

Abstract
NMR titration experiments with labeled human ubiquitin were employed in concert with chromatographic data obtained with a library of ubiquitin mutants to study the nature of protein adsorption in multimodal (MM) chromatography. The elution order of the mutants on the MM resin was significantly different from that obtained by ion-exchange chromatography. Further, the chromatographic results with the protein library indicated that mutations in a defined region induced greater changes in protein affinity to the solid support. Chemical shift mapping and determination of dissociation constants from NMR titration experiments with the MM ligand and isotopically enriched ubiquitin were used to determine and rank the relative binding affinities of interaction sites on the protein surface. The results with NMR confirmed that the protein possessed a distinct preferred binding region for the MM ligand in agreement with the chromatographic results. Finally, coarse-grained ligand docking simulations were employed to study the modes of interaction between the MM ligand and ubiquitin. The use of NMR titration experiments in concert with chromatographic data obtained with protein libraries represents a previously undescribed approach for elucidating the structural basis of protein binding affinity in MM chromatographic systems.
Keywords: ligand binding site, mixed mode chromatography, protein-ligand interactions, binding site mapping, pseudoaffinity
The development of efficient bioseparation processes for the production of high-purity biopharmaceuticals is one of the most pressing challenges facing the pharmaceutical and biotechnology industries today. In addition, high-resolution separations for complex bioanalytical applications are becoming increasingly important. Although it is generally accepted that nonspecific interactions can often complicate single mode chromatographic separations (e.g., ion-exchange, reversed-phase), these additional interactions can also result in unexpected selectivities (1, 2).
Recent advances in the design of multimodal (MM) chromatographic systems have produced previously undescribed classes of chromatographic materials that can provide alternative and improved selectivities as compared to traditional single mode chromatographic materials (3–9). Johansson et al. have developed a library of MM ligands that can be employed for the capture of charged proteins under high salt conditions (4, 5). Liu et al. have developed a silica-based MM resin capable of weak anion-exchange and reversed-phase interactions for the simultaneous separation of acidic, basic, and neutral pharmaceutical compounds (9). Small ligand pseudoaffinity chromatographic materials such as those used for hydrophobic charge induction chromatography have resulted in previously undescribed classes of MM ligands that offer unique selectivities due more to multiple low affinity MM interactions than to specific binding to certain classes of proteins (10). In addition, several libraries of MM ligands have been recently developed and employed on chromatographic resins for screening with biological mixtures. Applications of this technology range from preparative protein purifications (11, 12) to front end separations for mass-spectrometry analysis (13, 14).
Although MM resins offer significant potential for bioseparations, there is a lack of fundamental understanding of the nature of binding of these ligands to protein surfaces. We have recently employed a homologous library of cold shock protein B mutants to examine differences in protein binding behavior on an ion-exchange (SP Sepharose FF) and MM chromatographic surface (Capto MMC) (15). Chromatographic experiments showed stronger retention for the majority of the proteins on Capto MMC as compared to SP Sepharose FF. Although that study provided implicit information about protein binding behavior in MM chromatographic systems, there is a need to more directly interrogate the underlying nature of MM-protein binding in these systems.
In this study, we employ NMR titration experiments with 15N/13C-enriched human ubiquitin as a model protein in concert with chromatographic data obtained with a library of ubiquitin mutants to carry out an in-depth analysis of the nature of MM ligand binding to ubiquitin. Binding sites are identified and characterized in a series of 1H-15N-correlated spectra at varying ligand concentrations to observe ligand-induced changes in the chemical shifts of the various residues. The primary sites of specific protein-ligand interaction for the MM ligand are determined, and the dissociation constants of the residues within the binding regions are calculated to determine and rank the relative binding affinities of these regions on the protein surface. Finally, coarse-grained ligand docking simulations are employed to study the modes of interaction between the MM ligand and the identified preferred binding region on the protein surface. The use of NMR titration experiments in concert with chromatographic data obtained with protein libraries represents a previously undescribed approach for probing the structural basis of protein adsorption in MM chromatographic systems.
