

Abstract
OBJECTIVE: To determine clinical features, natural history, and outcome of a well-defined cohort of 25 consecutive patients with idiopathic systemic capillary leak syndrome (SCLS) evaluated at a tertiary care center.
PATIENTS AND METHODS: Records of patients diagnosed as having SCLS from November 1, 1981, through April 30, 2008, were reviewed. Descriptive statistics were used to analyze patient demographics, clinical features, complications, and therapeutic interventions.
RESULTS: Of the 34 patients whose records were reviewed, 25 fulfilled all diagnostic criteria for SCLS. The median age at diagnosis of SCLS was 44 years. Median follow-up of surviving patients was 4.9 years, and median time to diagnosis from symptom onset was 1.1 years (interquartile range, 0.5-4.1 years). Flulike illness or myalgia was reported by 14 patients (56%) at onset of an acute attack of SCLS, and rhabdomyolysis developed in 9 patients (36%). Patients with a greater decrease in albumin level had a higher likelihood of developing rhabdomyolysis (p=.03). Monoclonal gammopathy, predominantly of the IgG-κ type, was found in 19 patients (76%). The progression rate to multiple myeloma was 0.7% per person-year of follow-up. The overall response rate to the different therapies was 76%, and 24% of patients sustained durable (>2 years) complete remission. The estimated 5-year overall survival rate was 76% (95% confidence interval, 59%-97%).
CONCLUSION: Systemic capillary leak syndrome, a rare disease that occurs in those of middle age, is usually diagnosed after a considerable delay from onset of symptoms. The degree of albumin decrement during an attack correlates with development of rhabdomyolysis. A reduction in the frequency and/or the severity of attacks was seen in nearly three-fourths of patients who were offered empirical therapies. The rate of progression to multiple myeloma appears to be comparable to that of monoclonal gammopathy of undetermined significance.
A review of records of patients with this rare disease, which occurs during middle age, showed that systemic capillary leak syndrome is usually diagnosed after a considerable delay from onset of symptoms.
BM = bone marrow; IQR = interquartile range; MGUS = monoclonal gammopathy of undetermined significance; MM = multiple myeloma; OS = overall survival; PC = plasma cell; SCLS = systemic capillary leak syndrome; VEGF = vascular endothelial growth factor
Idiopathic systemic capillary leak syndrome (SCLS), also known as Clarkson's disease and spontaneous periodic edema, is a potentially fatal disorder characterized by stereotypic “attacks”of varying intensity of hypovolemic shock and generalized edema in association with hemoconcentration (as detected by an elevated hematocrit concentration) and hypoalbuminemia, typically occurring in the absence of albuminuria.1 Presence of serum M protein, a notable hallmark, is the only pertinent laboratory abnormality during the quiescent phase between the attacks.2 Although the exact incidence of this disease is unknown, a relative surge in the reported cases was noted during the past 15 years, possibly as a result of increased awareness and successful identification of patients presenting with a plethora of complex symptoms and laboratory findings that could be unified by a common diagnosis of idiopathic SCLS.3,4 Although the initial accounts emerged from the United States and Europe, cases were later reported from around the world, including India, Japan, and Kuwait.5-8
The goals of our study were to determine the clinical features, natural history, and outcome of a well-defined cohort of consecutive patients with SCLS and to compare our findings with those of previously reported cases and smaller retrospective studies.
The path from the first description of the cluster of symptoms in a female patient to their formal recognition as a clinical syndrome of systemic capillary leak is instructive and merits discussion. In 1960, Clarkson et al9 described a case of a 32-year-old woman with an unremarkable family history who experienced “a strange cyclical illness in which she intermittently had a sudden massive movement of plasma from her vascular bed.” The illness was characterized by recurrent episodes of edema, hypovolemic shock, hypoalbuminemia, hemoconcentration, and persistence of serum M protein.9 This first account of SCLS was followed 2 years later by another case of a 29-year-old man with episodes of edema, weight gain, urticaria, fever, and malaise, in addition to increased serum γ-globulin levels, splenomegaly, and marked eosinophilia (50%-70%).10 Although originally perceived to be similar to the index case, this patient lacked the characteristic hypotension and was eventually diagnosed as having episodic angioedema with eosinophilia, now known as Gleich syndrome.11,12
