

Abstract
Exposure to organophosphorus nerve agents induces brain seizures, which can cause profound brain damage resulting in death or long-term cognitive deficits. The amygdala and the hippocampus are two of the most seizure-prone brain structures, but their relative contribution to the generation of seizures after nerve agent exposure is unclear. Here, we report that application of 1 µM soman for 30 min, in coronal brain slices containing both the hippocampus and the amygdala, produces prolonged synchronous neuronal discharges (10 to 40 sec duration, 1.5 to 5 min interval of occurrence) resembling ictal activity in the basolateral nucleus of the amygdale (BLA), but only interictal-like activity (“spikes” of 100 to 250 msec duration; 2 to 5 sec interval) in the pyramical cell layer of the CA1 hippocampal area. BLA ictal- and CA1 interictal-like activity were synaptically driven, as they were blocked by the AMPA/kainate receptor antagonist CNQX. As the expression of the GluR5 subunit of kainate receptors is high in the amygdala, and kainate receptors containing this subunit (GluR5KRs) play an important role in the regulation of neuronal excitability in both the amygdala and the hippocampus, we tested the efficacy of a GluR5KR antagonist against the epileptiform activity induced by soman. The GluR5KR antagonist UBP302 reduced the amplitude of the hippocampal interictal-like spikes, and eliminated the seizure-like discharges in the BLA, or reduced their duration and frequency, with no significant effect on the evoked field potentials. This is the first study reporting in vitro ictal-like activity in response to a nerve agent. Our findings, along with previous literature, suggest that the amygdala may play a more important role than the hippocampus in the generation of seizures following soman exposure, and provide the first evidence that GluR5KR antagonists may be an effective treatment against nerve agent-induced seizures.
Keywords: basolateral amygdala, CA1 area, ictogenesis, GluR5 receptors, nerve agents
Organophosphorus compounds are potent neurotoxic chemicals that are widely used in industry and agriculture. Used as insecticides, worldwide, they are responsible for millions of poisonings annually (Abou-Donia, 1981; Singh and Sharma, 2000). Nerve agents are also organophosphorus compounds, and are the most lethal chemical warfare agents (Bajgar, 2005). Their primary action is the irreversible inhibition of acetylcholinesterase (AChE), leading to accumulation of acetylcholine (ACh) and overstimulation of nicotinic and muscarinic receptors in the central nervous system and the periphery (Bajgar, 2005; Barthold and Schier, 2005). Clinical manifestations after exposure develop rapidly, and can result in death or brain damage, with long-term neurological and behavioral consequences (McDonough et al., 1986; Brown and Brix, 1998; Bajgar et al., 2004). Nerve agent-induced neuronal cell death in the brain is primarily due to excitotoxicity and oxidative stress produced by excessive seizure activity (McDonough et al., 1987, 1997; Shih et al., 2003; Baille et al., 2005). Therefore, control of seizures after exposure to a nerve agent is crucial for protection against acute lethality or brain pathology (Shih et al., 2003). Current medical countermeasures against nerve agent poisoning are not always effective in preventing seizure-induced brain damage (Layish et al., 2005). Knowledge of which brain regions are primarily responsible for generating seizures in response to nerve agents, combined with knowledge of the biochemistry and physiology of these regions, can facilitate the development of effective antidotes against nerve agent-induced seizures. Limbic structures, particularly the hippocampus, piriform cortex, and the amygdala appear to play an important role in the generation of seizures by nerve agents, as suggested by the rapid increases in extracellular glutamate in these brain regions after nerve agent exposure (Lallement et al., 1991a, 1991b, 1992), and the profound damage they suffer following exposure (Hayward et al., 1990; Baze, 1993; Kadar et al., 1995; Shih et al., 2003). The amygdala, in particular, displays the earliest and most rapid increase in extracellular glutamate (Lallement et al., 1991a,b) and the most extensive damage after nerve agent exposure (Shih et al., 2003).
Soman is a highly toxic nerve agent (Boskovic, 1981), with deleterious effects that are very difficult to counteract (Shih and McDonough, 2000; Shih et al., 2003; Bajgar, 2005) due, in part, to the rapid aging of soman-inhibited AChE, which severely limits the reactivatability of the enzyme (Boskovic, 1981; Bajgar, 2005). In the present study, we investigated the in vitro effects of soman on the spontaneous and evoked neuronal activity in the CA1 hippocampal area and the basolateral nucleus of the amygdala (BLA), which, of the more than 10 nuclei comprising the amygdala (Pitkanen, 2000; McDonald, 2003; Sah et al., 2003), plays the most central role in the generation and spread of seizure activity (White and Price, 1993a, 1993b; Mohapel et al., 1996; Pitkanen et al., 1998).
Although soman induces seizures primarily via muscarinic receptor hyperstimulation (following the inhibition of AChE; Harrison et al., 2004), muscarinic receptor antagonists are effective against soman-induced seizures only when administered soon after exposure (McDonough and Shih, 1993; McDonough et al., 2000). This is part of the evidence that has led to the view that nerve agent-induced seizures are initiated by muscarinic receptor hyperstimulation, but they are sustained and reinforced primarily by glutamatergic activity (McDonough and Shih, 1997). Consistent with this view is the finding that glutamate receptor antagonists, specifically antagonists of kainate receptors containing the GluR5 subunit (GluR5KRs) block hippocampal epileptiform activity in vitro and limbic seizures in vivo induced by the muscarinic agonist pilocarpine (Smolders et al., 2002). GluR5KRs are present in the hippocampus and are exceptionally high in the BLA (Bettler et al., 1990; Li et al., 2001; Braga et al., 2003), where they modulate GABAergic and glutamatergic synaptic transmission (Huettner, 2003; Braga et al., 2003, 2004; Rogawski et al., 2003; Gryder and Rogawski, 2003; Aroniadou-Anderjaska et al., 2007), and are involved in synaptic plasticity (Li et al., 2001; Bortolotto et al., 2005) and epilepsy (Smolders et al., 2002). The potential anticonvulsant properties of GluR5KR antagonists have attracted interest because these agents are expected to have minimal side effects, as they do not affect normal synaptic transmission (Smolders et al., 2002), and their distribution in the brain is limited (Bettler et al., 1990; Li et al., 2001; Braga et al., 2003). For these reasons, in the present study we also tested the effectiveness of a GluR5KR antagonist against epileptiform activity induced by soman, in the hippocampus and the amygdala.
Experimental Procedures
Coronal slices containing both the amygdala and the hippocampus were prepared from male Sprague-Dawley rats, weighing 445 to 570 g (498.1 ± 8.5, mean ± SE; n = 25; age range: 4 to 5.5 months). The rats were deeply anesthetized with isoflurane and decapitated. The brain was rapidly removed and placed, for 1 to 2 min, in ice-cold artificial cerebrospinal fluid (ACSF) consisting of (in mM) 125 NaCl, 3 KCl, 2.0 CaCl2, 2 MgCl2, 25 NaHCO3, 1.25 NaH2PO4, and 10 glucose, and bubbled with 95% O2 and 5% CO2 to maintain a pH of 7.4. A block of the brain was prepared, and 400 µm thick coronal slices were cut with a Vibratome (Ted Pella, Redding, CA). Slices were placed in a holding chamber at room temperature. After 1 to 2 h, individual slices were transferred to a submerged type chamber, where they were superfused at 5 ml/min with pre-warmed ACSF, maintained at 32.5 – 33.5°C. The composition of the recording buffer was as above, except for KCl and MgCl2 which were 5 mM and 1 mM, respectively, in order to elevate neuronal excitability, which might facilitate induction of epileptiform activity by soman. In a group of slices, we also recorded in medium containing 3 mM KCl (as noted in the Results). Slices were allowed to equilibrate in the recording chamber for about 20 min before initiation of recordings. Extracellular recordings were obtained simultaneously from the BLA and the stratum pyramidale of hippocampal area CA1, with ACSF-filled glass micropipettes (5 to 10 MΩ resistance). Electric stimulation was applied to the Schaffer collaterals and the external capsule to evoke field potentials in the CA1 area and the BLA, respectively, using concentric, bipolar stimulating electrodes (inner diameter 25 µm, total diameter 200 µm; FHC, Bowdoinham, ME). Spontaneous and evoked analog signals from the two recorded channels were filtered at 1 Hz and 5 kHz (high and low-pass filter settings), and digitized at 3 kHz, using the pClamp10 software (Molecular Devices, Union City, CA). Soman (pinacoyl methylphosphonofluoridate) was obtained from Edgewood Chemical Biological Center diluted in saline, at the concentration of 1.9 mg/ml (10.4 mM). It was stored at −80 °C. On the day of the experiment it was thawed slowly on ice and diluted to final concentration of 1 µM with ice-cold ACSF. CNQX (an AMPA/kainate receptor antagonist) was obtained from Sigma-Aldrich (St. Louis, MO). UPB302 (a GluR5KR antagonist) was obtained from Tocris Bioscience (Ellisville, MO). Data are presented as mean ± standard error of the mean. Sample size “n” refers to the number of slices. Statistical significance was determined with the use of paired t-test. In conducting the research described in this report, the investigators adhered to the Guide for the Care and Use of Laboratory Animals by the Institute of Laboratory Animal Resources, National Research Council, in accordance with the stipulation mandated for an AAALAC accredited facility.
