity to the neurotransmitter dopamine, on previous reports of association to neuropsychiatric disorders, and on Michal’s publication: Biochemical Pathways: An Atlas of Biochemistry and Molecular Biology” (Michal, 1999). SNPs were genotyped using the ABI 7900 Taqman system (Oliveira et al., 2003). Laboratory personnel were blinded to pedigree structure, affection status, and location of quality control samples. Duplicate quality control samples were placed both within and across 384-well plates, and equivalent genotypes were required for all quality control samples to ensure accurate genotyping. Hardy-Weinberg calculations were performed for each marker, and Mendelian inconsistencies (in the multiplex families) were identified using PedCheck (O'Connell et al., 1998). Suspect genotypes were re-read or retested. All SNPs were required to pass 95% efficiency when genotyped.
Table 2.
Tested Genes and Single Nucleotide Polymorphisms (SNPs)
| SNPa | Gene | Chrb | NCBI built 36 | MAFc | MAd |
|---|---|---|---|---|---|
| rs37020 | SLC6A3 | 5 | 1471374 | 0.42 | C |
| rs1611115 | DBH | 9 | 133530069 | 0.22 | T |
| rs2797849 | DBH | 9 | 133531495 | 0.35 | C |
| rs3025388 | DBH | 9 | 133532810 | 0.17 | G |
| rs1108581 | DBH | 9 | 133534795 | 0.21 | G |
| rs1611125 | DBH | 9 | 133538866 | 0.47 | C |
| rs2797853 | DBH | 9 | 133542069 | 0.34 | T |
| rs2073833 | DBH | 9 | 133549836 | 0.44 | C |
| rs2073837 | DBH | 9 | 133552482 | 0.31 | A |
| rs2070762 | TH | 11 | 2142911 | 0.48 | G |
| rs7129973 | TYR | 11 | 88555218 | 0.41 | C |
| rs12419949 | TYR | 11 | 88562111 | 0.11 | T |
| rs2000554 | TYR | 11 | 88575589 | 0.41 | G |
| rs7123206 | TYR | 11 | 88590980 | 0.12 | A |
| rs3819331 | PTS | 11 | 111604249 | 0.14 | C |
| rs1801153 | PAH | 12 | 101735233 | 0.35 | T |
| rs1522307 | PAH | 12 | 101800984 | 0.34 | G |
| rs1006556 | ALDH3A1 | 17 | 19580307 | 0.24 | C |
| rs2072330e | ALDH3A1 | 17 | 19585064 | 0.40 | A |
| rs4646787 | ALDH3A1 | 17 | 19589841 | 0.34 | A |
| rs2239535 | YWHAB | 20 | 42949570 | 0.24 | A |
| rs2425672 | YWHAB | 20 | 42959551 | 0.43 | A |
| rs2425675 | YWHAB | 20 | 42968348 | 0.28 | A |
| rs2020917 | COMT | 22 | 18303438 | 0.30 | T |
| rs1055503 | COMT | 22 | 18316065 | 0.13 | A |
| rs4646312 | COMT | 22 | 18322891 | 0.40 | C |
| rs4633e | COMT | 22 | 18324789 | 0.48 | C |
| rs165774 | COMT | 22 | 18327115 | 0.34 | A |
Single Nucleotide Polymorphism designation
Chromosome
Minor Allele Frequency
Minor Allele
synonymous changes
Statistical Analysis
Genotype efficiency, Hardy-Weinberg Equilibrium and linkage disequilibrium were checked using Haploview (Barrett et al., 2005). If any SNP fell below 95% genotype efficiency, a SNP in high LD with the failed SNP was added to ensure complete coverage of the gene. Linkage analysis was conducted using two-point heterogeneity LOD scores (HLOD) calculated using FASTLINK and HOMOG (Ott, 1999). Both recessive and dominant models with disease allele frequencies of 0.01 and 0.001, respectively, were analyzed. This approach is robust for detecting linkage signals when the underlying model is unknown or complex (Hodge, 1994). The Pedigree Disequilibrium Test (PDT) for single-locus analysis (Martin et al., 2003) assessed the family based association and the Genotype-PDT (GenoPDT) tested genotypic association to the risk of autism (Martin et al., 2003). Taking into account the 4:1 ratio of males to females affected with autism, the HLOD, PDT, and Geno-PDT were also run in a subset of families containing only affected males (male-only, N=303).