Results and Discussion
Chromatographic Retention Behavior of Mutants Under Linear Gradient Conditions.
A library of ubiquitin mutant variants was employed to examine differences in protein binding behavior in ion-exchange and MM chromatography (note: representative ligands for these chromatographic materials are shown in Fig. 1). These mutants included single point mutations involving reductions in surface hydrophobicity, removal of negative charge, and replacement of lysines with arginines. The retention times of these mutants obtained under linear gradient conditions in cation-exchange (SP Sepharose FF) and MM (Capto MMC) chromatographic systems were determined, and the results are presented in Fig. 2 A and B, respectively. As seen in Fig. 2A, all variants (with the exception of mutant D58A) exhibited chromatographic behavior similar to the native ubiquitin on the cation exchanger. Mutant D58A showed a slightly stronger retention due to reduced electrostatic repulsion arising from the loss in negative surface charge. These results are expected because the net charge and charge distribution of these proteins (with the exception of D58A) were the same.
Fig. 1.

Structures of (A) representative cation-exchange chromatographic ligand, 1-Propanesulfonic acid; (B) representative multimodal chromatographic ligand, N-Benzoyl-DL-Methionine. (Note: the point where these ligands would be immobilized to a resin is indicated by a box.)
Fig. 2.

Retention times of mutant variants in a linear salt gradient on (A) SP Sepharose FF and (B) Capto MMC compared to the native protein (note: the error associated with the native protein (gray) and variants (bars) was determined from the average standard deviation).
In contrast to the relatively weak retention of the protein library on the cation-exchange column, the proteins exhibited strong retention on the MM Capto MMC column under the same pH conditions. For example, although the proteins eluted from the ion exchanger at roughly 0.22 M NaCl, significantly higher salt concentrations (0.9–1.4 M) were required to elute the proteins from the MM system. In addition, there were significant variations in protein binding affinities to the MM surface in contrast to the behavior observed with the ion-exchange system.
As can be seen in Fig. 2B, significant variation in the elution times was observed for the mutants on the Capto MMC surface. For the subclass of mutants with decreased surface hydrophobicity, whereas mutant F4A exhibited similar affinity as the native protein, mutants L8A, I44A, and V70A showed reductions in adsorption strength. These results indicate that hydrophobic moieties at Leu 8, Ile 44, and Val 70 are playing an important role in the binding affinity of the native protein to Capto MMC. On the other hand, because the loss in hydrophobicity at Phe 4 does not impact protein binding affinity, it is likely that this residue does not interact strongly with the resin surface.
As described above, mutant D58A exhibited increased affinity in the ion-exchange system due to a loss in repulsive electrostatic interactions. However, the same effect was not observed in the MM system. In fact, no change in variant retention behavior was observed, indicating that the aspartate residue may be situated in a region of the protein that played a minimal role in affecting protein retention on the MM surface.
Another class of mutants was the one where a surface lysine was replaced with arginine. As can be seen in Fig. 2B, with the exception of Lys 29, these variants all exhibited an increased affinity for the MM surface. The effects of such a mutation are complex because the guanidinium group of arginine possesses a delocalized positive charge relative to lysine and has additional moieties that can form multiple hydrogen bonds. In addition, the side chain of arginine is considered to be more hydrophobic than that for lysine (16, 17). All of these interactions could potentially play a role in increasing the affinity of the protein surface for the MM ligand. Further, because the change in protein affinity is dependent upon the location of the lysine residue, these results also indicate that the locality and surrounding microenvironment of the amino acid plays an important role in determining the extent of interaction between the amino acid residue and the ligand moiety.
It is instructive to examine the location of residues on the protein surface that corresponded to either significant or minimal changes in protein retention in this MM system. As indicated in green (Fig. 3), mutations at sites Lys 6, Leu 8, Lys 11, Leu 44, Lys 48, and Val 70 resulted in significant changes in protein adsorption. On the other hand, mutations at sites Phe 4, Lys 29, Lys 33, Asp 58, and Lys 63 (indicated in blue) had little or no effect. As can be seen in Fig. 3, the sites that resulted in changes in protein retention all occurred in a distinct region on the protein surface, which may be indicative of a preferred binding region for interactions with this MM ligand.