A year later (1963), I. Weinbren13 suggested the term spontaneous periodic edema instead of cyclical edema for this syndrome in a 45-year-old Englishman with recurrent attacks of edema that “…developed rapidly: within hours he felt unwell, became intensely thirsty, and had an aversion to food.” These attacks were comparable to those described in Clarkson's original case. Four years later in 1967, Melvin Horwith, one of the invited coauthors of the index case report, wrote yet another account of fatal hypovolemic shock of unknown etiology wherein the autopsy findings were suggestive of diffuse interstitial edema, pulmonary edema, and serous effusions.14 During the course of the next 2 decades, only 20 more cases appeared in the medical literature.15,16
In 1992 and 1999, 2 small case series from our institution reported on the efficacy of terbutaline and aminophylline in treating this disorder.17,18 In 1997, a larger, multicenter, retrospective study by Amoura et al19 outlined the course of SCLS and evaluated the efficacy of the available therapy in a cohort of 13 patients.
PATIENTS AND METHODS
With approval from the Mayo Clinic Institutional Review Board, records of all patients seen at Mayo Clinic in Rochester, MN, from November 1, 1981, through April 30, 2008, with a diagnosis of SCLS, capillary hyperpermeability, Clarkson's disease, and angioedema with hypoalbuminemia were reviewed. The diagnosis of SCLS was based on unequivocal documentation of recurrent attacks of hypotension, elevated hematocrit concentrations, peripheral edema, and hypoalbuminemia without albuminuria. Monoclonal gammopathy was not considered a mandatory criterion, but its concurrent presence strengthened the diagnosis. Patients whose physical signs, symptoms, and laboratory findings could be explained by an alternative diagnosis were excluded if that alternative diagnosis was confirmed with further investigations.
Laboratory investigation reports obtained during the first prototypical “attack” and/or within 30 days of the patient encounter were included. Most patients had been managed at other medical centers; with their consent, data were retrieved from the documented records of the referring institutions. Follow-up data were obtained, and at least 2 attempts were made to contact the surviving patients by telephone. When feasible, an attempt was made to determine the cause of death. Within the constraints of a retrospective review, we endeavored to classify disease outcome in our cohort.
Statistical Analyses
Descriptive statistics and simple scatter plots were used to analyze patient demographics, clinical features, complications, and therapeutic interventions. Overall survival (OS) was estimated by the Kaplan-Meier method and defined as the time from the date of diagnosis until death or last follow-up. Patients alive at the last recorded follow-up were censored. Wilcoxon signed rank tests were used to test for changes in laboratory parameters at the time of attack relative to baseline, and Wilcoxon rank sum tests were used to test for associations between changes in albumin and hematocrit concentration during an attack relative to baseline and development of clinical complications. All tests were 2-sided, and all analyses were carried out using SAS software, version 9.1 (SAS Institute, Cary, NC).
RESULTS
Of the 34 patients whose records were reviewed, 25 fulfilled all diagnostic criteria for SCLS. Table 1 outlines the demographic profile of the study cohort. The median time between the onset of symptoms and definitive diagnosis was 1.1 years (interquartile range [IQR], 0.5-4.1 years). In 3 patients, a formal diagnosis was made more than 5 years after the onset of symptoms (after 5.5, 8.0, and 32.0 years). The median number of acute attacks per year was 3 (IQR, 0.5-5.0 attacks per year).
TABLE 1.
Demographics of Study Patients at Diagnosis of SCLSa