Multifactor Dimensionality Reduction (MDR) analysis was used to detect multilocus interactions (Ritchie et al., 2001; Ritchie et al., 2007). Since MDR is designed for case-control data, we extracted from any family with a complete parent-child trio (one per family for multiplex families) the affected child. We constructed “pseudo” controls, using the non-transmitted alleles of the parents (Collins et al., 2006; Ma et al., 2005). We tested for all two way and three way interactions. All p-values are reported as nominal P values unless otherwise stated.
Results
A marginal association was detected for the DBH (dopamine-β-monooxygenase) p=0.03 in the overall dataset for rs161115. COMT (Catechol-O-methyl transferase) showed a marginal association p=0.05 (whole dataset) and p=0.03 (male only dataset). We also observed a marginal association (p=0.01) for PTS (6-pyruvoyl-tetrahydropterin synthase). The strongest association, p=0.008 (0.018 when corrected for multiple comparisons), was observed for rs2239535 (chromosome 20) for YWHAB (tyrosine 3-monooxygenase/tryptophan 5-monooxygenase activation protein, beta polypeptide), TH’s (tyrosine 3-monooxygenase) activation protein. Linkage analysis was performed and the only result of note was a LOD score of 2.49 (recessive model) for DBH in the male only subset (supplementary material). MDR was run, but it detected no gene-gene interaction (supplementary material).
Discussion
Compelling evidence has linked dysfunction in the dopamine pathway with autism, fostering studies of genes along this pathway. Variation in genes within the dopamine pathway has been implicated in various genetic linkage and/or association studies, but the results have been inconsistent. This lack of replication across the studies might be explained by the extreme genetic and phenotypic heterogeneity of the samples. To overcome this inconsistency, examination of families with only affected males has been suggested as a method of looking at a more homogeneous autism subset (Cantor et al., 2005; Stone et al., 2004). However, the subsetting of our samples into a male only subset did not improve the consistency of our results. Ultimately, our study does not provide overwhelming evidence of a main effect of any specific gene in the pathway.
This led us to hypothesize that interactions between genes might represent a more consistent genetic effect. To test the possibility that interactions might result in stronger joint effects, we performed MDR analysis. Although MDR has >80% power to detect both main and interactive effects in a dataset of this size, even in the presence of locus or genetic heterogeneity (Ritchie et al., 2007), this analysis did not identify any strong or moderate interactive effects..
That we found no significantly strong effects suggests that none of these genes, alone or in combination, carries a pervasive effect in autism. . There could be variation in regulatory elements for these genes outside of the coding regions covered by our analysis or an extreme level of locus heterogeneity that has a significant negative impact on power both for PDT and MDR analyses. Since we only examined common variations, the underlying effects could arise from multiple rare variants in one or more of these genes. Furthermore, genes part of the dopamine pathway but not genotyped in this study could harbor susceptibility alleles.
Supplementary Material
Table 3.
