

Abstract
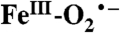 intermediates are well known in heme enzymes, but none have been characterized in the nonheme mononuclear FeII enzyme family. Many steps in the O2 activation and reaction cycle of FeII-containing homoprotocatechuate 2,3-dioxygenase are made detectable by using the alternative substrate 4-nitrocatechol (4NC) and mutation of the active site His200 to Asn (H200N). Here, the first intermediate (Int-1) observed after adding O2 to the H200N-4NC complex is trapped and characterized using EPR and Mössbauer (MB) spectroscopies. Int-1 is a high-spin (S1 = 5/2) FeIII antiferromagnetically (AF) coupled to an S2 = 1/2 radical (J ≈ 6 cm-1 in
intermediates are well known in heme enzymes, but none have been characterized in the nonheme mononuclear FeII enzyme family. Many steps in the O2 activation and reaction cycle of FeII-containing homoprotocatechuate 2,3-dioxygenase are made detectable by using the alternative substrate 4-nitrocatechol (4NC) and mutation of the active site His200 to Asn (H200N). Here, the first intermediate (Int-1) observed after adding O2 to the H200N-4NC complex is trapped and characterized using EPR and Mössbauer (MB) spectroscopies. Int-1 is a high-spin (S1 = 5/2) FeIII antiferromagnetically (AF) coupled to an S2 = 1/2 radical (J ≈ 6 cm-1 in 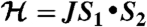 ). It exhibits parallel-mode EPR signals at g = 8.17 from the S = 2 multiplet, and g = 8.8 and 11.6 from the S = 3 multiplet. These signals are broadened significantly by
). It exhibits parallel-mode EPR signals at g = 8.17 from the S = 2 multiplet, and g = 8.8 and 11.6 from the S = 3 multiplet. These signals are broadened significantly by 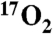 hyperfine interactions (A17O ≈ 180 MHz). Thus, Int-1 is an AF-coupled
hyperfine interactions (A17O ≈ 180 MHz). Thus, Int-1 is an AF-coupled 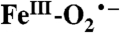 species. The experimental observations are supported by density functional theory calculations that show nearly complete transfer of spin density to the bound O2. Int-1 decays to form a second intermediate (Int-2). MB spectra show that it is also an AF-coupled FeIII-radical complex. Int-2 exhibits an EPR signal at g = 8.05 arising from an S = 2 state. The signal is only slightly broadened by
species. The experimental observations are supported by density functional theory calculations that show nearly complete transfer of spin density to the bound O2. Int-1 decays to form a second intermediate (Int-2). MB spectra show that it is also an AF-coupled FeIII-radical complex. Int-2 exhibits an EPR signal at g = 8.05 arising from an S = 2 state. The signal is only slightly broadened by 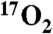 (< 3% spin delocalization), suggesting that Int-2 is a peroxo-FeIII-4NC semiquinone radical species. Our results demonstrate facile electron transfer between FeII, O2, and the organic ligand, thereby supporting the proposed wild-type enzyme mechanism.
(< 3% spin delocalization), suggesting that Int-2 is a peroxo-FeIII-4NC semiquinone radical species. Our results demonstrate facile electron transfer between FeII, O2, and the organic ligand, thereby supporting the proposed wild-type enzyme mechanism.
Keywords: oxygen activation, oxygenase, spectroscopy, superoxide
Most mononuclear nonheme iron-containing oxidases and oxygenases are proposed to initiate their oxygen activation cycles by binding O2 to an active site FeII (1–8). Internal electron transfer to form an FeIII-superoxo species converts the kinetically inert triplet ground state of O2 to a doublet that can participate in the many types of chemistry characteristic of this mechanistically diverse group of enzymes. The same strategy is usually employed by heme-containing oxygenases and oxidases, leading in some cases to comparatively stable FeIII-superoxo intermediates that have been structurally and spectroscopically characterized (9–12). Instability of the putative superoxo intermediate in all mononuclear nonheme iron-containing enzymes has prevented similar characterization, although a superoxide level species has been reported for the dinuclear iron site of myo-inositol oxygenase (13).
In recent studies of the nonheme FeII-containing homoprotocatechuate 2,3-dioxygenase (2,3-HPCD), we have shown that three intermediates of the catalytic cycle can be trapped in one crystal for structural analysis (14). One of these intermediates has been proposed to be an FeII-superoxo species based on the long Fe-O bond distances and an unexpected lack of planarity of the aromatic ring of the alternative substrate 4-nitrocatechol (4NC), which chelates the iron in ligand sites adjacent to that of the O2. In accord with the mechanism postulated for this enzyme class as illustrated in Scheme 1 (1, 8, 15–21), we have proposed that net electron transfer from 4NC through the FeII to O2 forms adjacent substrate and oxygen radicals (Scheme 1B). Recombination of the radicals would begin the ring cleavage and oxygen insertion reactions of this enzyme that eventually yield a muconic semialdehyde adduct as the product. A localized radical on the 4NC semiquinone at the incipient position of oxygen attack would account for the lack of ring planarity. Although this is the only structurally characterized nonheme Fe-superoxo species, the iron oxidation state differs from all of the other postulated Fe-superoxo intermediates.
Scheme 1.

Proposed mechanism for extradiol dioxygenases. In the case of 2,3-HPCD, R is -CH2COO- and B is His200. When R is -NO2 and His200 is changed to Asn, the reaction stalls before reaching intermediate C. Peroxide is slowly released and the product is 4NC quinone.
The mechanism that emerges from the structural and kinetic studies does not require a change in metal oxidation state to form a reactive intermediate (22). However, our studies of 2,3-HPCD in which FeII is replaced with MnII suggest that transient formation of an oxidized metal center may occur (23). The MnII-replaced enzyme is fully active and has no detectable change in structure. Our studies showed that, upon addition of O2 to the enzyme complexed with the normal substrate [homoprotocatechuate or 3,4 dihydroxyphenylacetate (HPCA)], an intermediate is formed with a lifetime of a few milliseconds. The EPR characteristics of this intermediate are consistent with a MnIII-superoxo formulation. This observation suggests that a similar intermediate might exist in the native enzyme as a transient FeIII-superoxo species (Scheme 1A).