Results
In all of the experiments described below, the recording slice medium consisted of (in mM) 125 NaCl, 5 KCl, 2.0 CaCl2, 1 MgCl2, 25 NaHCO3, 1.25 NaH2PO4, and 10 glucose. All extracellularly recorded signals were collected in the gap-free mode to capture spontaneous events. Control recordings were obtained for 20 to 40 min before application of soman. Single stimulus pulses were delivered every 30 sec to the external capsule and the Schaffer collaterals to evoke field potentials in the BLA and the CA1 pyramidal cell layer, respectively. Stimulus intensity was adjusted to evoke a BLA field potential that was 60 to 80% of the maximal response amplitude, and a population spike in the CA1 area (PS1; Fig. 1a) that was 1 to 2 mV (peak amplitude). BLA field potentials consisted of one major negative component (N1; Fig. 1a); the characteristics of this component have been described previously (Aroniadou-Anderjaska et al., 2001). N1 was often followed by one to three low amplitude negative components, and was preceded by an early (less than 3 msec peak latency) low amplitude negative component generated by non-synaptic (antidromic or axonal) activity (see it isolated in Fig 2d). In the CA1 area, a second, smaller amplitude population spike followed PS1 in 65 % of the slices, while a third small component was also present in 25% of the slices. A non-synaptic early spike (fiber volley or neuronal firing generated by direct stimulation of dendrites or axon collaterals) sometimes preceded PS1 (see Fig. 2d). The late components present in the evoked BLA and hippocampal field potentials were probably indicative of hyperexcitability due to the relatively high potassium (5 mM) and low magnesium (1mM) in the perfusing medium. Nevertheless, there was no spontaneous activity during control recordings in any of the slices, except for one slice in which the hippocampus displayed persistent low-amplitude, high-frequency activity; this slice was not used.
FIG. 1. Soman-induced ictal activity in the BLA and interictal activity in the hippocampus are synaptically driven.
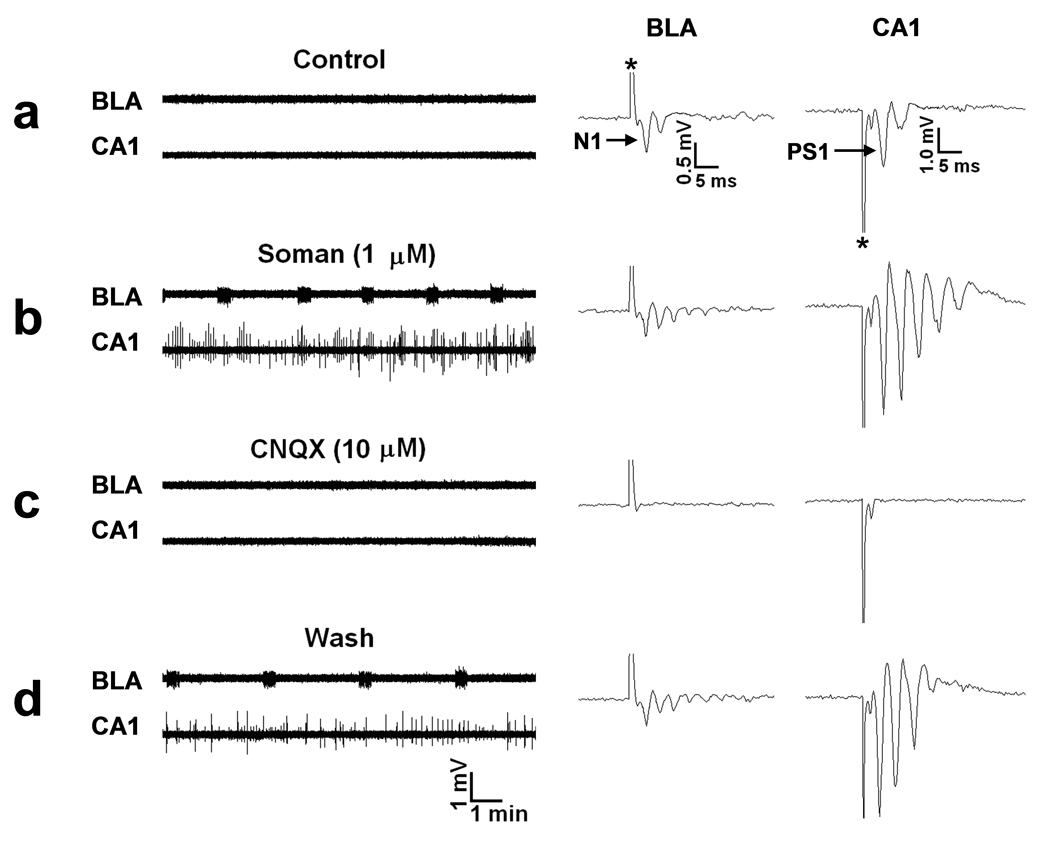
Extracellular field recordings, in gap-free mode, were simultaneously obtained in the BLA and the stratum pyramidale of the CA1 hippocampal area, in slices containing both regions. In this and the subsequent figures, the stimulus artifacts in the evoked field potentials (right panels) are indicated with an asterisk. (a) Field potentials in the BLA, evoked by stimulation of the external capsule, consisted of one major negative component (N1), followed by one or more lower-amplitude, late components. In the CA1 area, field potentials evoked by stimulation of the Schaffer collaterals consisted of a large population spike (PS1), which was often followed by one or two smaller amplitude, negative components. No spontaneous activity was present in the BLA or the CA1 area. (b) Exposure to 1 µM soman for 30 min reduced the amplitude of N1 in the BLA, and induced spontaneous, prolonged episodes of synchronous neuronal discharges resembling brain seizures. In response to soman exposure, the CA1 area produced additional population spikes in the enhanced evoked response, as well as spontaneous, interictal-like bursts. (c) Bath application of 10 µM CNQX (an AMPA/kainite receptor antagonist) blocked all synaptically-evoked components of the field potentials, as well as the BLA seizures and the CA1 interictal spikes. (d) The effects of CNQX were reversible.
FIG. 2. Soman-induced ictal activity in the BLA is blocked by a GluR5KR antagonist.