Pedigree Disequilibrium Test (PDT) and Geno-PDT Results
| Overall | MO* | ||||
|---|---|---|---|---|---|
| PDT | PDT | ||||
| SNP | Gene | SUM** | GENO** | SUM | GENO |
| rs37020 | SLC6A3 | 0.655 | 0.324 | 0.304 | 0.151 |
| rs161115 | DBH | 0.070 | 0.033 | 0.239 | 0.134 |
| rs2797849 | DBH | 0.084 | 0.237 | 0.089 | 0.242 |
| rs3025388 | DBH | 1.000 | 0.668 | 0.265 | 0.220 |
| rs1108581 | DBH | 0.852 | 0.050 | 0.456 | 0.125 |
| rs1611125 | DBH | 0.429 | 0.768 | 0.897 | 0.951 |
| rs2797853 | DBH | 0.472 | 0.489 | 0.164 | 0.376 |
| rs2073833 | DBH | 0.558 | 0.274 | 0.827 | 0.216 |
| rs2073837 | DBH | 0.492 | 0.329 | 0.850 | 0.816 |
| rs2070762 | TH | 0.297 | 0.590 | 0.478 | 0.768 |
| rs7129973 | TYR | 0.591 | 0.150 | 0.898 | 0.781 |
| rs12419949 | TYR | 0.273 | 0.456 | 0.848 | 0.963 |
| rs2000554 | TYR | 0.306 | 0.509 | 0.967 | 0.951 |
| rs7123206 | TYR | 0.876 | 0.973 | 0.948 | 0.997 |
| rs3819331 | PTS | 0.012 | 0.029 | 0.087 | 0.157 |
| rs1801153 | PAH | 0.906 | 0.927 | 0.621 | 0.795 |
| rs1522307 | PAH | 0.412 | 0.143 | 0.131 | 0.024 |
| rs1006556 | ALDH3A1 | 0.928 | 0.740 | 0.673 | 0.685 |
| rs17857757 | ALDH3A1 | 0.798 | 0.926 | 0.582 | 0.700 |
| rs4646787 | ALDH3A1 | 0.365 | 0.255 | 0.268 | 0.136 |
| rs2239535 | YWHAB | 0.008 | 0.017 | 0.013 | 0.028 |
| rs2425672 | YWHAB | 0.687 | 0.721 | 0.559 | 0.756 |
| rs2425675 | YWHAB | 0.072 | 0.178 | 0.083 | 0.209 |
| rs2020917 | COMT | 0.501 | 0.468 | 1.000 | 0.517 |
| rs1055503 | COMT | 0.152 | 0.192 | 0.060 | 0.090 |
| rs4646312 | COMT | 0.405 | 0.729 | 0.528 | 0.833 |
| rs4633 | COMT | 0.704 | 0.676 | 0.371 | 0.675 |
| rs165774 | COMT | 0.075 | 0.046 | 0.028 | 0.083 |
Families where only males are affected
The Sum-PDT is an allelic test; the Geno-PDT is a genotype test
Acknowledgements
We wish to thank both the patients with autism and their family members who agreed to participate in this study, as well as the personnel of the Center for Human Genetics Research at Vanderbilt University and the Miami Institute for Human Genomics at the University of Miami. We would like to thank M.J. Allen for her excellent technical support. This research was supported in part by National Institutes of Health (NIH) program project grant NS026630 (MPV, JLH) and NIH R01 grant MH080647.