Ongoing rapid freeze quench (RFQ) studies of the WT 2,3-HPCD with HPCA as a substrate and the native FeII in the active site have not revealed accumulation of an FeIII-superoxo species that can be detected by EPR or Mössbauer experiments. These results suggest that if such a species is formed, it is very short-lived, so a strategy to slow the reaction is required to detect the putative ferric-superoxo intermediate. Two methods have been described to slow the oxygen activation and substrate attack portions of the reaction cycle. The first is to use an alternative substrate with electron withdrawing substituents, such as 4NC (17). The second is to change the key active site acid-base catalyst and hydrogen bonding residue His200 to an Asn residue (2,3-HPCD variant H200N) (19). Using either one of these strategies results in a slow reaction that gives the expected ring-cleaved product at the end of the cycle. When both strategies are used simultaneously, the reaction slows further and the 4NC aromatic ring is not opened; rather, it is converted to 4NC quinone and H2O2 is released (19). Independent of whether ring cleavage (with HPCA) or ring oxidation (with 4NC) is catalyzed by H200N, a common blue intermediate (λmax = 610 nm) is formed. The rate constant for the formation of this intermediate depends linearly on the O2 concentration (19). We speculate that this species is the initial oxy intermediate, potentially the elusive FeIII-superoxo species of the mononuclear nonheme iron oxidase/oxygenase family. The transient kinetic, EPR, and Mössbauer studies reported here show this to be the case.
Results
Absorption Spectra Show That an Intermediate Precedes Substrate Oxidation.
The stoichiometric H200N-4NC complex exhibits an absorption spectrum characteristic of the dianionic form of 4NC at 518 nm. Mixing this complex with O2 causes a shift to 506 nm within 25 ms at 4 °C showing that an intermediate (Int-1) is formed (Fig. S1) (17, 19). The small magnitude of the shift suggests that 4NC is still in the aromatic, dianion form. Int-1 decays over a period of > 200 s, to form an intermediate with an absorption spectrum maximizing at 405 nm (Int-2) (Fig. S1). This spectrum is characteristic of bound 4NC quinone product complex (19). 4NC quinone is more tightly bound than the ring-cleaved product derived from the reaction of 4NC with the WT enzyme such that complete dissociation to form the resting enzyme requires hours. The released quinone product has a λmax at 380 nm, suggesting that an alternative form of the quinone is bound in Int-2.
Int-1 and Int-2 Exhibit EPR Signals Originating from S1 = 5/2 FeIII Antiferromagnetically Coupled to Different S2 = 1/2 Species.
The anaerobic FeII H200N-4NC complex shows EPR signals from minor S = 5/2 contaminants of FeIII and MnII with g values near 4.27 (0.05 spins/subunit) and 2 (< 0.07 spins/subunit), respectively (Fig. S2). Fig. 1 A and B show EPR spectra recorded in parallel mode for a sample frozen 10 s after the anaerobic H200N-4NC complex was rapidly mixed with 1 eq of O2 at 4 °C. This sample exhibits a pronounced new spectrum (Int-1) with integer-spin resonances at g = 8.17, 8.8, and 11.6, which are observed in maximum yield in RFQ experiments at the earliest time point collected (32 ms) (Fig. 2). Int-1 decays very slowly (k = 0.015 s-1, Fig. 2, Inset) to yield a long-lived intermediate (Int-2), which exhibits, also in parallel mode, a new EPR signal at g = 8.05 (Figs. 1D and 2).
Fig. 1.

Parallel-mode EPR spectra (colored lines) and simulations (thin black lines). (A) 2 K spectrum 10 s after mixing H200N-4NC complex with O2 saturated buffer at 4 °C. (B) Sample of A at 9 K. (C) Spectrum at 10 K for a sample prepared as in A but with 70% enriched 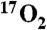 . (D) Sample from A at 9 K after 10 min incubation at 4 °C. Conditions prior to mixing: 1.64 mM H200N-4NC, 200 mM MOPS buffer pH 7.5. Simulation parameters: B, S1 = 5/2, S2 = 1/2, J = +6 cm-1, D1 = -0.48 cm-1, E/D1 = 0.20, g1 = 2.01, g2 = (2.02,1.98,2.04), r = 0.29 nm, rθ = 90°, and rφ = 55°; C, same as B but with A17O ≈ 180 MHz (I = 5/2); D, S1 = 5/2, S2 = 1/2, J = +40 cm-1, D1 = +0.5 cm-1, E/D1 = 0.13, g1 = 2.01, and g2 = 2.00. EPR conditions: frequency, 9.24 GHz; power, 20 mW; and modulation amplitude, 10 G.
. (D) Sample from A at 9 K after 10 min incubation at 4 °C. Conditions prior to mixing: 1.64 mM H200N-4NC, 200 mM MOPS buffer pH 7.5. Simulation parameters: B, S1 = 5/2, S2 = 1/2, J = +6 cm-1, D1 = -0.48 cm-1, E/D1 = 0.20, g1 = 2.01, g2 = (2.02,1.98,2.04), r = 0.29 nm, rθ = 90°, and rφ = 55°; C, same as B but with A17O ≈ 180 MHz (I = 5/2); D, S1 = 5/2, S2 = 1/2, J = +40 cm-1, D1 = +0.5 cm-1, E/D1 = 0.13, g1 = 2.01, and g2 = 2.00. EPR conditions: frequency, 9.24 GHz; power, 20 mW; and modulation amplitude, 10 G.
Fig. 2.

Time dependence of Int-1 EPR signal decay. Parallel-mode EPR spectra of the g = 8 region for samples frozen at the indicated times after mixing anaerobic 1.7 mM stoichiometric H200N-4NC complex 1∶1 with buffer containing 1 eq of O2 at 4 °C in MOPS buffer pH 7.5. The bottom trace is the result of including 2 eq of 4NC and incubating anaerobically for 1.5 h at 25 °C. (Inset) Exponential fit to the decay of the g = 8.17 EPR signal. EPR conditions: frequency 9.35 GHz; microwave power, 1 mW; modulation amplitude, 10 G; and temperature, 2 K.
The EPR spectra of Int-1 over a range of temperatures (spectra at 2 and 9 K are shown in Fig. 1 A and B) show that the g = 8.17 resonance originates from a ground doublet, and the g = 8.8 and 11.6 resonances from excited states. The Mössbauer spectra, shown below, reveal that the iron in both Int-1 and Int-2 is high-spin (S1 = 5/2) FeIII. Fig. 1B shows a simulation of the Int-1 spectrum for an S1 = 5/2 center antiferromagnetically (AF) coupled to an S2 = 1/2 center, using the spin Hamiltonian of Eq. 1 for the parameters given in the caption and Table 1.
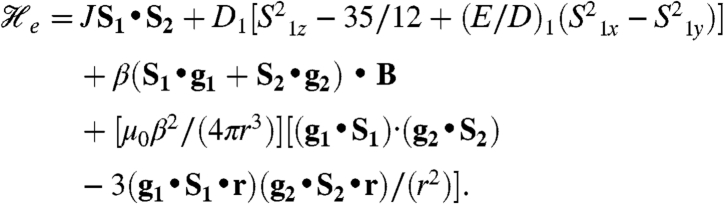 |
[1] |
Table 1.