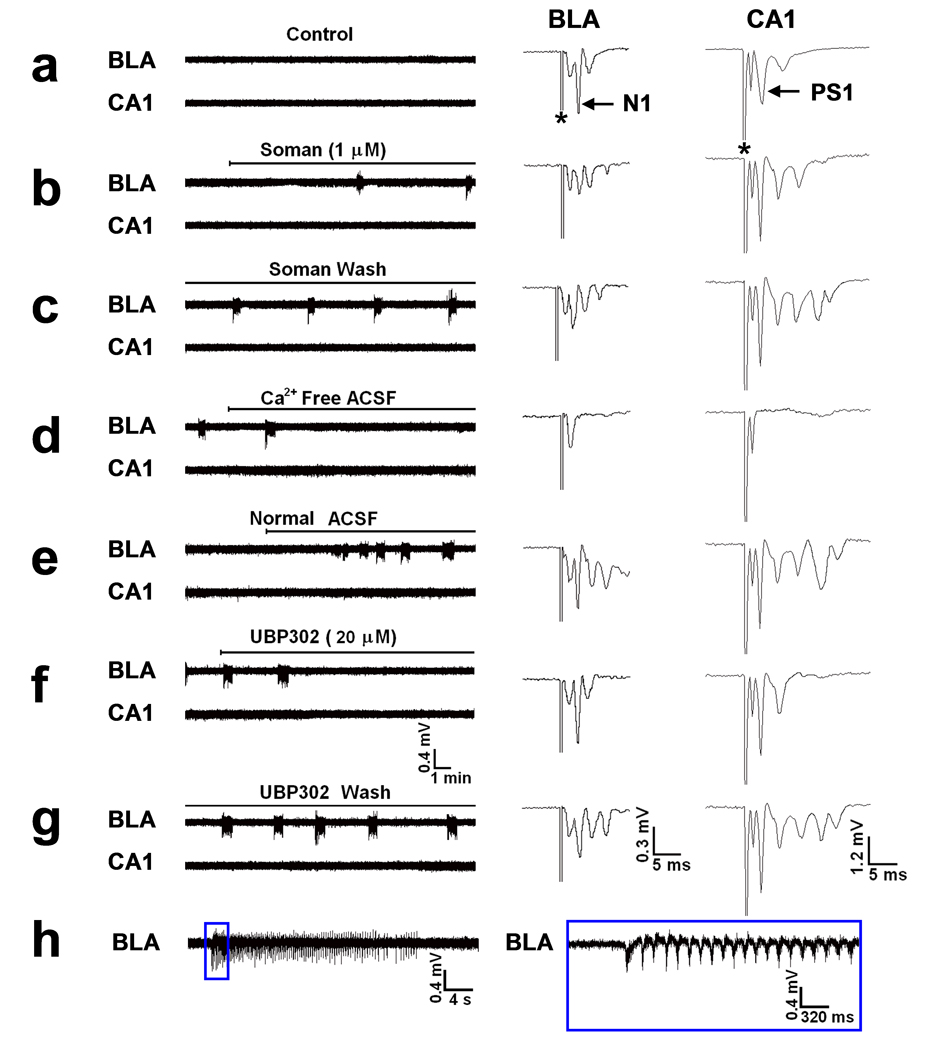
Extracellular field recordings were simultaneously obtained in the BLA and the stratum pyramidale of the CA1 hippocampal area, in slices containing both regions. (a) There was no spontaneous activity during control recordings in either the BLA or the CA1 area. N1 and PS1 of the BLA and CA1 field potentials, evoked by external capsule and Schaffer collateral pathway stimulation, respectively, were each followed by a smaller amplitude negative component, while a short latency fast component (non-synaptic; see d) was relatively pronounced in both regions, in this slice. (b) Exposure to 1 µM soman reduced the amplitude of N1 in the BLA, and produced seizure-like activity within 13 min of soman exposure. The first 3 seizures appeared at a 6 min interval, which was soon reduced to 5 min and then 4 min. Additional late negative components appeared in both the BLA and CA1 evoked field potentials, but no spontaneous activity was induced in the hippocampus in this slice. (c) The BLA seizures were not affected after soman washout. (d) Perfusion with Ca++ -free ACSF blocked both the BLA seizures and the synaptically-evoked components of the field potentials, and revealed the non-synaptic nature (fiber volley or antidromic) of the earliest, short-latency components of the evoked responses. (e) The BLA seizures and the evoked field responses returned upon reperfusion with normal medium (2 mM Ca++). (f) Bath application of 20 µM UBP302 (a GluR5KR antagonist) blocked the BLA seizures without affecting significantly the evoked field potentials, except for a reduction in the late components. (g) The effects of UBP302 were reversible. (h) A BLA seizure on an expanded time base. Oscillations had a higher frequency and amplitude at the beginning of the seizure, and dissipated gradually over the course of the seizure. The section of the trace within the blue rectangle is shown on the right with the time base further expanded.
Soman (1 µM) was bath applied for 30 min. Within 3 to 5 min of exposure to soman, the N1 component of the BLA field potential was consistently reduced (53.6 ± 3.1% reduction, n = 24; Figs. 1b and 2b), and remained at the reduced level throughout the 30 min exposure, in all slices. Spontaneous ictal-like activity (as operationally defined by Lebeda et al. 1990; hereafter referred to as ictal activity or seizures) in the BLA was induced in response to soman in 14 out of 24 (61%) slices (Figs. 1b and 2b,c), while there was no significant effect on spontaneous activity in the remaining 39% of the slices. Seizures appeared within 5 to 26 min of soman exposure (14.3 ± 2.7 min, n = 12), and in 2 slices seizures started at 4 and 7 min after washout of soman. The frequency of seizure occurrence increased somewhat during the first 15 to 20 min of their appearance, and then remained relatively stable within a slice, but varied in different slices from 0.2 /min to 0.7 /min (interval of occurrence: 1.5 to 5 min; mean interval: 3.4 ± 0.3 min, n = 14). Seizures that occurred at lower frequencies tended to have longer durations. The duration of seizures ranged from 10 to 40 sec in different slices, while within a slice it varied by 5 to 10 sec. The frequency of oscillations within a seizure was greatest within the first few seconds of seizure onset; then the frequency stabilized and subsequently decreased as the seizure dissipated (see Fig. 2h). Measured during a 5 sec period in about the mid-point of a seizure, the frequency of oscillations ranged from 5.5 Hz to 8 Hz, in different slices. Seizures were not triggered by the stimulus pulses (which were delivered every 30 sec to sample the evoked field responses), and the frequency of seizures was not affected when stimulation was turned off. In all experiments in which seizures were induced in the BLA, seizure activity continued after soman washout and throughout the recording period (up to 4 hours). After washout of soman, the amplitude of N1 recovered, partially or fully, while low amplitude, long-latency components remained more pronounced than in control (Fig. 2c,e). There was no clear correlation between recovery of the field potential and expression of seizures, as seizures appeared in some of the slices in which recovery of the field potential after soman washout was modest, and did not appear in some of the slices where recovery was nearly complete.
In the hippocampus, at the same time that the field potential in the BLA was decreased upon exposure to soman, the PS1 of the hippocampal field potential increased gradually, while additional population spikes were developing during the course of soman exposure (Fig. 1b, 2c, 3b). This was observed in all slices. The field potential remained large with multiple population spikes after wash-out of soman, for the duration of the recordings. Spontaneous, interictal-like bursts (as defined by Lebeda et al., 1990; hereafter referred to as interictal spikes or interictal activity) in response to soman exposure were observed in 17 out of 30 (57%) slices, while there was no significant effect of soman on spontaneous activity in the remaining 43% of the slices. Interictal spikes appeared within 5 to 18 min of soman exposure (11.1 ± 1.1 min; n = 17). Their frequency, duration, and amplitude increased during the first 10 to 20 min following their appearance. Although in most slices (9 out of 17) the frequency and amplitude of the interictal bursts were remarkably stable (see for example Fig. 3b), in the remaining responsive slices (n=8), brief epochs of high frequency interictal activity were interrupted by lower frequency and lower amplitude interictal spikes (for example, Fig. 1b). Interictal spike frequency, measured when stable (9 slices) or during the epochs of high frequency activity (8 slices), ranged from 0.2 Hz to 0.5 Hz. The duration of the interictal spikes ranged from 100 to 250 msec in different slices. There was no correlation between the appearance of interictal activity in the CA1 hippocampal area and the occurrence of seizures in the BLA. Furthermore, seizures in the CA1 area and interictal activity in the BLA, after soman exposure, were not observed in any of the slices. Finally, in those slices that did not display spontaneous activity in either the CA1 or the BLA area after exposure to 1 µM soman, increasing the concentration of soman to 10 µM for another 30 min had no additional effect (spontaneous epileptiform activity was not induced; n = 5).
FIG. 3. Soman-induced interictal spikes in the hippocampus are reduced by a GluR5KR antagonist.