Fig. 3.

Color-coded surface representation of ubiquitin based on the multimodal chromatographic results. The green residues (6, 8, 11, 44, 48, and 70) showed significant change in protein adsorption upon mutation, whereas the blue residues (4, 29, 33, 58, and 63) showed little or no effect.
Clearly, these chromatographic results indicate that both binding affinity and selectivity differ markedly between the cation and the MM cation-exchange systems. The fact that the ubiquitin library was significantly more strongly bound in the MMC column indicates that synergistic interactions may be taking place at the ligand-protein interaction sites. In order to gain further insight into the interactions between the two ligand types and the protein surface, heteronuclear sequential quantum correlation (HSQC) titration experiments were performed.
1H-15N HSQC-NMR Titration Experiments with Representative Chromatographic Ligands.
Protein-ligand titrations were monitored by 1H-15N HSQCs and carried out using a representative analogue of the MM cation-exchange ligand, N-benzoyl-DL-methionine (Fig. 1A). As described in Materials and Methods, aliquots of the MM ligand were titrated against a fixed concentration of isotopically labeled ubiquitin to study protein-MM ligand interactions in solution. In this NMR experiment, labeled amide groups on the protein that came into close proximity and interacted with the ligand experienced a change in chemical shift due to perturbations in the local electronic environment. These amide groups serve as reporters of the local environment for each residue.
In the resulting series of 2D 1H-15N HSQC spectra, a single resonance peak for both the unbound and ligand bound form of the protein was observed for each amide group. The linewidths were largely free of exchange broadening and had ligand-dependent chemical shift values characteristic of “fast exchange” behavior for protein-ligand interactions. As a result, the observed resonance frequencies were assumed to be time averaged and were fit with the single-site binding model as presented in Materials and Methods.
The observed changes in chemical shifts exhibited four distinct patterns (Fig. 4). There were those that exhibited no change in their position as a function of ligand concentration (Fig. 4A), indicating that these residues were not associated with a binding event. Other residues showed migrations that did not trend toward a saturation point (Fig. 4B). This behavior is likely due to nonspecific and/or weak interactions. Of the sites that had ligand-induced changes in chemical shift that trended toward saturation, two classes of behavior were observed. This included sites that displayed simple 2-state behavior, indicative of the chemical shift perturbations being induced by ligand binding to a single proximal site (Fig. 4C), and those that displayed more complex multisite behavior (Fig. 4D). When a spin experiences chemical shift perturbations due to multiple noncompeting binding events, migration of chemical shift most often occurs in a nonlinear fashion as shown in Fig. 4D. This is indicative of multiphasic behavior occurring at the boundary between sites (Fig. S1).
Fig. 4.

Examples of 15N-1H HSQC peaks from the Capto MMC titration experiments at 0 (red), 0.64 (orange), 1.28 (yellow), 1.92 (green), 2.56 (cyan), and 3.2 mM (blue) ligand concentrations. (A) Asn 60, a nonbinding residue, (B) Gln 2, a nonbinding residue that shows chemical shift changes, (C) Lys 6, a binding residue, and (D) Phe 45, a residue that exhibits multiphasic behavior.
It is of value to observe the distribution of chemical shift changes and how these map on the protein surface. Fig. S2a shows a plot of the maximum observed chemical shift change (Δδ) of the ubiquitin residues that was obtained during the titration experiments. A structural representation of these results is illustrated in Fig. S2b, which shows how the greatest chemical shift changes concentrate in a single distinct area on the protein surface. Although chemical shift perturbation data can be potentially useful in determining ligand-protein binding interactions, interpretation of these data can be complicated by a number of factors. Most notably, residues on the periphery of an interaction site that may not be directly interacting with the ligand may encounter a chemical shift perturbation due to their proximity to the interaction site. In addition, a residue in direct contact with the ligand may not necessarily undergo a significant change in chemical shift. Further, it is difficult to identify discrete binding sites when there are multiple interactions in the immediate vicinity.