Precipitating Factors
Flulike illness or myalgias heralded a crisis in most patients, but considerable variation in the prodromata was noted (Table 2). Most patients reported fatigue and profound exhaustion along with light-headedness at the onset of an attack.
TABLE 2.
Clinical Symptoms of 25 Patients with Systemic Capillary Leak Syndrome

Family history was mostly noncontributory. One patient with disease progression to multiple myeloma (MM) had a sibling who died of MM at the age of 44 years. In addition, a neonate of an SCLS patient died of cardiorespiratory dysfunction in the setting of profound hypotension, hemoconcentration, and leukocytosis on the 17th day of life. The postmortem findings were suggestive of SCLS. However, electron microscopy showed no widening of endothelial cell junctional gaps.
During quiescent periods between attacks, findings on physical examination were essentially unremarkable, except in 2 patients with chronic SCLS who had progressive generalized edema with pleural and pericardial effusions. Residual damage as a consequence of compartment syndrome or stroke during a previous attack was noted in a few patients (Table 3). A precipitous decline in the blood pressure was seen in all the patients in crises. The median systolic blood pressure, available for 16 patients (64%), was 72 mm Hg (IQR, 57.0-90.5 mm Hg) during an attack.
TABLE 3.
Frequency of Complications in 25 Patients with Systemic Capillary Leak Syndrome

Rhabdomyolysis, diagnosed by an elevated creatine kinase level in the appropriate clinical setting, was noted in 9 patients (36%) during at least 1 attack, with 5 patients (20%) developing compartment syndrome. Most patients with compartment syndrome (60%) required fasciotomy. A Wilcoxon rank sum test comparing the distributions of albumin in those with vs those without rhabdomyolysis suggests that the greater the difference in albumin from baseline to attack, the higher the likelihood of associated rhabdomyolysis (P=.03; Figure 1). However, no association between rhabdomyolysis and the change in hematocrit concentration was observed (P=.73).
FIGURE 1.

Albumin difference (from baseline to attack) and development of rhabdomyolysis. The degree of albumin decrement during an attack correlates with the development of rhabdomyolysis.
Two patients presented with cardiac tamponade, necessitating urgent pericardiocentesis, followed by pericardial window placement. Pulmonary edema during the recruitment phase is an iatrogenic complication of SCLS management that results from normalization of the capillary permeability during the recovery phase of the disease, in the face of ongoing vigorous fluid resuscitation.5,20 It developed in 10 (40%) of the patients, a few of whom required mechanical ventilation.
Laboratory Findings
All patients had well-documented elevation of hematocrit concentrations during an attack (Table 4). The median absolute increase in hematocrit was 19.8% (IQR, 14.8%-28.0%) from the baseline (Wilcoxon signed rank test, P<.001). Leukocytosis was observed in 36% of the patients, an abnormality that, along with the concomitant increase in the hemoglobin concentration, led to misdiagnosis of polycythemia vera in 12% of patients. No evidence of eosinophilia, a cardinal finding in Gleich syndrome, or thrombocytosis was noted in any case.
TABLE 4.
Laboratory Values at Baseline and During Attacks in Patients with Systemic Capillary Leak Syndromea,b