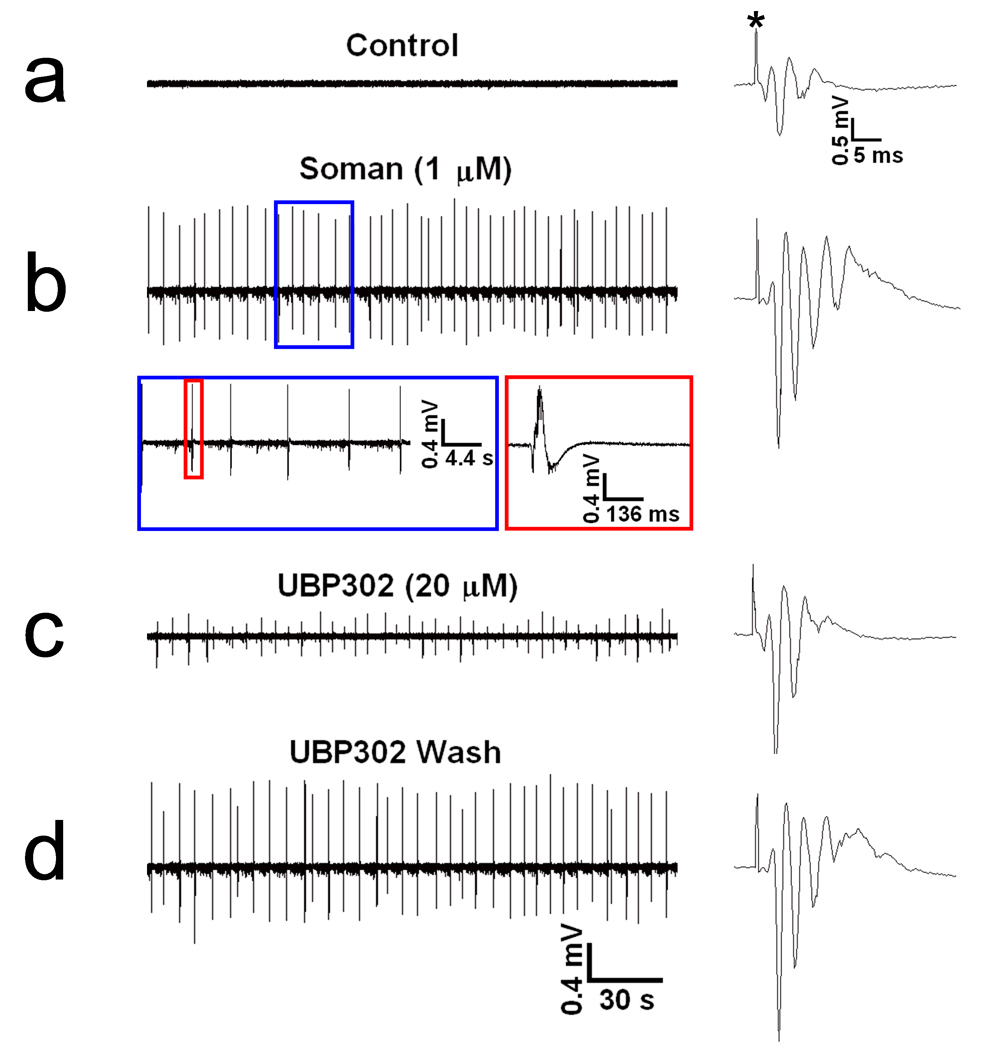
The extracellular field recordings presented here are from the pyramidal cell layer of the CA1 hippocampal area; in this experiment the BLA did not produce spontaneous activity in response to soman and, for this reason, it is not shown. (a) In control conditions there was no spontaneous activity, while the evoked field potential consisted of PS1 and a second, smaller population spike. (b) Exposure to 1 µM soman for 30 min increased the number of population spikes in the evoked field potential and induced spontaneous interictal-like spikes, within 7 min of exposure; these spikes were unaffected by soman washout. The section of trace b within the blue rectangle is shown with an expanded time base, and the spike within the red rectangle is shown on the right with the time base further expanded. (c) Bath application of 20 µM UBP302 reduced the amplitude of the spontaneous spikes, without affecting their frequency. (d) The effects of UBP302 were reversible.
The BLA seizures were synaptically driven, as they were blocked by bath application of 20 µM CNQX (n = 6, Fig. 1c) and by perfusion with Ca++-free medium (ACSF that did not include CaCl2; n = 3, Fig 2d). Similarly, the soman-induced interictal activity in the CA1 area was blocked by CNQX (n = 6, Fig. 1c), indicating its dependence on synaptic glutamatergic transmission. Bath application of the selective GluR5KR antagonist UBP302 (More et al., 2004; Partovi and Frerking, 2006) at 20 µM concentration in 5 of the slices in which soman induced seizures in the BLA, reversibly reduced the duration of the seizures (from 25 ~ 35 sec to 5 ~ 8 sec) and their frequency of occurrence (from 2 ~ 3 min to 6 ~ 7 min interval of occurrence) in 2 slices, and completely blocked the seizures in 3 slices (Fig. 2f,g). We also tested the effects of UBP302 in some of the slices in which the frequency and amplitude of the soman-induced interictal spikes in area CA1 were relatively stable. UBP302, at 20 µM, had no significant effect on the frequency of interictal spikes, but it reversibly reduced their amplitude (absolute amplitude measured from the positive peak to the negative peak and averaged within a 60 sec period) from 0.88 ± 0.07 mV to 0.46 ± 0.07 mV (n = 5, P < 0.01, Fig. 3c,d). At the concentration of 20 µM used in these experiments, UBP302 had no effect on N1 or PS1.
It has been previously shown that the pattern of epileptiform activity in vitro (ictal versus interictal-like) is influenced by the concentration of K+ in the slice medium. In the CA3 region of the hippocampus, both ictal and interictal activity were present in 5 mM K+, but the probability of occurrence of ictal activity was significantly increased when the concentration of K+ was raised to 7.5 mM (Rutecki and Yang, 1998). In the present study, it was not our aim to determine if the distinct pattern of epileptiform activity induced by soman in the BLA versus the CA1 area was still present when the K+ concentration in the slice medium is substantially above normal. However, as the potassium-dependence of epileptiform activity observed in the CA3 area may also apply to other brain regions, we wanted to determine if the ictal discharges observed in the BLA are also observed when the concentration of K+ in the recording ACSF is reduced to 3 mM, which is the K+ concentration in physiological cerebrospinal fluid (Reed et al., 1967). In 5 out of 8 slices, ictal activity was induced by 1 µM soman in the BLA, while interictal activity was observed in area CA1 of 3 slices. The effects of soman on the evoked field potentials were very similar to those in the experiments described above, which were performed in 5 mM K+. Thus, the different forms of epileptiform activity induced by soman in the BLA versus the CA1 area are observed whether 3 mM or 5 mM K+ is used in the slice recording buffer.
Discussion
Previous studies in guinea pig hippocampal slices have shown that in the CA1 region, paraoxon (Endres et al., 1989; Harrison et al., 2004) and soman (Harrison et al., 2004, 2005; Apland, 2001) induce interictal activity. There are no previous reports of the effects of nerve agents on amygdala slices. The in vitro generation of seizure-like prolonged neuronal discharges by the amygdala in response to a nerve agent, when the hippocampus, under the same conditions, generates only interictal-like bursts is demonstrated for the first time in the present study. However, similar observations have been reported when epileptiform activity is induced by 4-aminopyridine; when the BLA was independent from hippocampal inputs, it generated ictal activity in response to 4-aminopyridine, at a time when the CA3 hippocampal region generated only interictal activity (Benini et al., 2003). This suggests that the difference in the pattern of epileptiform activity generated by the BLA versus the hippocampus is not associated exclusively with soman and the mechanisms by which soman induces epileptiform activity, but, rather, the BLA circuitry may have an inherent propensity to generate ictal neuronal discharges in response to convulsants. It is presently unclear what features of the anatomy and synaptic organization of the BLA neuronal network, and/or its biochemical and physiological substrates, favor the generation of ictal activity. It seems unlikely that the drive for the generation of the BLA seizures, in the present study, originated from another brain region, because such connectivity is limited in coronal brain slices. It is also unlikely that the lack of connectivity with the entorhinal cortex, or the concentration of soman that we used influenced the form of epileptiform activity we observed in the CA1 area, as interictal-only activity is also seen in horizontal guinea pig slices that maintain the hippocampus-entorhinal cortex connections, at concentrations of soman ranging from 100 nM to 10 µM (Apland, 2001).
More than half of the slices developed spontaneous epileptiform activity in response to soman. In previous studies, application of nerve agents or other anticholinesterases to hippocampal slices has yielded a much lower percentage of responsive slices in rats (Cole and Nicoll, 1984; Williamson and Sarvey, 1985), while in guinea pigs the reported percentage of slices that developed epileptiform activity has been lower (Apland, 2001) or higher (Endres et al., 1989; Harrison et al., 2004, 2005) than in the present study. This difficulty that investigators have experienced in inducing consistently epileptiform activity in vitro by AChE inhibitors may relate to the mechanism by which these agents initiate seizure activity (increased concentration of extracellular acetylcholine), which necessitates the preservation of a sufficient number of cholinergic afferent fibers in the slices. Other factors that may have affected the percentage of responsive slices in different studies are the gender of the animals used, their age, or possible differences between rats and guinea pigs. The concentration of certain ions in the slice medium can also affect the propensity for generation of epileptiform activity (Rutecki and Yang, 1998).