To address these complications it is instructive to examine the changes in chemical shift during the titration experiment. In Fig. S3 the change in chemical shift as a function of ligand concentration is shown for several residues comprising two specific binding sites as well as a nonbinding residue for the sake of comparison. As can be seen in Fig. S3, the residues have distinct dissociation constants and can be grouped accordingly into distinct binding sites.
Examination of the ligand-induced chemical shift data taken from this series of 1H-15N HSQC spectra resulted in the determination of dissociation constants (KD) according to the single-site model (Materials and Methods: Eq. 2). Residues within a given interaction site were assumed to have similar KD values. Based on these values, spatial restrictions determined by the ligand size, and the boundaries defined by sites exhibiting multiphasic behavior, the residues were grouped together to identify explicit ligand interaction sites on the protein surface. A list of interaction sites obtained during the MM ligand-protein titration experiments is presented in Fig. 5A, which includes a color code for the identified interaction sites, the residues present within each site, and the corresponding average KD values. (Note: limitations in the solubility of the MM ligand limited the ability to determine explicit KD values greater than 15 mM.) As can be seen in Fig. 5A, one distinct high affinity site was measured with a KD of 0.4 mM, along with several intermediate affinity sites (1–4 mM). The following residues displayed multiphasic behavior in the NMR analysis: F4, L8, F45, K48, H68, and V70. Although these residues could not be fit to a single KD value, they served to highlight the boundaries between the postulated binding sites.
Fig. 5.

Multimodal binding sites determined by NMR. (A) Data include color-coded binding sites, the associated residues, the average dissociation constants (KD), and the standard error of the fitted KD values. (B–D) Color-coded surface representations of ubiquitin-MM ligand interaction sites determined by NMR. Boxed residues indicate those that showed significant change in protein adsorption upon mutation, whereas circled residues indicate those that showed little or no effect. Residues that displayed multiphasic behavior are depicted in gray. The proposed preferred binding region is outlined in black.
The location of residues on the protein surface that corresponded to the interaction sites identified from the NMR titration experiments are presented in Fig. 5. As can be seen in this figure, the strongest interaction site (site 1) on the middle front face of the protein is surrounded by several other intermediate affinity interaction sites. The fact that these five binding sites are all concentrated in a discrete region on the protein surface indicates that this is a preferred binding region for interacting with the MM ligand.
Although these results are intriguing, it is important to note that the NMR experiments were carried out in free solution with the MM ligand end group from the resin. When interacting with a chromatographic surface containing immobilized MM ligands, it is likely that only one face of the protein can interact at a given time. Accordingly, a proposed binding region of the protein that could interact with the chromatographic surface based on both the NMR results and geometrical considerations is presented in Fig. 5. It was of interest to compare the ligand interaction results from NMR to the chromatographic retention data obtained with the ubiquitin mutants. The amino acid residues that were mutated to produce the ubiquitin protein library are indicated in Fig. 5 by either squares or circles. It was observed that the mutants in the protein library that showed measurable variation in protein retention time on the MM column (K6R, L8A, K11R, K48R, I44A, and V70A) had modifications to residues (indicated by squares) that were located within the preferred binding region. On the other hand, mutants with modified residues (indicated by circles) that were located away from the preferred binding region (F4A, D58A, K29R, K33R, and K63R) resulted in little or no change in protein retention time in the chromatography experiments. Interestingly, mutant L8A, which showed the largest reduction in protein retention time on the MM column, exhibited multiphasic behavior on the periphery of the preferred binding region. In addition, as can be seen in Fig. 5C, Leu 8 protrudes from the surface of the protein, which may give it a more important role in binding to the MM chromatographic surface. Although mutant F4A also exhibited multiphasic behavior in the NMR, it lies outside the proposed preferred binding region indicated in Fig. 5. Mutant K11R did not produce a measurable signal in the NMR experiments; however, this residue resides at the periphery of the proposed binding region, which may explain its modest increase in chromatographic retention behavior. Clearly, steric effects arising from the immobilization of the ligand onto the solid support may affect the relative binding affinities of all of the variants on the resin surface. Future work involving the use of EPR and solid-state NMR will be performed to further investigate the avidity and steric effects of ligand immobilization on protein binding affinity in detail.