Reference List
- Abrahams BS, Geschwind DH. Advances in autism genetics: on the threshold of a new neurobiology. Nature Review Genetics. 2008;9:341–355. doi: 10.1038/nrg2346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arking DE, Cutler DJ, Brune CW, Teslovich TM, West K, Ikeda M, et al. A common genetic variant in the neurexin superfamily member CNTNAP2 increases familial risk of autism. American Journal of Human Genetics. 2008;82:160–164. doi: 10.1016/j.ajhg.2007.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett JC, Fry B, Maller J, Daly MJ. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics. 2005;21:263–265. doi: 10.1093/bioinformatics/bth457. [DOI] [PubMed] [Google Scholar]
- Barrett S, Beck JC, Bernier R, Bisson E, Braun TA, Casavant TL, et al. An autosomal genomic screen for autism. Collaborative linkage study of autism. American Journal of Medical Genetics. 1999;88:609–615. doi: 10.1002/(sici)1096-8628(19991215)88:6<609::aid-ajmg7>3.3.co;2-c. [DOI] [PubMed] [Google Scholar]
- Buxbaum JD, Silverman JM, Smith CJ, Kilifarski M, Reichert J, Hollander E, et al. Evidence for a susceptibility gene for autism on chromosome 2 and for genetic heterogeneity. American Journal of Human Genetics. 2001;68:1514–1520. doi: 10.1086/320588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantor RM, Kono N, Duvall JA, varez-Retuerto A, Stone JL, Alarcon M, et al. Replication of autism linkage: fine-mapping peak at 17q21. American Journal of Human Genetics. 2005;76:1050–1056. doi: 10.1086/430278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins AL, Ma D, Whitehead PL, Martin ER, Wright HH, Abramson RK, et al. Investigation of autism and GABA receptor subunit genes in multiple ethnic groups. Neurogenetics. 2006;7:167–174. doi: 10.1007/s10048-006-0045-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fombonne E. The epidemiology of autism: a review. Psychological Medicine. 1999;29:769–786. doi: 10.1017/s0033291799008508. [DOI] [PubMed] [Google Scholar]
- Garreau B, Barthelemy C, Domenech J, Sauvage D, Muh JP, Lelord G, et al. Disturbances in dopamine metabolism in autistic children: results of clinical tests and urinary dosages of homovanilic acid (HVA) Acta Psychiatrica Belgica. 1980;80:249–265. [PubMed] [Google Scholar]
- Hodge SE. What association analysis can and cannot tell us about the genetics of complex disease. American Journal of Medical Genetics. 1994;54:318–323. doi: 10.1002/ajmg.1320540408. [DOI] [PubMed] [Google Scholar]
- International Molecular Genetic Study Autism Consortium (IMGSAC) A genomewide screen for autism: strong evidence for linkage to chromosomes 2q, 7q, and 16p. American Journal of Human Genetics. 2001;69:570–581. doi: 10.1086/323264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Nyholt DR, Magnussen P, Parano E, Pavone P, Geschwind D, et al. A genomewide screen for autism susceptibility loci. American Journal of Human Genetics. 2001;69:327–340. doi: 10.1086/321980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma DQ, Whitehead PL, Menold MM, Martin ER, shley-Koch AE, Mei H, et al. Identification of significant association and gene-gene interaction of GABA receptor subunit genes in autism. American Journal of Human Genetics. 2005;77:377–388. doi: 10.1086/433195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malone RP, Gratz SS, Delaney MA, Hyman SB. Advances in drug treatments for children and adolescents with autism and other pervasive developmental disorders. CNS. Drugs. 2005;19:923–934. doi: 10.2165/00023210-200519110-00003. [DOI] [PubMed] [Google Scholar]
- Martin ER, Bass MP, Gilbert JR, Pericak-Vance MA, Hauser ER. Genotype-based association test for general pedigrees: the genotype-PDT. Genetic. Epidemiology. 2003;25:203–213. doi: 10.1002/gepi.10258. [DOI] [PubMed] [Google Scholar]
- Michal G. Biochemical Pathways: an atlas of biochemistry and molecular biology. Spektrum: Wiley; 1999. p. 277. [Google Scholar]
- Nieoullon A. Dopamine and the regulation of cognition and attention. Progress in Neurobiology. 2002;67:53–83. doi: 10.1016/s0301-0082(02)00011-4. [DOI] [PubMed] [Google Scholar]