How did soman induce the observed epileptiform activity? Muscarinic rather than nicotinic receptor activation appears to be responsible for induction of epileptiform activity following inhibition of AChE by soman, at least in the hippocampus (Harrison et al., 2004). Muscarinic receptors are present at postsynaptic sites where they mediate the excitatory effects of ACh, such as blockade of various potassium conductances (Cole and Nicoll, 1984; Madison et al., 1987; Washburn and Moises, 1992; Womble and Moises, 1992) or activation of a calcium-sensitive non-specific cation current (Egorov et al., 2006), but also on presynaptic terminals where they modulate the release of glutamate (Yajeya et al., 2000; Fernández de Sevilla and Buño, 2003) and GABA (Fukudome et al., 2004; Salgado et al., 2007). Thus, overstimulation of muscarinic receptors disrupts the balance of glutamatergic and GABAergic activity (Wade et al., 1987; Lallement et al., 1991b; McDonough and Shih, 1997). The effects of soman on GABA release are not quite clear, as suppression of GABAergic transmission (Santos et al., 2003), but also an increase in extracellular concentrations of GABA (Grasshoff et al., 2003) have been reported in different brain regions. In regard to glutamate release, intracranial microdialysis studies have shown increases in extracellular glutamate during soman-induced seizures, in the amygdala (Lallement et al., 1991a), hippocampus (Lallement et al., 1991a, 1991b) and piriform cortex (Wade et al., 1987), which are brain regions that suffer extensive damage by nerve agent exposure (McDonough et al., 1987, 1997; Shih et al., 2003). Thus, the current view is that cholinergic hyperactivity mediated by muscarinic receptors initiates nerve agent-induced seizures, and triggers glutamatergic hyperactivity, which sustains and reinforces seizures, and is ultimately responsible for excitotoxic neuronal damage (Lallement et al., 1991c; McDonough and Shih, 1997).
It is not clear how the increase in glutamate release in the amygdala (Lallement et al., 1991a) and hippocampus (Lallement et al., 1991a, 1991b) following soman exposure reconciles with the suppressive, presynaptic effects of muscarinic activation on evoked glutamatergic transmission in the BLA (Yajeya et al., 2000) and hippocampal area CA1 (Fernández de Sevilla and Buño, 2003). Consistent with the muscarinic receptor-mediated suppression of excitatory postsynaptic potentials in the BLA (Yajeya et al., 2000), the major component of the BLA field potential (N1) was suppressed by soman; however, in most slices, this was accompanied by the appearance of ictal activity. Perhaps the rapid massive increase in glutamate release in the amygdala following soman exposure (Lallement et al., 1991a), which could be primarily due to postsynaptic effects of the increased ACh, reduces the available glutamate at synaptic sites. This effect, along with a presynaptic inhibition of glutamate release (Yajeva et al., 2000) suppresses evoked excitatory synaptic transmission, at the same time that spontaneous activity is increased due to high levels of extracellular glutamate. In contrast to the effects of soman on the BLA field potential, the population spike in area CA1 (PS1) was increased by soman; similar observations have been reported in response to paraoxon (Endres et al., 1989). This seems consistent with the persistent enhancement of glutamatergic transmission in area CA1 when the levels of ACh are increased, in vivo, by administration of muscarinic autoreceptor antagonists (Li et al., 2007; Hayes et al., 2008). Thus, the excitatory postsynaptic effects of increased ACh may override the inhibitory presynaptic (Fernández de Sevilla and Buño, 2003) effects, in the CA1 area.
In the present study, the blockade of both the BLA and the CA1 soman-induced epileptiform activity by CNQX supports the view that glutamatergic mechanisms play a major role in driving nerve agent-induced epileptiform activity, but does not exclude the involvement of cell-intrinsic membrane conductances, whether these conductances come into play by muscarinic stimulation, or by other mechanisms triggered secondarily. The generation of ictal and interictal activity involves a complex interplay between a number of parameters, such as recurrent glutamatergic synaptic interactions, GABAergic inhibition, and intrinsic membrane conductances that participate in or are responsible for dendritic calcium spikes, generation of bursting activity, or sustained depolarization (Traub and Jefferys, 1994; Rutecki and Yang, 1998; McCormick and Contreras, 2001; Hadar et al., 2002; Rutecki et al., 2002). However, the relative contribution of these parameters to the generation of epileptiform activity, how it differs in different brain regions, and the factors determining the generation of ictal versus interictal activity are still not well understood.
The suppression of epileptiform activity in both the BLA and the CA1 area by the GluR5KR antagonist UBP302 also supports the view that glutamatergic hyperactivity sustains soman-induced seizures. GluR5KRs modulate excitatory and inhibitory transmission in both the BLA (Braga et al. 2003, 2004; Gryder and Rogawski, 2003; Aroniadou-Anderjaska et al., 2007) and the hippocampus (Clarke et al., 1997; Huettner, 2003; Lerma, 2003), and their expression is particulalry high in the BLA (Braga et al., 2003; Li et al., 2001). These receptors are also involved in epilepsy (Smolders et al., 2002; Rogawski et al., 2003; Braga et al., 2004; Kaminski et al., 2004). Most relevant to the present study is the demonstration that GluR5KR antagonists block pilocarpine-induced epileptiform activity in hippocampal slices, and limbic seizures in vivo. Pilocarpine, like soman, initiates seizures by excessive muscarinic receptor activation (Smolders et al., 2002), which, as discussed above, is followed by glutamatergic hyperactivity.
How can the effectiveness of GluR5KR antagonists against epileptiform/seizure activity be explained? A complete answer is not available because not all of the functions of the GluR5KRs are clear at present. Based on existing knowledge we can suggest that during epileptiform activity, elevated levels of glutamate acting via presynaptic GluR5KRs will reduce GABA release in the amygdala (Braga et al., 2003) and the hippocampus (Clarke et al., 1997), and will contribute to hyperexcitation of pyramidal cells, at least in the BLA where it is known that principal cells have functional somatodendritic GluR5KRs (Gryder and Rogawski, 2003). Somatodendritic GluR5KRs are also present on GABAergic neurons (Clarke et al., 1997; Braga et al., 2003), however, at least in the BLA, the net effect of strong GluR5KR activation is excitatory, producing strong epileptiform activity (Aroniadou-Anderjaska et al., 2008). Blockade of the excitatory postsynaptic effects on principal neurons and the suppressive, presynaptic effect of GluR5KR activation on GABA release by a GluR5KR antagonist – which will enhance inhibition- could produce or contribute to the reduction in the amplitude of the CA1 interictal spikes, observed in the present study, and the reduction in the frequency and duration or the complete elimination of the BLA seizures. In addition, GluR5KRs are permeable to Ca++, particularly when they contain the unedited version of the GluR5 subunit, which confers high Ca++ permeability (Burnashev et al., 1996, Savidge et al., 1997; Chittajallu et al., 1999); approximately 30% of the GluR5 mRNA remains in the unedited form in the adult hippocampus (Bernard et al., 1994) and amygdala (Li et al., 2001). Therefore, Ca++ influx through GluR5KRs could contribute to epileptiform activity; this may occur by various mechanisms, including stimulation of Ca++ release from intracellular stores which appears to be necessary for ictal activity (Hadar et al., 2002; Rutecki et al., 2002).
One of the reasons that GluR5KRs have attracted strong interest as a potential pharmacological target for the prevention and treatment of epilepsy is that GluR5KR antagonists are unlikely to have significant side effects (see for example Sang et al., 2004). This is because the distribution of GluR5KRs in the brain is relatively limited (Bettler et al., 1990; Li et al., 2001; Braga et al., 2003), and GluR5KR antagonists have no significant effect on normal excitatory synaptic transmission (Smolders et al., 2002). Indeed, in the present study, 20 µM UBP302 had no significant effect on N1 or PS1, although it reduced the late components of the evoked field potentials, an effect that seems consistent with the enhancement of GABAergic synaptic transmission suggested above.