A similar set of NMR studies was carried out with the ion-exchange ligand. The results indicated that the KD values were up to 2 orders of magnitude greater than those obtained with the MM ligand. The ion-exchange ligand possessed binding constants greater than 50 mM, whereas those for the multimodal ligand ranged from 0.4 to 12 mM. The weaker binding constants with the ion-exchange ligand qualitatively agree with the lower retention observed on the ion exchanger (Fig. 2A). Further, the interaction sites obtained with the ion-exchange ligand were distributed throughout the protein surface in contrast to the MM system.
Coarse-grained simulations were carried out to determine the most likely docked conformation of the MM ligand within the strongest interaction site shown in Fig. 6 involving residues Lys 6, Ile 44, and His 68. The resulting lowest energy conformation determined for this interaction site is shown in Fig. 6. As seen in this figure, multiple interactions were involved including a charge–charge interaction with Lys 6, hydrophobic interaction with Ile 44, and a pi–pi stacking with His 68. It is likely that these multiple interactions are responsible for the high affinity of the ligand for this region of the protein surface. The multipoint attachment of the ligand is also likely to be important in the cooperativity of proximal sites when the protein is adsorbing to a solid substrate.
Fig. 6.

Multimodal ligand binding conformation predicted by coarse-grained modeling of the strongest interaction site identified from NMR. Surface colors denote NMR determined binding sites (Fig. 5).
Although the simulation indicated a single low energy binding conformation at this site, there were other possible binding orientations with lower affinities that were also identified in the simulations. This multiplicity of potential binding orientations with the MM ligand may play an important role in the adsorption of proteins to MM chromatographic systems where the ligand is immobilized on the resin. Under these conditions, the protein interacts with a ligand that is sterically constrained and may not be able to attain a minimum energy contact with the protein. However, because several other low energy conformations can be employed, the protein may still be able to interact with immobilized MM ligand via an alternative mode of interaction.
Conclusions
In this paper, a multifaceted approach was employed to study the nature of protein adsorption in MM chromatographic systems. The chromatographic results with the protein library indicated that significant changes in protein affinity to the solid support occurred for mutations in a defined region on the protein surface. The results with NMR provided further insight into the specific MM ligand binding sites that clustered to form a distinct preferred binding region, in agreement with the chromatography results. The clustering of binding sites in a preferred region on the protein surface suggests that binding in a MM chromatographic system occurs through multiple epitopes. However, it is important to note that difficulties in the interpretation of the NMR data make it challenging to define the exact boundaries of these binding sites. Coarse-grained ligand docking simulations between the MM ligand and the preferred binding region indicated that multiple interactions were involved including charge–charge, hydrophobic contacts, and pi–pi stacking. These results indicate that chemical diversity plays an important role in creating both the high affinity and unique selectivity of MM chromatographic systems.
This work provides an improved understanding of protein binding affinity in MM chromatography as well as an elucidation of ligand interaction sites on the protein surface. Moreover, it is likely that other proteins that show significant binding to a MM surface possess similar pseudoaffinity behavior, with distinct preferred binding regions on their surface. Further, whereas this work has focused on the protein ubiquitin, we believe that the approach of using NMR, protein libraries, and molecular modeling can be employed to provide insight into a wide range of systems where protein-ligand interactions play an important role (e.g., bioseparations, biomaterials, and drug delivery). In particular, this approach may be useful in identifying appropriate MM ligands and operating conditions for separating proteins from very similar variants, a key challenge in bioprocessing. Finally, although the NMR and docking studies presented here have focused on ligand-protein binding in solution, it will be important in the future to examine protein binding to solid resin systems containing the MM ligand in order to elucidate avidity and steric effects in these systems.