Electronic and nuclear parameters of Int-1 and Int-2 obtained from EPR (italics) and Mössbauer spectroscopy
| Species | J, cm-1 | D1, cm-1 | (E/D)1 | A0/gnβn, T | ΔEQ, mm/s | η | δ, mm/s |
| Int-1 | −0.59 | 0.20 | −21.4(2) | −0.33(2) | −3 | 0.50(1) | |
| +6(2)* | −0.48 | 0.20 | |||||
| Int-2 | +0.67 | 0.11 | −21.5(2) | 0.87(2) | −7.2 | 0.49(1) | |
| +40(10) | +0.50 | 0.13 |
*Numbers in parentheses give estimate of uncertainty.
The exchange, zero-field splitting, Zeeman, and dipole–dipole terms have their common definitions. The simulation is quantitative, i.e., the signal intensities are correctly predicted by simulation for the protein concentration. The agreement unambiguously established the spin centers of Int-1 as S1 = 5/2 and S2 = 1/2 and determines J = +6 cm-1. The energy of the exchange interaction is larger than the zero-field splitting energy, and consequently the spin system approximates isolated S = 2 and 3 multiplets (see energy diagram, Fig. S3). The EPR resonances are from the transitions indicated in the diagram. Subtle shifts from the expected positions of these transitions (g = 8 and 12) require the introduction of dipole–dipole interaction between the two spin centers with a distance of ≈0.3 nm.
The parallel-mode EPR spectrum of Int-2 is distinctly different from that of Int-1. Specifically, the g = 11.6 and 8.8 features are not present, the main signal is shifted to g = 8.05, and its temperature dependence indicates that it arises from an excited doublet (Fig. S4A). The simulation displayed in Fig. 1D is for an S1 = 5/2 center AF coupled to an S2 = 1/2 center with parameters given in the figure caption. From the temperature dependence of the signal, we obtained J ≈ +40 cm-1. The simulation is also quantitative, showing that the spin concentration approximately matches the concentration of the protein.
Mössbauer Spectroscopy Shows That the S = 2 Int-1 is an S1 = 5/2 FeIII AF Coupled to an S2 = 1/2 Radical.
Mössbauer spectra of resting H200N recorded at 4.2 K in zero applied field (B = 0) exhibit a doublet with quadrupole splitting ΔEQ = 3.01 mm/s and isomer shift δ = 1.24 mm/s, consistent with six-coordinate high-spin FeII with N/O coordination (24, 25). The crystal structure of 2,3-HPCD has shown that substrate binding converts the FeII site from a six-coordinate octahedral geometry to a five-coordinate square pyramidal geometry through release of solvent (26). Accordingly, after anaerobic addition of 1 eq of 4NC to H200N, the parameters of the FeII site changed to ΔEQ = 3.57 mm/s and δ = 1.12 mm/s (Fig. 3A). The decreased δ is consistent with conversion of the metal site to five-coordinate or a six-coordinate site with one loosely bound solvent (5, 24).
Fig. 3.

Mössbauer spectra of H200N variant recorded at 4.2 K for B = 0. (A) Anaerobic H200N-4NC complex. (B) Int-1 prepared by freezing the sample from A 10 s after mixing with 1 eq of O2. The sample contains a small amount of Int-2 (blue, ∼20% of Fe). (C) Sample of Int-2 frozen 10 min after mixing with O2. (D) Endpoint complex in the presence of one additional equivalent of 4NC but stoichiometric O2. This sample was prepared as described in Materials and Methods. The black, red, or blue lines are fits using parameters of Table 1. Concentrations before mixing: 1.62 mM H200N-4NC (except in D, where an additional equivalent of 4NC is added), 50 mM MOPS pH 7.5 at 4 °C. The 57Fe enrichment of H200N was approximately 95%.
Fig. 3 B and C shows spectra of samples from the single turnover reaction in which preformed H200N-4NC complex was mixed rapidly with 1 eq of O2 at 4 °C. The sample of Fig. 3B, frozen 10 s after mixing, exhibits at 4.2 K a doublet (representing 80% of Fe) with ΔEQ = 0.33 mm/s and δ = 0.50 mm/s assigned to Int-1; the remainder of the Fe belongs to Int-2. The observation of a quadrupole doublet for B = 0, rather than a magnetically split spectrum, implies a species with integer (or zero) electronic spin. A δ value of 0.50 mm/s, on the other hand, unambiguously shows that the iron of Int-1 is high-spin FeIII, rather than FeII.
The spectra recorded in strong applied magnetic fields (between 0.6 and 8 T), see Fig. 4 A and B, have features typical of high-spin FeIII sites. We have analyzed these spectra with the spin Hamiltonian 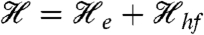 (omitting the dipole–dipole term in
(omitting the dipole–dipole term in 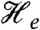 ) where
) where 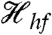 describes the 57Fe hyperfine interactions.
describes the 57Fe hyperfine interactions.
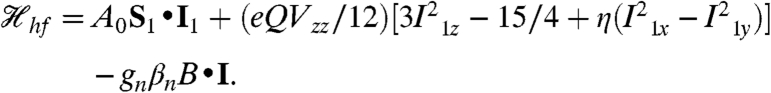 |
[2] |
Fig. 4.

Mössbauer spectra of H200N intermediates Int-1 and Int-2 recorded at 4.2 K in parallel applied magnetic fields indicated. (A and B) Spectrum of Int-1. The 20% contribution of Int-2 has been subtracted from the spectra. Red solid lines are simulations based on Eqs. 1 and 2 using the parameters listed in Table 1. (C) Spectrum of Int-2. Red line is a spectral simulation using the parameters of Table 1.
In Eq. 2 all symbols have their conventional meanings. The high-field Mössbauer spectra are associated with the lowest spin doublet (roughly the MS = ± 2 doublet); the g = 8.17 EPR feature originates from this doublet. The observed magnetic hyperfine field is along z, which is also the direction for which the g = 8.17 feature is observed. For the 57Fe magnetic hyperfine coupling constant of the ferric ion, we obtained A0/gnβn = -21.4(2) T, which compares well with A0 values reported for sites with octahedral FeIII with N/O coordination (27). The parameters obtained from the Mössbauer analysis are listed in Table 1; the D and E/D values obtained from EPR and Mössbauer spectroscopy agree within the uncertainties.
17O-Hyperfine Coupling Shows That an Oxygen Radical is Present in Int-1.