Previous findings have hinted that the amygdala may play the most central role in the generation of brain seizures after nerve agent exposure, by showing that after in vivo exposure to soman, the amygdala displays the earliest and most rapid increase in extracellular glutamate (suggesting an early involvement in the development of seizures; Lallement et al., 1991a,b), and suffers the most extensive damage (Shih et al., 2003). The present findings that the amygdala generates spontaneous, prolonged, synchronous neuronal discharges resembling brain seizures following soman exposure, while the hippocampus generates only interictal-like activity, may suggest that it is primarily from the amygdala rather than from the hippocampus that seizures spread to other brain regions culminating to status epilepticus after soman exposure, and potentially identify the most important brain structure that should be targeted pharmacologically for the suppression of soman-induced seizures. However, it remains to be determined if the in vitro findings also apply in vivo. The significance of the soman-induced interictal-like activity in the hippocampus, seen in our in vitro conditions, and its potential contribution to triggering seizure activity requires investigation, particularly considering that hippocampal interictal discharges restrain rather than facilitate ictal activity in the BLA and entorhinal cortex (Benini et al., 2003).
Acknowledgments
This work was supported by the Uniformed Services University of the Health Sciences Grant H070SG and by the National Institutes of Health CounterACT Program through the National Institute of Neurological Disorders and Stroke (award # U01 NS058162-01). The opinions or assertions contained herein are the private views of the authors and are not to be construed as official or as reflecting the views of the Department of the Army or the Department of Defense. This work was also supported by the Defense Threat Reduction Agency-Joint Science and Technology Office, Medical S&T Division grant # 1.E0021_07_US_C.
Abbreviations
- BLA
basolateral amygdala
- ACSF
artificial cerebrospinal fluid
- GluR5KRs
kainate receptors containing the GluR5 subunit
- CNQX
6-cyano-7-nitroquinoxaline-2,3-dione
- ACh
acetylcholine
- AChE
acetylcholinesterase
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abou-Donia MB. Organophosphorus ester-induced delayed neurotoxicity. Annu Rev Pharmacol Toxicol. 1981;21:511–548. doi: 10.1146/annurev.pa.21.040181.002455. [DOI] [PubMed] [Google Scholar]
- Apland J. Effects of anticonvulsant drugs on soman-induced epileptiform activity in guinea pig hippocampal-entorhinal slices. USAMRICD-TR-01-02. Aberdeen Proving Ground, MD: U.S. Army Medical Research Institute of Chemical Defense; 2001. [Google Scholar]
- Aroniadou-Anderjaska V, Post RM, Rogawski MA, Li H. Input-specific LTP and depotentiation in the basolateral amygdala. NeuroReport. 2001;2:635–640. doi: 10.1097/00001756-200103050-00041. [DOI] [PubMed] [Google Scholar]
- Aroniadou-Anderjaska V, Qashu F, Braga MF. Mechanisms regulating GABAergic inhibitory transmission in the basolateral amygdala: Implications for epilepsy and anxiety disorders. Amino Acids. 2007;32:305–315. doi: 10.1007/s00726-006-0415-x. [DOI] [PubMed] [Google Scholar]
- Aroniadou-Anderjaska V, Fritsch B, Qashu F, Braga MF. Pathology and pathophysiology of the amygdala in epileptogenesis and epilepsy. Epilepsy Res. 2008;78:102–116. doi: 10.1016/j.eplepsyres.2007.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baille V, Clarke PG, Brochier G, Dorandeu F, Verna JM, Four E, Lallement G, Carpentier P. Soman-induced convulsions: the neuropathology revisited. Toxicology. 2005;215:1–24. doi: 10.1016/j.tox.2005.05.028. [DOI] [PubMed] [Google Scholar]
- Bajgar J. Complex view on poisoning with nerve agents and organophosphates. Acta Medica (Hradec Kralove) 2005;48:3–21. [PubMed] [Google Scholar]
- Bajgar J, Sevelova L, Krejcova G, Fusek J, Vachek J, Kassa J, Herink J, de Jong LP, Benschop HP. Biochemical and behavioral effects of soman vapors in low concentrations. Inhal Toxicol. 2004;16:497–507. doi: 10.1080/08958370490442430. [DOI] [PubMed] [Google Scholar]
- Barthold CL, Schier JG. Organic phosphorus compounds-nerve agents. Crit Care Clin. 2005;21:673–689. doi: 10.1016/j.ccc.2005.05.010. [DOI] [PubMed] [Google Scholar]
- Baze WB. Soman-induced morphological changes: an overview in the nonhuman primate. J Appl Toxicol. 1993;13:173–177. doi: 10.1002/jat.2550130306. [DOI] [PubMed] [Google Scholar]
- Benini R, D'Antuono M, Pralong E, Avoli M. Involvement of amygdala networks in epileptiform synchronization in vitro. Neuroscience. 2003;120:75–84. doi: 10.1016/s0306-4522(03)00262-8. [DOI] [PubMed] [Google Scholar]
- Bernard A, Khrestchatisky M. Assessing the extent of RNA editing in the TMII regions of GluR5 and GluR6 kainate receptors during rat brain development. J Neurochem. 1994;62:2057–2060. doi: 10.1046/j.1471-4159.1994.62052057.x. [DOI] [PubMed] [Google Scholar]
- Bettler B, Boulter J, Hermans-Borgmeyer I, O'Shea-Greenfield A, Deneris ES, Moll C, Borgmeyer U, Hollmann M, Heinemann S. Cloning of a novel glutamate receptor subunit, GluR5: expression in the nervous system during development. Neuron. 1990;5:583–595. doi: 10.1016/0896-6273(90)90213-y. [DOI] [PubMed] [Google Scholar]
- Bortolotto ZA, Nistico R, More JC, Jane DE, Collingridge GL. Kainate receptors and mossy fiber LTP. Neurotoxicology. 2005;26:769–777. doi: 10.1016/j.neuro.2005.02.004. [DOI] [PubMed] [Google Scholar]
- Boskovic B. The treatment of soman poisoning and its perspectives. Toxicol Sci. 1981;1:203–213. doi: 10.1016/s0272-0590(81)80059-0. [DOI] [PubMed] [Google Scholar]
- Braga MF, Aroniadou-Anderjaska V, Xie J, Li H. Bidirectional modulation of GABA release by presynaptic glutamate receptor 5 kainate receptors in the basolateral amygdala. J Neurosci. 2003;23:442–452. doi: 10.1523/JNEUROSCI.23-02-00442.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braga MF, Aroniadou-Anderjaska V, Li H. The physiological role of kainate receptors in the amygdala. Mol Neurobiol. 2004;30:127–141. doi: 10.1385/MN:30:2:127. [DOI] [PubMed] [Google Scholar]
- Brown MA, Brix KA. Review of health consequences from high, intermediate, and low-level exposure to organophosphorus nerve agents. J Appl Toxicol. 1998;6:393–408. doi: 10.1002/(sici)1099-1263(199811/12)18:6<393::aid-jat528>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- Burnashev N, Villarroel A, Sakmann B. Dimensions and ion selectivity of recombinant AMPA and kainate receptor channels and their dependence on Q/R site residues. J Physiol (Lond) 1996;496:165–173. doi: 10.1113/jphysiol.1996.sp021674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chittajallu R, Braithwaite SP, Clarke VR, Henley JM. Kainate receptors: subunits, synaptic localization and function. Trends Pharmacol Sci. 1999;20:26–35. doi: 10.1016/s0165-6147(98)01286-3. [DOI] [PubMed] [Google Scholar]
- Clarke VR, Ballyk BA, Hoo KH, Mandelzys A, Pellizzari A, Bath CP, Thomas J, Sharpe EF, Davies CH, Ornstein PL, Schoepp DD, Kamboj RK, Collingridge GL, Lodge D, Bleakman D. A hippocampal GluR5 kainate receptor regulating inhibitory synaptic transmission. Nature. 1997;389:599–603. doi: 10.1038/39315. [DOI] [PubMed] [Google Scholar]