- Nieoullon A, Coquerel A. Dopamine: a key regulator to adapt action, emotion, motivation and cognition. Current Opinion in Neurology. 2003;16 Suppl 2:S3–S9. [PubMed] [Google Scholar]
- O'Connell JR, Weeks DE. PedCheck: a program for identification of genotype incompatibilities in linkage analysis. American Journal of Human Genetics. 1998;63:259–266. doi: 10.1086/301904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliveira SA, Scott WK, Nance MA, Watts RL, Hubble JP, Koller WC, et al. Association study of Parkin gene polymorphisms with idiopathic Parkinson disease. Archives of Neurology. 2003;60:975–980. doi: 10.1001/archneur.60.7.975. [DOI] [PubMed] [Google Scholar]
- Oswald DP, Sonenklar NA. Medication use among children with autism spectrum disorders. J. Child Adolesc. Psychopharmacol. 2007;17:348–355. doi: 10.1089/cap.2006.17303. [DOI] [PubMed] [Google Scholar]
- Ott J. Analysis of human Genetic Linkage. 3rd ed. Baltimore: Johns Hopkins University Press; 1999. [Google Scholar]
- Philippe A, Martinez M, Guilloud-Bataille M, Gillberg C, Rastam M, Sponheim E, et al. Paris Autism Research International Sibpair Study. Genome-wide scan for autism susceptibility genes. Human Molecular Genetics. 1999;8:805–812. doi: 10.1093/hmg/8.5.805. [DOI] [PubMed] [Google Scholar]
- Rice CE, Baio J, Van Naarden BK, Doernberg N, Meaney FJ, Kirby RS. A public health collaboration for the surveillance of autism spectrum disorders. Paediatrics and Perinatal Epidemiology. 2007;21:179–190. doi: 10.1111/j.1365-3016.2007.00801.x. [DOI] [PubMed] [Google Scholar]
- Risch N, Spiker D, Lotspeich L, Nouri N, Hinds D, Hallmayer J, et al. A genomic screen of autism: evidence for a multilocus etiology. American Journal of Human Genetics. 1999;65:493–507. doi: 10.1086/302497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie MD, Edwards TL, Fanelli TJ, Motsinger AA. Genetic heterogeneity is not as threatening as you might think. Genetic Epidemiology. 2007;31:797–800. doi: 10.1002/gepi.20256. [DOI] [PubMed] [Google Scholar]
- Ritchie MD, Hahn LW, Roodi N, Bailey LR, Dupont WD, Parl FF, et al. Multifactor-dimensionality reduction reveals high-order interactions among estrogen-metabolism genes in sporadic breast cancer. American Journal of Human Genetics. 2001;69:138–147. doi: 10.1086/321276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao Y, Wolpert CM, Raiford KL, Menold MM, Donnelly SL, Ravan SA, et al. Genomic screen and follow-up analysis for autistic disorder. American Journal of Medical Genetics. 2002;114:99–105. doi: 10.1002/ajmg.10153. [DOI] [PubMed] [Google Scholar]
- Stone JL, Merriman B, Cantor RM, Yonan AL, Gilliam TC, Geschwind DH, et al. Evidence for sex-specific risk alleles in autism spectrum disorder. American Journal of Human Genetics. 2004;75:1117–1123. doi: 10.1086/426034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szatmari P, Paterson AD, Zwaigenbaum L, Roberts W, Brian J, Liu XQ, et al. Mapping autism risk loci using genetic linkage and chromosomal rearrangements. Nature Genetics. 2007;39:319–328. doi: 10.1038/ng1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veenstra-Vanderweele J, Christian SL, Cook EH., Jr Autism as a paradigmatic complex genetic disorder. Annual Review of Genomics and Human Genetics. 2004a;5:379–405. doi: 10.1146/annurev.genom.5.061903.180050. [DOI] [PubMed] [Google Scholar]
- Veenstra-Vanderweele J, Cook EH., Jr Molecular genetics of autism spectrum disorder. Molecular Psychiatry. 2004b;9:819–832. doi: 10.1038/sj.mp.4001505. [DOI] [PubMed] [Google Scholar]
- Williams JG, Higgins JP, Brayne CE. Systematic review of prevalence studies of autism spectrum disorders. Archives of Diseases in Childhood. 2006;91:8–15. doi: 10.1136/adc.2004.062083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yonan AL, Alarcon M, Cheng R, Magnusson PK, Spence SJ, Palmer AA, et al. A genomewide screen of 345 families for autism-susceptibility loci. American Journal of Human Genetics. 2003;73:886–897. doi: 10.1086/378778. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.