Fig. 1C shows parallel-mode EPR spectra for a sample of Int-1 prepared with 70% enriched 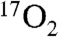 (I = 5/2). It is evident that 17O broadens the spectrum considerably. Fig. 1C also shows a simulation of the spectrum of the 17O-enriched sample using the same set of electronic parameters as that shown in Fig. 1B, with the inclusion of a term for the hyperfine interaction with 17O. For the 70% enrichment of this sample, the fit to the data gives A17O ≈ 180 MHz. The simulations show that at least one 17O A-tensor component for Int-1 must be as large as 180 MHz. This large hyperfine constant is therefore indicative of > 80% radical electron density on the oxygen, identifying Int-1 as an
(I = 5/2). It is evident that 17O broadens the spectrum considerably. Fig. 1C also shows a simulation of the spectrum of the 17O-enriched sample using the same set of electronic parameters as that shown in Fig. 1B, with the inclusion of a term for the hyperfine interaction with 17O. For the 70% enrichment of this sample, the fit to the data gives A17O ≈ 180 MHz. The simulations show that at least one 17O A-tensor component for Int-1 must be as large as 180 MHz. This large hyperfine constant is therefore indicative of > 80% radical electron density on the oxygen, identifying Int-1 as an 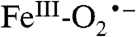 radical species.
radical species.
Int-2 May Contain a Bound Peroxo and a Product Radical.
The zero-field Mössbauer spectrum of Int-2, shown in Fig. 3C, consists of a doublet with ΔEQ = 0.87 mm/s and δ = 0.49 mm/s. As for Int-1, the system reflects a high-spin FeIII ion that is exchange coupled to some S = 1/2 radical. Analysis of the Mössbauer spectra recorded in strong applied fields (a 1.0 T spectrum is shown in Fig. 4C), demonstrates AF coupling with a J > 6 cm-1.
In contrast to Int-1, formation of Int-2 using 70% enriched 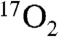 resulted in only a slight hyperfine broadening of the EPR signal in the g = 8 region (Fig. S4B). Simulations for Int-2 indicate A17O ≈ 5 MHz. This small A value indicates little spin density (< 3%) on the oxygen, showing that the quantitative radical must be localized elsewhere. Because the absorption spectra show that the quinone product of the reaction is present in the active site of Int-2, it is possible that this is the site of the radical, which was indirectly tested by displacing the quinone product with 4NC. The reaction was started with 1 eq of O2 and 2 eq of 4NC relative to the active site FeII. The samples from the reaction time course were aged and frozen anaerobically in order to limit O2 to 1 eq. The samples progressed normally through Int-1 (Fig. S5A) and Int-2, but at 80 s (Fig. S5B), the spectrum of the FeII H200N-4NC substrate complex began to reappear, representing 20% of the total iron. These results suggest slow replacement of the quinone product by the excess 4NC. After 30 min (anaerobically) at room temperature (Fig. 3D and Fig. S5C), the reaction mixture contained 20% Int-2, whereas 80% of the sample was the FeII H200N-4NC complex. It is notable that in this complete cycle, the active site iron returns to the FeII state without the introduction of external reducing equivalents. This observation suggests Int-2 is formally a peroxo-FeII-4NC quinone complex but internal electron transfer forms the quasi-stable peroxo-FeIII-4NC quinone radical species actually observed. The presence of the peroxo would account for the slight broadening of the EPR spectrum due to 17O. As the peroxide and quinone dissociate, the electron from the quinone radical is retained by the iron to restore the resting FeII state (Fig. 3D) ready for another turnover cycle.
resulted in only a slight hyperfine broadening of the EPR signal in the g = 8 region (Fig. S4B). Simulations for Int-2 indicate A17O ≈ 5 MHz. This small A value indicates little spin density (< 3%) on the oxygen, showing that the quantitative radical must be localized elsewhere. Because the absorption spectra show that the quinone product of the reaction is present in the active site of Int-2, it is possible that this is the site of the radical, which was indirectly tested by displacing the quinone product with 4NC. The reaction was started with 1 eq of O2 and 2 eq of 4NC relative to the active site FeII. The samples from the reaction time course were aged and frozen anaerobically in order to limit O2 to 1 eq. The samples progressed normally through Int-1 (Fig. S5A) and Int-2, but at 80 s (Fig. S5B), the spectrum of the FeII H200N-4NC substrate complex began to reappear, representing 20% of the total iron. These results suggest slow replacement of the quinone product by the excess 4NC. After 30 min (anaerobically) at room temperature (Fig. 3D and Fig. S5C), the reaction mixture contained 20% Int-2, whereas 80% of the sample was the FeII H200N-4NC complex. It is notable that in this complete cycle, the active site iron returns to the FeII state without the introduction of external reducing equivalents. This observation suggests Int-2 is formally a peroxo-FeII-4NC quinone complex but internal electron transfer forms the quasi-stable peroxo-FeIII-4NC quinone radical species actually observed. The presence of the peroxo would account for the slight broadening of the EPR spectrum due to 17O. As the peroxide and quinone dissociate, the electron from the quinone radical is retained by the iron to restore the resting FeII state (Fig. 3D) ready for another turnover cycle.
Discussion
Past studies from our laboratories and several others have shown that O2 activation at nonheme iron centers in oxidases and oxygenases is usually a highly regulated process designed to produce a reactive form of oxygen only when it can react rapidly with a substrate (1, 5, 8, 13, 18, 28–30). This type of regulation promotes regiospecificity and prevents adventitious reactions with other biological components, but often leads to fleeting intermediates that can be detected only after the development of strategies to slow the reaction (31, 32). The study described here employs both mutagenesis of a key active site acid/base catalyst and the use of a slow substrate to kinetically resolve the half reactions of what we believe are very fast processes in the catalytic cycle of the WT enzyme. The first intermediate in this process is an active site 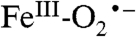 , a species that has not been stabilized in the large nonheme mononuclear iron oxygenase enzyme class. Below, the characteristics of this species and its relevance to catalysis are discussed.
, a species that has not been stabilized in the large nonheme mononuclear iron oxygenase enzyme class. Below, the characteristics of this species and its relevance to catalysis are discussed.
Electronic Structure of Int-1.
The above spectroscopic data clearly indicate that Int-1 is well described as a high-spin FeIII ion exchanged coupled to a superoxo radical (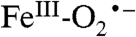 ). The 57Fe hyperfine interaction (A0/gnβn = -21.4 T) compares well with that of the mononuclear FeIII site (-21.2 T) of protocatechuate 3,4 dioxygenase from Brevibacterium fuscum (27), and the isomer shift (δ = 0.50 mm/s) falls squarely into the range of high-spin FeIII (25). The 17O hyperfine interaction of the superoxide radical is large (A17O = 180 MHz) as in superoxide adsorbed onto various surfaces (33, 34) and oxycobaltomyoglobin (35).