Materials and Methods
Details of materials and equipment used in this study are provided in SI Text.
Linear Gradient Experiments.
The mutant variants were analyzed on the Capto MMC column at pH 5. Linear gradient elution runs were carried out from 100% buffer A (20 mM sodium acetate, pH 5) to 100% buffer B (20 mM sodium acetate containing 1.5 M of sodium chloride, pH 5) in 60 column volumes at a flow rate of 1 mL/ min to obtain retention time data on each mutant variant. Injection volumes ranging from 25 to 50 μL of sample were used depending on the initial concentration of each mutant variant. The column effluent was monitored at UV wavelength 280 nm. The variants were also analyzed on the SP Sepharose column under the same experimental conditions, with the exception of buffer B (20 mM sodium acetate with 0.5 M sodium chloride, pH 5).
13C/15N Isotopically Labeled Protein Expression and Purification.
13C/15N ubiquitin samples were obtained by growing the BL21 (DE3) strain of Escherichia coli, containing the plasmid for the ubiquitin variant in MOPS-based minimal media (18), with 0.2% 13C glucose as the carbon source and 0.2% 15N ammonium chloride as the nitrogen source. The media was also supplemented with 10 mg/L thiamine and 100 mg/L ampicillin. Ubiquitin expression was induced when the cell broth reached an optical density of 0.6 (600 nm) by adding 1 mM IPTG. The cells were harvested by centrifugation and resuspended in a lysis buffer (50 mM Tris/HCl pH 7.5, protease inhibitor cocktail containing DNAse). Lysis was achieved by passing the cell suspension through the French press three times. The cell debris was removed by centrifugation at 12,000 rpm for 1 h at 4 °C (Sorvall SS-34 fixed angle rotor). Acid precipitation was performed by adding acetic acid to the supernatant until the pH of the solution reached 5.0. The mixture was then centrifuged again at 12,000 rpm for 1 h at 4 °C to remove the precipitated protein contaminants. The supernatant was filtered through a 0.2-μm pore diameter syringe filter. The protein solution was loaded onto a 16-mL SP Sepharose FF cation-exchange column and partially purified using a linear gradient elution run from 100% buffer A (50 mM sodium acetate, pH 5) to 100% buffer B (50 mM sodium acetate containing 0.5 M of sodium chloride, pH 5) in 60 column volumes at a flow rate of 1 mL/ min. Eluent fractions containing ubiquitin were collected, pooled, and concentrated using centrifugation with Centriprep tubes with a molecular weight cutoff of 3,000 Da. The final purification step for the protein was carried out using a Sephadex G-50 gel filtration column equilibrated with a buffer containing 10 mM sodium acetate pH 5.0 and 0.02% sodium azide. Eluent fractions containing ubiquitin were again collected, pooled, and concentrated using the Centriprep tubes in preparation for the NMR experiments.
NMR Experiments.
All NMR spectra were obtained at 25 °C using a Bruker 800-MHz spectrometer equipped with a 1H/13C/15N cryoprobe and z-axis gradients. Data were acquired and processed using Bruker TopSpin 2.1 software and the software package Sparky (19). Confirmation of backbone assignments was guided using published chemical shift values (BioMagResBank accession no. 6457). Samples had an initial volume of 400 μL and contained 0.1 mM isotopically labeled ubiquitin in NMR buffer (10 mM sodium acetate, pH 5.0, 0.02% sodium azide, 1 μM 3-(trimethylsilyl)propionic acid-d4 sodium salt, and 5% D2O). For the titration experiments with the ion-exchange ligand (1-propanesulfonic acid, Fig. 1A), the ligand that was available in a concentrated solution form was diluted to a concentration of 1.0 M in the NMR buffer and the pH adjusted to 5.0 with NaOH before titrating against the protein solution. For the titration experiments with the MM ligand (N-benzoyl-DL-methionine, Fig. 1B), lyophilized ligand was added to the 0.1 mM isotopically labeled ubiquitin solution to obtain a concentration of 3.2 mM prior to titration (note: due to limited solubility of this ligand, this was the maximum concentration that was able to be obtained). Because the commercial MM cation-exchange material (Capto MMC) provided by GE Healthcare is composed of immobilized enantiomers, we also used a similar racemic mixture in our studies.