- Cole AE, Nicoll RA. Characterization of a slow cholinergic postsynaptic potential recorded in vitro from rat hippocampal pyramidal cells. J Physiol (Lond) 1984;352:173–188. doi: 10.1113/jphysiol.1984.sp015285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egorov AV, Unsicker K, von Bohlen und Halbach O. Muscarinic control of graded persistent activity in lateral amygdala neurons. Eur J Neurosci. 2006;24:3183–3194. doi: 10.1111/j.1460-9568.2006.05200.x. [DOI] [PubMed] [Google Scholar]
- Endres W, Spuler A, ten Bruggencate G. Acetylcholinesterase reactivators antagonize epileptiform bursting induced by paraoxon in guinea pig hippocampal slices. Pharmacol Exp Ther. 1989;251:1181–1186. [PubMed] [Google Scholar]
- Fernández de Sevilla D, Buño W. Presynaptic inhibition of Schaffer collateral synapses by stimulation of hippocampal cholinergic afferent fibres. Eur J Neurosci. 2003;17:555–558. doi: 10.1046/j.1460-9568.2003.02490.x. [DOI] [PubMed] [Google Scholar]
- Fukudome Y, Ohno-Shosaku T, Matsui M, Omori Y, Fukaya M, Tsubokawa H, Taketo MM, Watanabe M, Manabe T, Kano M. Two distinct classes of muscarinic action on hippocampal inhibitory synapses: M2-mediated direct suppression and M1/M3-mediated indirect suppression through endocannabinoid signalling. Eur J Neurosci. 2004;19:2682–2692. doi: 10.1111/j.0953-816X.2004.03384.x. [DOI] [PubMed] [Google Scholar]
- Grasshoff C, Gillessen T, Thiermann H, Wagner E, Szinicz L. The effect of acetylcholinesterase-inhibition on depolarization-induced GABA release from rat striatal slices. Toxicology. 2003;184:149–156. doi: 10.1016/s0300-483x(02)00571-1. [DOI] [PubMed] [Google Scholar]
- Gryder DS, Rogawski MA. Selective antagonism of GluR5 kainate-receptor-mediated synaptic currents by topiramate in rat basolateral amygdala neurons. J Neurosci. 2003;23:7069–7074. doi: 10.1523/JNEUROSCI.23-18-07069.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadar EJ, Yang Y, Sayin U, Rutecki PA. Suppression of pilocarpine-induced ictal oscillations in the hippocampal slice. Epilepsy Res. 2002;49:61–71. doi: 10.1016/s0920-1211(02)00016-5. [DOI] [PubMed] [Google Scholar]
- Harrison PK, Sheridan RD, Green AC, Scott IR, Tattersall JE. A guinea pig hippocampal slice model of organophosphate-induced seizure activity. J Pharmacol Exp Ther. 2004;310:678–686. doi: 10.1124/jpet.104.065433. [DOI] [PubMed] [Google Scholar]
- Harrison PK, Sheridan RD, Green AC, Tattersall JE. Effects of anticonvulsants on soman-induced epileptiform activity in the guinea-pig in vitro hippocampus. Eur J Pharmacol. 2005;518:123–132. doi: 10.1016/j.ejphar.2005.06.032. [DOI] [PubMed] [Google Scholar]
- Hayes J, Li S, Anwyl R, Rowan MJ. A role for protein kinase A and protein kinase M zeta in muscarinic acetylcholine receptor-initiated persistent synaptic enhancement in rat hippocampus in vivo. Neuroscience. 2008;151:604–612. doi: 10.1016/j.neuroscience.2007.10.016. [DOI] [PubMed] [Google Scholar]
- Hayward IJ, Wall HG, Jaax NK, Wade JV, Marlow DD, Nold JB. Decreased brain pathology in organophosphate-exposed rhesus monkeys following benzodiazepine therapy. Neurol Sci. 1990;98:99–106. doi: 10.1016/0022-510x(90)90185-p. [DOI] [PubMed] [Google Scholar]
- Huettner JE. Kainate receptors and synaptic transmission. Prog Neurobiol. 2003;70:387–407. doi: 10.1016/s0301-0082(03)00122-9. [DOI] [PubMed] [Google Scholar]
- Kadar T, Shapira S, Cohen G, Sahar R, Alkalay D, Raveh L. Sarin-induced neuropathology in rats. Hum Exp Toxicol. 1995;14:252–259. doi: 10.1177/096032719501400304. [DOI] [PubMed] [Google Scholar]
- Kaminski RM, Banerjee M, Rogawski MA. Topiramate selectively protects against seizures induced by ATPA, a GluR5 kainate receptor agonist. Neuropharmacology. 2004;46:1097–1104. doi: 10.1016/j.neuropharm.2004.02.010. [DOI] [PubMed] [Google Scholar]
- Lallement G, Carpentier P, Collet A, Pernot-Marino I, Baubichon D, Sentenac-Roumanou H, Blanchet G. Involvement of glutamatergic system of amygdala in generalized seizures induced by soman: comparison with the hippocampus. C R Acad Sci III. 1991a;313:421–426. [PubMed] [Google Scholar]
- Lallement G, Carpentier P, Collet A, Pernot-Marino I, Baubichon D, Blanchet G. Effects of soman-induced seizures on different extracellular amino acid levels and on glutamate uptake in rat hippocampus. Brain Res. 1991b;563:234–240. doi: 10.1016/0006-8993(91)91539-d. [DOI] [PubMed] [Google Scholar]
- Lallement G, Carpentier P, Pernot-Marino I, Baubichon D, Collet A, Blanchet G. Involvement of the different rat hippocampal glutamatergic receptors in development of seizures induced by soman: an autoradiographic study. Neurotoxicology. 1991c;12:655–664. [PubMed] [Google Scholar]
- Lallement G, Carpentier P, Collet A, Baubichon D, Pernot-Marino I, Blanchet G. Extracellular acetylcholine changes in rat limbic structures during soman-induced seizures. Neurotoxicology. 1992;13:557–567. [PubMed] [Google Scholar]
- Layish I, Krivoy A, Rotman E, Finkelstein A, Tashma Z, Yehezkelli Y. Pharmacologic prophylaxis against nerve agent poisoning. Isr Med Assoc J. 2005;7:182–187. [PubMed] [Google Scholar]
- Lebeda F, Ton T, Rutecki P. Synaptic inhibition and soman-induced epileptiform activity in the hippocampus. Proceedings of the Workshop on Convulsions and Related Brain Damage Induced by Organophosphorus Agents. Aberdeen Proving Ground, MD: U.S. Army Medical Research Institute of Chemical Defense; 1990. [Google Scholar]
- Lerma J. Roles and rules of kainate receptors in synaptic transmission. Nat Rev Neurosci. 2003;4:481–495. doi: 10.1038/nrn1118. [DOI] [PubMed] [Google Scholar]
- Li H, Chen A, Xing G, Wei ML, Rogawski MA. Kainate receptor-mediated heterosynaptic facilitation in the amygdala. Nat Neurosci. 2001;4:612–620. doi: 10.1038/88432. [DOI] [PubMed] [Google Scholar]
- Li S, Cullen WK, Anwyl R, Rowan MJ. Muscarinic acetylcholine receptor-dependent induction of persistent synaptic enhancement in rat hippocampus in vivo. Neuroscience. 2007;144:754–761. doi: 10.1016/j.neuroscience.2006.10.001. [DOI] [PubMed] [Google Scholar]
- Madison DV, Lancaster B, Nicoll RA. Voltage clamp analysis of cholinergic action in the hippocampus. J Neurosci. 1987;7:733–741. doi: 10.1523/JNEUROSCI.07-03-00733.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormick DA, Contreras D. On the cellular and network bases of epileptic seizures. Annu Rev Physiol. 2001;63:815–846. doi: 10.1146/annurev.physiol.63.1.815. [DOI] [PubMed] [Google Scholar]
- McDonald AJ. Is there an amygdala and how far does it extend? An anatomical perspective. Ann N Y Acad Sci. 2003;985:1–21. doi: 10.1111/j.1749-6632.2003.tb07067.x. [DOI] [PubMed] [Google Scholar]
- McDonough JH, Jr, Smith RF, Smith CD. Behavioral correlates of soman-induced neuropathology: deficits in DRL acquisition. Toxicol Teratol. 1986;8:179–187. [PubMed] [Google Scholar]
- McDonough JH, Jr, McLeod CG, Jr, Nipwoda MT. Direct microinjection of soman or VX into the amygdala produces repetitive limbic convulsions and neuropathology. Brain Res. 1987;435:123–137. doi: 10.1016/0006-8993(87)91593-9. [DOI] [PubMed] [Google Scholar]