). The 57Fe hyperfine interaction (A0/gnβn = -21.4 T) compares well with that of the mononuclear FeIII site (-21.2 T) of protocatechuate 3,4 dioxygenase from Brevibacterium fuscum (27), and the isomer shift (δ = 0.50 mm/s) falls squarely into the range of high-spin FeIII (25). The 17O hyperfine interaction of the superoxide radical is large (A17O = 180 MHz) as in superoxide adsorbed onto various surfaces (33, 34) and oxycobaltomyoglobin (35).
The electronic structure of Int-1 developed from spectroscopy is supported by density functional theory (DFT) calculations for a truncated system, consisting of the Fe, its coordinated ligands, and residues of the second coordination sphere in 2,3-HPCD (see SI Text and Fig. S6A). The calculations yield an 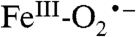 ground state with a superoxo radical bound end-on to a high-spin FeIII (Fe-O-O = 117°) (Fig. 5 and Fig. S6A). The calculated values for δ and ΔEQ are typical for high-spin FeIII and agree well with the experiment (Table S1). The calculated A values for the distal 17O, A1,2,3 = (-226,+59,+81) MHz, are quite similar to those observed in matrix-isolated
ground state with a superoxo radical bound end-on to a high-spin FeIII (Fe-O-O = 117°) (Fig. 5 and Fig. S6A). The calculated values for δ and ΔEQ are typical for high-spin FeIII and agree well with the experiment (Table S1). The calculated A values for the distal 17O, A1,2,3 = (-226,+59,+81) MHz, are quite similar to those observed in matrix-isolated 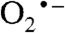 (33, 34), with the largest component perpendicular to the O–O axis in the plane of the π* orbital containing the unpaired electron (note that because the largest 17O splitting is observed along the z axis of Eq. 1, it follows that this axis is in the plane of the π* orbital and perpendicular to O–O bond). The calculated O–O bond length (1.38 Å) is close to that calculated for
(33, 34), with the largest component perpendicular to the O–O axis in the plane of the π* orbital containing the unpaired electron (note that because the largest 17O splitting is observed along the z axis of Eq. 1, it follows that this axis is in the plane of the π* orbital and perpendicular to O–O bond). The calculated O–O bond length (1.38 Å) is close to that calculated for 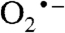 (1.41 Å), and quite different from that for O2 (1.26 Å). The computations show nearly complete transfer of the electron to the dioxygen with some polarization such that the distal oxygen has a higher spin population (Table S2). This oxygen points toward the catechol carbon para to the nitro group, whereas structural studies show that the side-on bound FeII-superoxo species of the WT 2,3-HPCD-4NC complex points the adjacent hydroxyl bearing carbon (14). The calculated J value is small (J = -5.8 cm-1), as observed experimentally (J = +6 cm-1), but ferromagnetic. Preliminary calculations suggest that the orientation of the radical-containing π* orbital (Fig. S6B) and the exchange-coupling constant are sensitive to hydrogen bonding interactions between
(1.41 Å), and quite different from that for O2 (1.26 Å). The computations show nearly complete transfer of the electron to the dioxygen with some polarization such that the distal oxygen has a higher spin population (Table S2). This oxygen points toward the catechol carbon para to the nitro group, whereas structural studies show that the side-on bound FeII-superoxo species of the WT 2,3-HPCD-4NC complex points the adjacent hydroxyl bearing carbon (14). The calculated J value is small (J = -5.8 cm-1), as observed experimentally (J = +6 cm-1), but ferromagnetic. Preliminary calculations suggest that the orientation of the radical-containing π* orbital (Fig. S6B) and the exchange-coupling constant are sensitive to hydrogen bonding interactions between 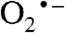 and the protein residues, and that minor changes in protein conformation may change the J value by as much as 100 cm-1. The nature of these correlations is currently under investigation.
and the protein residues, and that minor changes in protein conformation may change the J value by as much as 100 cm-1. The nature of these correlations is currently under investigation.
Fig. 5.

DFT geometry optimization of Int-1. Dashed lines indicate hydrogen bonds. Blue numbers indicate distance in Angstroms or angle in degrees. All possible orientations of Asn200 are beyond hydrogen bonding range to the O2.
Comparison of Int-1 with 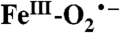 Species from Heme-Containing Enzymes.
Species from Heme-Containing Enzymes.
As in the case of the nonheme iron-containing oxidases and oxygenases, the reaction of O2 with heme-containing systems usually involves a reaction with iron in a five-coordinate S = 2 state. An electron is transferred to O2 in both the heme and nonheme systems. However, the reactions diverge at this point because the iron in the heme system generally becomes a low-spin 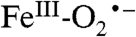 system, with a DFT-calculated singlet–triplet gap of ≈1200 cm-1 for P-450 (36). The
system, with a DFT-calculated singlet–triplet gap of ≈1200 cm-1 for P-450 (36). The 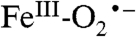 species of hemes is rarely proposed as a reactive species in oxygenase reactions unless the substrate is highly activated as, for example, in the final step of the nitric oxide synthase cycle (37). It is generally thought that the heme
species of hemes is rarely proposed as a reactive species in oxygenase reactions unless the substrate is highly activated as, for example, in the final step of the nitric oxide synthase cycle (37). It is generally thought that the heme 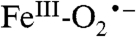 state is first converted to a peroxo, hydroperoxo, or high-valent oxo intermediate. Whereas similar conversions are postulated for many nonheme systems, others are thought to use the superoxo state directly as described below.
state is first converted to a peroxo, hydroperoxo, or high-valent oxo intermediate. Whereas similar conversions are postulated for many nonheme systems, others are thought to use the superoxo state directly as described below.
Significance for Nonheme Mononuclear Iron-Containing Enzymes.
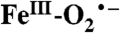 intermediates have been proposed to serve important roles in the reaction cycles of nonheme mononuclear iron-containing enzymes. For example, it has been proposed that this type of intermediate abstracts a hydrogen atom from the beta carbon of iron-bound cysteinyl moiety of the tripeptide δ-(Lα-aminoadipoyl)-L-cysteinyl-D-valine during the isopenicillin N-synthase catalyzed formation of the β-lactam ring of penicillin N (38). In the reaction cycle of the myriad of α-ketoglutarate linked dioxygenases, an
intermediates have been proposed to serve important roles in the reaction cycles of nonheme mononuclear iron-containing enzymes. For example, it has been proposed that this type of intermediate abstracts a hydrogen atom from the beta carbon of iron-bound cysteinyl moiety of the tripeptide δ-(Lα-aminoadipoyl)-L-cysteinyl-D-valine during the isopenicillin N-synthase catalyzed formation of the β-lactam ring of penicillin N (38). In the reaction cycle of the myriad of α-ketoglutarate linked dioxygenases, an 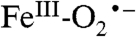 intermediate is proposed to attack the alpha carbon of the iron-bound α-keto glutarate to form an alkylperoxo intermediate prior to the release CO2 and the formation of a high-valent intermediate that ultimately hydroxylates the substrate (5). In these and all other examples, the putative iron-superoxo species has not been directly detected. Int-1, on the other hand, is detectable at 4 °C for several minutes and is shown here to contain oxygen from O2, presenting an opportunity to characterize this intermediate.
intermediate is proposed to attack the alpha carbon of the iron-bound α-keto glutarate to form an alkylperoxo intermediate prior to the release CO2 and the formation of a high-valent intermediate that ultimately hydroxylates the substrate (5). In these and all other examples, the putative iron-superoxo species has not been directly detected. Int-1, on the other hand, is detectable at 4 °C for several minutes and is shown here to contain oxygen from O2, presenting an opportunity to characterize this intermediate.