Ligand-induced changes in amide chemical shifts were analyzed in a series of 12 1H-15N HSQC spectra acquired at a fixed ubiquitin concentration (0.1 mM) and various protein-ligand ratios (1∶1.6-1∶32). Because amide resonances during the titration studies were observed largely free of exchange broadening and at the population weighted average of the bound and unbound chemical shifts, ligand-induced changes in chemical shift are in fast exchange and can be modeled as follows:
| [1] |
where δ and n represent the chemical shifts (in ppm) and mole fractions, respectively, of the bound (b) and unbound (u) states. As a result, the apparent dissociation constants (KD) were calculated by fitting 1H and 15N chemical shift changes (Δδ), as a function of ligand concentration using a one-site binding model (20, 21) defined in Eq. 2:
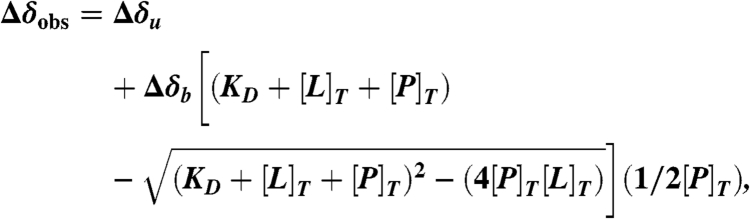 |
[2] |
where KD is the dissociation constant, [L]T and [P]T are the total ligand and protein concentrations, and Δδ is the chemical shift change. From fitting the curves, a KD value was obtained for each amide group identified in the NMR experiment. Because binding events are coordinated between the various residues involved, all residues within an interaction site should possess similar KD values. By combining the KD values with protein surface maps of average chemical shift changes, residues that are interacting with the ligands can be identified and clustered by KD values into interaction sites as discussed in Results and Discussion. Curve fitting and calculations were performed with Origin 8 (OriginLab) and protein visualization was carried out with PyMol (Schrödinger).
Coarse-Grained Docking Simulations.
Coarse-grained docking simulations were performed using the Autodock package, details of which are provided in SI Text.
Supplementary Material
Acknowledgments.
We also thank George Makhatadze, Mayank Patel, and Werner Streicher for their assistance with the expression of the labeled ubiquitin. This work was supported by National Science Foundation Grant CBET 0933169.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1002347107/-/DCSupplemental.
References
- 1.Melander WR, El Rassi Z, Horvath C. Interplay of hydrophobic and electrostatic interactions in biopolymer chromatography: Effect of salts on the retention of proteins. J Chromatogr A. 1989;469:3–27. doi: 10.1016/s0021-9673(01)96437-4. [DOI] [PubMed] [Google Scholar]
- 2.Hancock WS, Sparrow JT. Use of mixed-mode, high-performance liquid-chromatography for the separation of peptide and protein mixtures. J Chromatogr. 1981;206:71–82. doi: 10.1016/s0021-9673(00)82606-0. [DOI] [PubMed] [Google Scholar]
- 3.McLaughlin LW. Mixed-mode chromatography of nucleic-acids. Chem Rev. 1989;89:309–319. [Google Scholar]
- 4.Johansson BL, et al. Preparation and characterization of prototypes for multi-modal separation aimed for capture of positively charged biomolecules at high-salt conditions. J Chromatogr A. 2003;1016:35–49. doi: 10.1016/s0021-9673(03)01141-5. [DOI] [PubMed] [Google Scholar]
- 5.Johansson BL, et al. Preparation and characterization of prototypes for multi-modal separation media aimed for capture of negatively charged biomolecules at high salt conditions. J Chromatogr A. 2003;1016:21–33. doi: 10.1016/s0021-9673(03)01140-3. [DOI] [PubMed] [Google Scholar]