- McDonough JH, Jr, Shih TM. Pharmacological modulation of soman-induced seizures. Neurosci Biobehav Rev. 1993;17:203–215. doi: 10.1016/s0149-7634(05)80151-4. [DOI] [PubMed] [Google Scholar]
- McDonough JH, Jr, Shih TM. Neuropharmacological mechanisms of nerve agent-induced seizure and neuropathology. Neurosci Biobehav Rev. 1997;21:559–579. doi: 10.1016/s0149-7634(96)00050-4. [DOI] [PubMed] [Google Scholar]
- McDonough JH, Jr, Zoeffel LD, McMonagle J, Copeland TL, Smith CD, Shih TM. Anticonvulsant treatment of nerve agent seizures: anticholinergics versus diazepam in soman-intoxicated guinea pigs. Epilepsy Res. 2000;38:1–14. doi: 10.1016/s0920-1211(99)00060-1. [DOI] [PubMed] [Google Scholar]
- Mohapel P, Dufresne C, Kelly ME, McIntyre DC. Differential sensitivity of various temporal lobe structures in the rat to kindling and status epilepticus induction. Epilepsy Res. 1996;23:179–187. doi: 10.1016/0920-1211(95)00084-4. [DOI] [PubMed] [Google Scholar]
- More JC, Nistico R, Dolman NP, Clarke VR, Alt AJ, Ogden AM, Buelens FP, Troop HM, Kelland EE, Pilato F, Bleakman D, Bortolotto ZA, Collingridge GL, Jane DE. Characterisation of UBP296: a novel, potent and selective kainate receptor antagonist. Neuropharmacology. 2004;47:46–64. doi: 10.1016/j.neuropharm.2004.03.005. [DOI] [PubMed] [Google Scholar]
- Partovi D, Frerking M. Presynaptic inhibition by kainate receptors converges mechanistically with presynaptic inhibition by adenosine and GABAB receptors. Neuropharmacology. 2006;51:1030–1037. doi: 10.1016/j.neuropharm.2006.06.010. [DOI] [PubMed] [Google Scholar]
- Pitkänen A, Tuunanen J, Kalviainen R, Partanen K, Salmenpera T. Amygdala damage in experimental and human temporal lobe epilepsy. Epilepsy Res. 1998;32:233–253. doi: 10.1016/s0920-1211(98)00055-2. [DOI] [PubMed] [Google Scholar]
- Pitkänen A. Connectivity of the rat amygdaloid complex. In: Aggleton JP, editor. The Amygdala: A functional analysis. New York: Oxford UP; 2000. pp. 31–115. [Google Scholar]
- Reed DJ, Withrow CD, Woodbury DM. Electrolyte and acid-base parameters of rat cerebrospinal fluid. Exper. Brain Res. 1967;3:212–219. doi: 10.1007/BF00235585. [DOI] [PubMed] [Google Scholar]
- Rogawski MA, Gryder D, Castaneda D, Yonekawa W, Banks MK, Lia H. GluR5 kainate receptors, seizures, and the amygdala. Ann N Y Acad Sci. 2003;985:150–162. doi: 10.1111/j.1749-6632.2003.tb07079.x. [DOI] [PubMed] [Google Scholar]
- Rutecki PA, Yang Y. Ictal epileptiform activity in the CA3 region of hippocampal slices produced by pilocarpine. J Neurophysiol. 1998;79:3019–3029. doi: 10.1152/jn.1998.79.6.3019. [DOI] [PubMed] [Google Scholar]
- Rutecki PA, Sayin U, Yang Y, Hadar E. Determinants of ictal epileptiform patterns in the hippocampal slice. Epilepsia. 2002;43:179–183. doi: 10.1046/j.1528-1157.43.s.5.34.x. [DOI] [PubMed] [Google Scholar]
- Sah P, Faber ES, Lopez De Armentia M, Power J. The amygdaloid complex: anatomy and physiology. Physiol Rev. 2003;83:803–834. doi: 10.1152/physrev.00002.2003. [DOI] [PubMed] [Google Scholar]
- Salgado H, Bellay T, Nichols JA, Bose M, Martinolich L, Perrotti L, Atzori M. Muscarinic M2 and M1 receptors reduce GABA release by Ca2+ channel modulation through activation of PI3K/Ca2+ -independent and PLC/Ca2+ -dependent PKC. J Neurophysiol. 2007;98:952–965. doi: 10.1152/jn.00060.2007. [DOI] [PubMed] [Google Scholar]
- Sang CN, Ramadan NM, Wallihan RG, Chappell AS, Freitag FG, Smith TR, Silberstein SD, Johnson KW, Phebus LA, Bleakman D, Ornstein PL, Arnold B, Tepper SJ, Vandenhende F. LY293558, a novel AMPA/GluR5KR antagonist, is efficacious and well-tolerated in acute migraine. Cephalalgia. 2004;24:596–602. doi: 10.1111/j.1468-2982.2004.00723.x. [DOI] [PubMed] [Google Scholar]
- Santos MD, Pereira EF, Aracava Y, Castro NG, Fawcett WP, Randall WR, Albuquerque EX. Low concentrations of pyridostigmine prevent soman-induced inhibition of GABAergic transmission in the central nervous system: involvement of muscarinic receptors. J Pharmacol Exp Ther. 2003;304:254–265. doi: 10.1124/jpet.102.043109. [DOI] [PubMed] [Google Scholar]
- Savidge JR, Bleakman D, Bristow DR. Identification of kainate receptor-mediated intracellular calcium increases in cultured rat cerebellar granule cells. J Neurochem. 1997;69:1763–1766. doi: 10.1046/j.1471-4159.1997.69041763.x. [DOI] [PubMed] [Google Scholar]
- Shih TM, McDonough JH. Efficacy of biperiden and atropine as anticonvulsant treatment for organophosphorus nerve agent intoxication. Arch Toxicol. 2000;74:165–172. doi: 10.1007/s002040050670. [DOI] [PubMed] [Google Scholar]
- Shih TM, Duniho SM, John H, McDonough JH. Control of nerve agent induced seizures is critical for neuroprotection and survival. Tox Applied Pharm. 2003;188:69–80. doi: 10.1016/s0041-008x(03)00019-x. [DOI] [PubMed] [Google Scholar]
- Singh S, Sharma N. Neurological syndromes following organophosphate poisoning. Neurol India. 2000;48:308–313. [PubMed] [Google Scholar]
- Smolders I, Bortolotto ZA, Clarke VR, Warre R, Khan GM, O'Neill MJ, Ornstein PL, Bleakman D, Ogden A, Weiss B, Stables JP, Ho KH, Ebinger G, Collingridge GL, Lodge D, Michotte Y. Antagonists of GLU(K5)-containing kainate receptors prevent pilocarpine-induced limbic seizures. Nat Neurosci. 2002;5:796–804. doi: 10.1038/nn880. [DOI] [PubMed] [Google Scholar]
- Traub RD, Jefferys JG. Are there unifying principles underlying the generation of epileptic afterdischarges in vitro? Prog Brain Res. 1994;102:383–394. doi: 10.1016/S0079-6123(08)60554-3. [DOI] [PubMed] [Google Scholar]
- Wade JV, Samson FE, Nelson SR, Pazdernik TL. Changes in extracellular amino acids during soman- and kainic acid-induced seizures. J Neurochem. 1987;49:645–650. doi: 10.1111/j.1471-4159.1987.tb02912.x. [DOI] [PubMed] [Google Scholar]
- Washburn MS, Moises HC. Muscarinic responses of rat basolateral amygdaloid neurons recorded in vitro. J Physiol. 1992;449:121–154. doi: 10.1113/jphysiol.1992.sp019078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White LE, Price JL. The functional anatomy of limbic status epilepticus in the rat. I. Patterns of 14C-2-deoxyglucose uptake and Fos immunocytochemistry. J Neurosci. 1993a;13:4787–4809. doi: 10.1523/JNEUROSCI.13-11-04787.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White LE, Price JL. The functional anatomy of limbic status epilepticus in the rat. II. The effects of focal deactivation. J Neurosci. 1993b;13:4810–4830. doi: 10.1523/JNEUROSCI.13-11-04810.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson AM, Sarvey JM. Effects of cholinesterase inhibitors on evoked responses in field CA1 of the rat hippocampus. J Pharmacol Exp Ther. 1985;235:448–455. [PubMed] [Google Scholar]
- Womble MD, Moises HC. Muscarinic inhibition of M-current and a potassium leak conductance in neurones of the rat basolateral amygdala. J Physiol. 1992;457:93–114. doi: 10.1113/jphysiol.1992.sp019366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yajeya J, De La Fuente A, Criado JM, Bajo V, Sánchez-Riolobos A, Heredia M. Muscarinic agonist carbachol depresses excitatory synaptic transmission in the rat basolateral amygdala in vitro. Synapse. 2000;38:151–160. doi: 10.1002/1098-2396(200011)38:2<151::AID-SYN6>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]