Electronic Structure of Int-2.
Based on the small hyperfine interaction from 17O, the loss of the g = 8.05 EPR signal upon displacement of the quinone product, and observation of an FeIII by Mössbauer spectroscopy, it seems likely that Int-2 is a peroxo-FeIII-4NC semiquinone complex. Small molecule transition metal ligand complexes with quinones are well studied (39, 40). Experiments have shown that charge distribution between a transition metal and the noninnocent quinone ligand depends largely on relative energy levels between the quinone frontier and metal orbitals (39). This insight has been used to explain the rich redox chemistry commonly observed when a quinone interacts with a divalent metal ion such as Fe(II), Mn(II), and Co(II) (41). In most cases, these complexes exhibit a resultant spin state consistent with electron transfer from the metal to the quinone, with AF coupling between the metal and the quinone radical as we propose for Int-2.
Implications for the Mechanism of Extradiol Dioxygenases.
We show here that the turnover of 4NC by H200N leads initially to generation of an 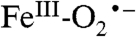 complex that cannot rapidly attack the substrate, which raises the question of whether Int-1 is relevant to normal ring-cleaving catalysis. Note that the transient absorption feature at 610 nm that we associate with Int-1 is observed when H200N catalyzes either ring cleavage of HPCA or quinone formation from 4NC. In the mechanistic model we have proposed (Scheme 1), the reaction becomes committed to ring cleavage by superoxo radical attack on the substrate semiquinone to form the structurally characterized alkylperoxo intermediate (14). Most probably, the highly electron withdrawing nitro substituent of 4NC greatly decreases the rate of electron transfer to the
complex that cannot rapidly attack the substrate, which raises the question of whether Int-1 is relevant to normal ring-cleaving catalysis. Note that the transient absorption feature at 610 nm that we associate with Int-1 is observed when H200N catalyzes either ring cleavage of HPCA or quinone formation from 4NC. In the mechanistic model we have proposed (Scheme 1), the reaction becomes committed to ring cleavage by superoxo radical attack on the substrate semiquinone to form the structurally characterized alkylperoxo intermediate (14). Most probably, the highly electron withdrawing nitro substituent of 4NC greatly decreases the rate of electron transfer to the 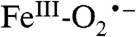 , especially in the absence of an acid-base catalyst and orienting residue at position 200. Consequently, the ability to see a long-lived Int-1 in H200N with 4NC may reflect a decrease in driving force for electron transfer from the substrate rather than the formation of a unique species in this enzyme variant.
, especially in the absence of an acid-base catalyst and orienting residue at position 200. Consequently, the ability to see a long-lived Int-1 in H200N with 4NC may reflect a decrease in driving force for electron transfer from the substrate rather than the formation of a unique species in this enzyme variant.
Conclusion and Perspective.
Most heme-containing oxidases and oxygenases that catalyze reactions involving cleavage of strong bonds of unactivated substrates generate FeIII-hydroperoxo or FeIV-oxo π cation radical reactive species. These are formed by first transferring two electrons in rapid succession to O2. In contrast, the nonheme FeII aromatic ring-cleaving dioxygenases appear to follow a different strategy involving a one-electron transfer to oxygen. The studies reported here and our previous studies suggest that this process proceeds by initial formation of a short-lived 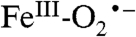 . We propose that this species is very rapidly converted in normal catalysis to a substrate radical-
. We propose that this species is very rapidly converted in normal catalysis to a substrate radical-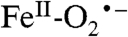 intermediate, which is the true reactive species from which oxygen attack on the substrate occurs. The initial formation of
intermediate, which is the true reactive species from which oxygen attack on the substrate occurs. The initial formation of 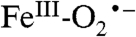 is likely to be a common theme throughout the remarkably diverse mononuclear nonheme FeII-containing superfamily, most members of which share the 2 His, 1 Asp/Glu FeII binding motif (42). After formation of the
is likely to be a common theme throughout the remarkably diverse mononuclear nonheme FeII-containing superfamily, most members of which share the 2 His, 1 Asp/Glu FeII binding motif (42). After formation of the 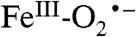 intermediate, the proposed reaction mechanisms diverge such that in some cases the
intermediate, the proposed reaction mechanisms diverge such that in some cases the 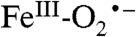 is the reactive species, whereas in other cases further activation steps are required. Thus, the
is the reactive species, whereas in other cases further activation steps are required. Thus, the 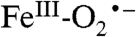 species described here is likely to be the intermediate from which emerges an exceptionally rich range of enzyme chemistry.
species described here is likely to be the intermediate from which emerges an exceptionally rich range of enzyme chemistry.
Materials and Methods
Overexpression of the H200 Variant, Purification, and 57Fe Incorporation.
The H200N variant of recombinant 2,3-HPCD (EC 1.13.11.15) from Brevibacterium fuscum was expressed and purified as previously described (43, 44). For enrichment of 57Fe in 2,3-HPCD, a media composed of 24 g/L casamino acids, 8 g/L yeast extract, 9.4 g/L K2HPO4, and 2.2 g/L KH2HPO4 was used. The cultures were grown at 37 °C to an optical density of 1 at 600 nm in 1 L shaker flasks, then supplemented with 9 mg/L 57Fe and induced with 280 μM IPTG for 4 h at 25 °C.
Preparation of Fully Reduced 2,3-HPCD.
Purified 2,3-HPCD was made anaerobic by mild stirring under argon at 4 °C then transferred to a Coy anaerobic glovebox. The sample was reduced with 1.5 eq of Na2S2O4 at 25 °C for ∼30 min. Excess Na2S2O4 was removed from the sample by passage through a Sephadex G-25 PD-10 column preequilibrated with anaerobic 200 mM MOPS buffer at pH 7.5. This procedure increased the specific activity, but the reaction cycle rate constants were unchanged.
Rapid Freeze Quench Methods.