- 6.Gao D, Lin DQ, Yao SJ. Protein adsorption kinetics of mixed-mode adsorbent with benzylamine as functional ligand. Chem Eng Sci. 2006;61:7260–7268. [Google Scholar]
- 7.Burton SC, Haggarty NW, Harding DRK. One step purification of chymosin by mixed mode chromatography. Biotechnol Bioeng. 1997;56:45–55. doi: 10.1002/(SICI)1097-0290(19971005)56:1<45::AID-BIT5>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- 8.Burton SC, Harding DRK. High-density ligand attachment to brominated allyl matrices and application to mixed mode chromatography of chymosin. J Chromatogr A. 1997;775:39–50. [Google Scholar]
- 9.Liu XD, Pohl C. A new weak anion-exchange/reversed-phase mixed-mode stationary phase for simultaneous separation of basic, acidic and neutral pharmaceuticals. Lc Gc Eur. 2007;20(3 Suppl):33. [Google Scholar]
- 10.Ghose S, Hubbard B, Cramer SM. Protein interactions in hydrophobic charge induction chromatography (HCIC) Biotechnol Prog. 2005;21:498–508. doi: 10.1021/bp049712+. [DOI] [PubMed] [Google Scholar]
- 11.Liu XD, Pohl C. A weak anion-exchange/reversed-phase mixed-mode HPLC column and its applications. Am Lab. 2007;39:22–25. [Google Scholar]
- 12.Gao D, Yao SJ, Lin DQ. Preparation and adsorption behavior of a cellulose-based, mixed-mode adsorbent with a benzylamine ligand for expanded bed applications. J Appl Polym Sci. 2008;107:674–682. [Google Scholar]
- 13.Bicker W, Lammerhofer M, Lindner W. Mixed-mode stationary phases as a complementary selectivity concept in liquid chromatography-tandem mass spectrometry-based bioanalytical assays. Anal Bioanal Chem. 2008;390:263–266. doi: 10.1007/s00216-007-1637-9. [DOI] [PubMed] [Google Scholar]
- 14.Boersema PJ, Divecha N, Heck AJR, Mohammed S. Evaluation and optimization of ZIC-HILIC-RP as an alternative MudPIT strategy. J Proteome Res. 2007;6:937–946. doi: 10.1021/pr060589m. [DOI] [PubMed] [Google Scholar]
- 15.Chung WK, et al. Investigation of protein binding affinity in multimodal chromatographic systems using a homologous protein library. J Chromatogr A. 2010;1217:191–198. doi: 10.1016/j.chroma.2009.08.005. [DOI] [PubMed] [Google Scholar]
- 16.Hirano A, Arakawa T, Shiraki K. Arginine increases the solubility of coumarin: Comparison with salting-in and salting-out additives. J Biochem. 2008;144:363–369. doi: 10.1093/jb/mvn078. [DOI] [PubMed] [Google Scholar]
- 17.Arakawa T, Tsumoto K, Nagase K, Ejima D. The effects of arginine on protein binding and elution in hydrophobic interaction and ion-exchange chromatography. Protein Expression Purif. 2007;54:110–116. doi: 10.1016/j.pep.2007.02.010. [DOI] [PubMed] [Google Scholar]
- 18.Neidhardt FC, Bloch PL, Smith DF. Culture medium for enterobacteria. J Bacteriol. 1974;119:736–747. doi: 10.1128/jb.119.3.736-747.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Goddard TD, Kneller DG. SPARKY 3. San Francisco: University of California; 2009. [Google Scholar]
- 20.Rule GS, Hitchens TK. Fundamentals of Protein NMR Spectroscopy. Dordrecht, Netherlands: Springer; 2005. [Google Scholar]
- 21.Roberts GCK. NMR of Macromolecules: A Practical Approach. Oxford, UK: Oxford Univ Press; 1993. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.