H200N-4NC anaerobic complex was prepared in the glovebox by mixing 1 eq of H200N with 1 or 2 eq of 4NC, as specified. RFQ syringes were loaded inside the anaerobic glovebox and then transferred to an Update Instrument model 1019 RFQ apparatus and allowed to equilibrate for 30 min at 4 °C. After rapid mixing and passage through a calibrated delay line, samples were collected by rapid freezing on counterrotating aluminum wheels at Liq N2 temperature. For samples at times > 6 s, the mixed sample was collected directly in an EPR tube or Mössbauer cup and frozen in a dry ice/methanol bath (EPR) or Liq N2 (Mössbauer) after the appropriate incubation time. For anaerobic aging of Mössbauer samples < 100 s, the sample was dispensed into a Mössbauer cup inside of an anaerobic vial preequilibrated at 4 °C. The vial and sample were then frozen in Liq N2 at the appropriate time.
Supplementary Material
Acknowledgments.
We thank Elena G. Kovaleva for assistance in analyzing the structural data used for computations. This work is supported by the National Institutes of Health Grant GM24689 (to J.D.L.), Grant EB-001475 (to E.M.), Grant GM77387 (to M.P.H.), and graduate traineeship GM08700 (to M.M.M.). This research was supported in part by the National Science Foundation Grant TG-CHE070073 through Teragrid.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1010015107/-/DCSupplemental.
References
- 1.Arciero DM, Lipscomb JD. Binding of 17O-labeled substrate and inhibitors to protocatechuate 4,5-dioxygenase-nitrosyl complex. Evidence for direct substrate binding to the active site Fe2+ of extradiol dioxygenases. J Biol Chem. 1986;261:2170–2178. [PubMed] [Google Scholar]
- 2.Baldwin JE, Bradley M. Isopenicillin N synthase: Mechanistic studies. Chem Rev. 1990;90:1079–1088. [Google Scholar]
- 3.Chen VJ, et al. Spectroscopic studies of isopenicillin N synthase. A mononuclear nonheme Fe2+ oxidase with metal coordination sites for small molecules and substrate. J Biol Chem. 1989;264:21677–21681. [PubMed] [Google Scholar]
- 4.Rocklin AM, et al. Mechanistic studies of 1-aminocyclopropane-1-carboxylic acid oxidase: Single turnover reaction. J Biol Inorg Chem. 2004;9:171–182. doi: 10.1007/s00775-003-0510-3. [DOI] [PubMed] [Google Scholar]
- 5.Price JC, et al. The first direct characterization of a high-valent iron intermediate in the reaction of an α-ketoglutarate-dependent dioxygenase: A high-spin Fe(IV) complex in taurine alpha-ketoglutarate dioxygenase (TauD) from Escherichia coli. Biochemistry. 2003;42:7497–7508. doi: 10.1021/bi030011f. [DOI] [PubMed] [Google Scholar]
- 6.Proshlyakov DA, et al. Direct detection of oxygen intermediates in the nonheme Fe enzyme taurine/alpha-ketoglutarate dioxygenase. J Am Chem Soc. 2004;126:1022–1023. doi: 10.1021/ja039113j. [DOI] [PubMed] [Google Scholar]
- 7.Mukherjee A, et al. Oxygen activation at mononuclear nonheme iron centers: A superoxo perspective. Inorg Chem. 2010;49:3618–3628. doi: 10.1021/ic901891n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kovaleva EG, Lipscomb JD. Versatility of biological nonheme Fe(II) centers in oxygen activation reactions. Nat Chem Biol. 2008;4:186–193. doi: 10.1038/nchembio.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Weiss JJ. Nature of the iron-oxygen bond in oxyhaemoglobin. Nature. 1964;202:83–84. doi: 10.1038/202083b0. [DOI] [PubMed] [Google Scholar]
- 10.Sharrock M, et al. Cytochrome P450cam and its complexes. Mössbauer parameters of the heme iron. Biochim Biophys Acta. 1976;420:8–26. doi: 10.1016/0005-2795(76)90340-8. [DOI] [PubMed] [Google Scholar]
- 11.Schlichting I, et al. The catalytic pathway of cytochrome P450cam at atomic resolution. Science. 2000;287:1615–1622. doi: 10.1126/science.287.5458.1615. [DOI] [PubMed] [Google Scholar]
- 12.Davydov R, et al. Hydroxylation of camphor by reduced oxy-cytochrome P450cam: Mechanistic implications of EPR and ENDOR studies of catalytic intermediates in native and mutant enzymes. J Am Chem Soc. 2001;123:1403–1415. doi: 10.1021/ja003583l. [DOI] [PubMed] [Google Scholar]
- 13.Xing G, et al. Evidence for C-H cleavage by an iron-superoxide complex in the glycol cleavage reaction catalyzed by myo-inositol oxygenase. Proc Natl Acad Sci USA. 2006;103:6130–6135. doi: 10.1073/pnas.0508473103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kovaleva EG, Lipscomb JD. Crystal structures of Fe2+ dioxygenase superoxo, alkylperoxo, and bound product intermediates. Science. 2007;316:453–457. doi: 10.1126/science.1134697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shu L, et al. X-ray absorption spectroscopic studies of the Fe(II) active site of catechol 2,3-dioxygenase. Implications for the extradiol cleavage mechanism. Biochemistry. 1995;34:6649–6659. doi: 10.1021/bi00020a010. [DOI] [PubMed] [Google Scholar]
- 16.Bugg TDH, Winfield CJ. Enzymic cleavage of aromatic rings: Mechanistic aspects of the catechol dioxygenases and later enzymes of bacterial oxidative cleavage pathways. Nat Prod Rep. 1998;15:513–530. [Google Scholar]
- 17.Groce SL, Miller-Rodeberg MA, Lipscomb JD. Single-turnover kinetics of homoprotocatechuate 2,3-dioxygenase. Biochemistry. 2004;43:15141–15153. doi: 10.1021/bi048690x. [DOI] [PubMed] [Google Scholar]
- 18.Vaillancourt FH, Bolin JT, Eltis LD. The ins and outs of ring-cleaving dioxygenases. Crit Rev Biochem Mol Biol. 2006;41:241–267. doi: 10.1080/10409230600817422. [DOI] [PubMed] [Google Scholar]
- 19.Groce SL, Lipscomb JD. Aromatic ring cleavage by homoprotocatechuate 2,3-dioxygenase: Role of His200 in the kinetics of interconversion of reaction cycle intermediates. Biochemistry. 2005;44:7175–7188. doi: 10.1021/bi050180v. [DOI] [PubMed] [Google Scholar]
- 20.Lipscomb JD. Mechanism of extradiol aromatic ring-cleaving dioxygenases. Curr Opin Struct Biol. 2008;18:644–649. doi: 10.1016/j.sbi.2008.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Siegbahn PEM, Haeffner F. Mechanism for catechol ring-cleavage by nonheme iron extradiol dioxygenases. J Am Chem Soc. 2004;126:8919–8932. doi: 10.1021/ja0493805. [DOI] [PubMed] [Google Scholar]
- 22.Emerson JP, et al. Swapping metals in Fe- and Mn-dependent dioxygenases: Evidence for oxygen activation without a change in metal redox state. Proc Natl Acad Sci USA. 2008;105:7347–7352. doi: 10.1073/pnas.0711179105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gunderson WA, et al. Electron paramagnetic resonance detection of intermediates in the enzymatic cycle of an extradiol dioxygenase. J Am Chem Soc. 2008;130:14465–14467. doi: 10.1021/ja8052255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Arciero DM, et al. EPR and Mössbauer studies of protocatechuate 4,5-dioxygenase. Characterization of a new Fe2+ environment. J Biol Chem. 1983;258:14981–14991. [PubMed] [Google Scholar]
- 25.Münck E. In: Physical Methods in Bioinorganic Chemistry. Que L Jr, editor. Sausalito, CA: University Science Books; 2000. pp. 287–319. [Google Scholar]
- 26.Vetting MW, et al. Crystallographic comparison of manganese- and iron-dependent homoprotocatechuate 2,3-dioxygenases. J Bacteriol. 2004;186:1945–1958. doi: 10.1128/JB.186.7.1945-1958.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Whittaker JW, Lipscomb JD, Kent TA, Münck E. Brevibacterium fuscum protocatechuate 3,4-dioxygenase. Purification, crystallization, and characterization. J Biol Chem. 1984;259:4466–4475. [PubMed] [Google Scholar]
- 28.Zhou J, et al. Substrate binding to the alpha-ketoglutarate-dependent nonheme iron enzyme clavaminate synthase 2: Coupling mechanism of oxidative decarboxylation and hydroxylation. J Am Chem Soc. 1998;120:13539–13540. [Google Scholar]
- 29.Wolfe MD, Parales JV, Gibson DT, Lipscomb JD. Single turnover chemistry and regulation of O2 activation by the oxygenase component of naphthalene 1,2-dioxygenase. J Biol Chem. 2001;276:1945–1953. doi: 10.1074/jbc.M007795200. [DOI] [PubMed] [Google Scholar]
- 30.Price JC, et al. Kinetic dissection of the catalytic mechanism of taurine:alpha-ketoglutarate dioxygenase (TauD) from Escherichia coli. Biochemistry. 2005;44:8138–8147. doi: 10.1021/bi050227c. [DOI] [PubMed] [Google Scholar]
- 31.Kovaleva EG, Neibergall MB, Chakrabarty S, Lipscomb JD. Finding intermediates in the O2 activation pathways of nonheme iron oxygenases. Acc Chem Res. 2007;40:475–483. doi: 10.1021/ar700052v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bollinger JM, Krebs C. Stalking intermediates in oxygen activation by iron enzymes: Motivation and method. J Inorg Biochem. 2006;100:586–605. doi: 10.1016/j.jinorgbio.2006.01.022. [DOI] [PubMed] [Google Scholar]
- 33.Che M, Tench AJ, Naccache C. Electron spin resonance studies of isotopically labeled oxygen species adsorbed on supported molybdenum. J Chem Soc Farad T 1. 1974;70:263–272. [Google Scholar]
-
34.Chiesa M, et al. Continuous wave electron paramagnetic resonance investigation of the hyperfine structure of
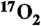 - adsorbed on the MgO surface. J Chem Phys. 2002;116:4266–4274. [Google Scholar]
- adsorbed on the MgO surface. J Chem Phys. 2002;116:4266–4274. [Google Scholar] - 35.Dickinson LC, Chien JCW. Electron paramagnetic resonance crystallography of oxygen-17-enriched oxycobaltomyoglobin: Stereoelectronic structure of the cobalt dioxygen system. Proc Natl Acad Sci USA. 1980;77:1235–1239. doi: 10.1073/pnas.77.3.1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shaik S, et al. P450 enzymes: Their structure, reactivity, and selectivities modeled by QM/MM calculations. Chem Rev. 2010;110:949–1017. doi: 10.1021/cr900121s. [DOI] [PubMed] [Google Scholar]
- 37.Jousserandot A, et al. Microsomal cytochrome P450 dependent oxidation of N-hydroxyguanidines, amidoximes, and ketoximes: Mechanism of the oxidative cleavage of their C∶N(OH) bond with formation of nitrogen oxides. Biochemistry. 1998;37:17179–17191. doi: 10.1021/bi981175c. [DOI] [PubMed] [Google Scholar]
- 38.Brown CD, et al. VTVH-MCD and DFT studies of thiolate bonding to {FeNO}7/{FeO2}8 complexes of isopenicillin N synthase: Substrate determination of oxidase versus oxygenase activity in nonheme Fe enzymes. J Am Chem Soc. 2007;129:7427–7438. doi: 10.1021/ja071364v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pierpont CG, Lange CW. The chemistry of transition metal complexes containing catechol and semiquinone ligands. In: Karlin KD, editor. Progress in Inorganic Chemistry. Vol. 41. New York: Wiley; 1994. pp. 331–442. [Google Scholar]
- 40.Pierpont CG, Buchanan RM. Transition metal complexes of o-benzoquinone, o-semiquinone, and catecholate ligands. Coord Chem Rev. 1981;38:45–87. [Google Scholar]
- 41.Kessel SL, Emberson RM, Debrunner PG, Hendrickson DN. Iron(III), manganese(III), and cobalt(III) complexes with single chelating o-semiquinone ligands. Inorg Chem. 1980;19:1170–1178. [Google Scholar]
- 42.Hegg EL, Que L. The 2-His-1-carboxylate facial triad: An emerging structural motif in mononuclear nonheme iron(II) enzymes. Eur J Biochem. 1997;250:625–629. doi: 10.1111/j.1432-1033.1997.t01-1-00625.x. [DOI] [PubMed] [Google Scholar]
- 43.Wang YZ, Lipscomb JD. Cloning, overexpression, and mutagenesis of the gene for homoprotocatechuate 2,3-dioxygenase from Brevibacterium fuscum. Protein Expres Purif. 1997;10:1–9. doi: 10.1006/prep.1996.0703. [DOI] [PubMed] [Google Scholar]
- 44.Miller MA, Lipscomb JD. Homoprotocatechuate 2,3-dioxygenase from Brevibacterium fuscum—A dioxygenase with catalase activity. J Biol Chem. 1996;271:5524–5535. doi: 10.1074/jbc.271.10.5524. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.