The attacks were characterized by a significant reduction in albumin (median reduction, 1.9 g/dL; IQR, 1.0-2.2 g/dL; Wilcoxon signed rank test, P<.001; Table 4). An M protein was detected by serum protein electrophoresis/immunofixation studies in 19 patients (76%) and was of the IgG type in all but 1 patient. Nearly three-fourths of predominant light chains were of the κ subtype. The M-protein level was classically low (median, 0.6 g/dL; IQR, 0-1.1 g/dL). Urinary M component was detected in approximately a third (n=7) of the 23 patients who were tested.
Of 18 patients who underwent a bone marrow (BM) biopsy during the course of SCLS, 9 (50%) had evidence of monoclonal plasma cell (PC) proliferation. One patient had 5% to 10% atypical, clonal PCs on the BM aspirate despite undetectable serum M protein, and molecular genetic studies using IgH polymerase chain reaction primers identified IgH gene rearrangements. Another patient had eosinophilic hyperplasia in the BM without peripheral blood eosinophilia. Only 2 patients had 10% or more bone marrow PCs at follow-up.
In the 20 patients tested for complement 1 esterase (antigen and function) assays, the absence of low values in all patients ruled out hereditary angioedema, a consideration in the differential diagnosis for SCLS.21,22 Levels of C3 and C4 were low in 28% and 22%, respectively, of the 18 tested patients. One patient exhibited low total hemolytic complement.
Twelve patients developed acute renal failure, with a median increase in creatinine of 1 mg/dL (IQR, 0.55-3.4 mg/dL). Acute renal failure was defined as an absolute increase in creatinine of 0.3 mg/dL or a value 50% above the baseline, or a decline in urine output to less than 0.5 mL/kg/h for more than 6 hours, manifested within 48 hours of onset of symptoms. Two patients temporarily required hemodialysis.
Follow-up
The median follow-up for living patients was 4.9 years (IQR, 3.2-8.5 years). After 17.0 years of close follow-up, progression to MM occurred in 1 patient with a baseline IgG-κ M-spike of 1.05 g/dL. This patient had a sibling who was diagnosed as having MM at the age of 44 years. The progression to MM was heralded by an abrupt increase in the M protein to 2.2 g/dL and Bence Jones proteinuria along with acute-on-chronic renal insufficiency and anemia. Fluorescence in situ hybridization identified a hyperdiploid clone of PCs. The patient died within 2 months of the progression to MM. Another patient with a 25-year history of monoclonal gammopathy of undetermined significance (MGUS) had BM biopsy findings suggestive of AL amyloidosis with BMPC of 10% to 15%. On presentation, her relapsing SCLS had evolved into a chronic form with persistent effusions requiring multiple thoracenteses and pericardiocenteses. Cardiac amyloidosis may also have been contributing to her symptoms; however, before this diagnosis could be conclusively ascertained through an endomyocardial biopsy, the patient died. An autopsy was not performed.
Treatment, Response, and Survival
Therapeutic intervention was recorded for all 25 patients. The most common first choice for long-term prophylactic therapy was a methylxanthine (theophylline or aminophylline) in 23 patients (92%), along with the β-agonist terbutaline in 21 patients (84%). A leukotriene inhibitor was used in 10 patients (40%). These patients often showed no meaningful response or intolerance to first-line therapies.
In a 4-year-old patient, montelukast therapy markedly reduced the frequency and severity of attacks. The combination of zafirlukast and an angiotensin-converting enzyme inhibitor, lisinopril, achieved greater depth of responses in 2 patients.
Both immediate and long-term treatment of SCLS involved anti-inflammatory therapy in some patients, including the use of prednisone in 12 patients (48%) and indomethacin in 1 (4%).
Successful management of severe acute attacks included judicious use of crystalloid/colloid volume expanders and diuretic agents. Additionally, epinephrine and plasmapheresis were used in 1 patient each, with resolution of symptoms. Overall, adverse effects of therapy were reported by 11 patients (44%) and were predominantly sympathomimetic, related to the use of β-agonists. One patient died of infectious complications of long-term corticosteroid therapy.
We developed a novel set of criteria to bring uniformity in determination of the clinical outcome of the patients in our retrospective analyses. Mild, self-limiting episodes did not require hospitalization, whereas moderate to severe attacks necessitated in-patient observation or admission with intravenous fluid resuscitation and often required intensive care. These criteria helped to objectively assess and compare the impact of therapeutic intervention (Table 5). Patients who achieved 100% freedom from attacks for 2 years or more were categorized as having a Mayo Clinic (MC) 100 response. Patients achieving a 50% or greater reduction in the frequency and/or severity of attacks were classified as having achieved an MC50 response.
TABLE 5.
Long-term Responses in 25 Study Patients

The range of therapies for the 6 patients in the MC100 group was varied. Notably, 2 patients who remain in MC100 were in the pediatric age group at diagnosis. One adult patient remained in MC100 therapy without maintenance therapy, whereas the others have maintained durable MC100 with ongoing therapy at the time of last follow-up.
Survival information was available for 24 patients who were followed up for a total of 140 person-years. Nine deaths were recorded. A steady decline in survival through the first 5 years after diagnosis followed by an apparent plateau from 6 to 12 years was noted. Progression to MM occurred in 1 patient. The rate of progression to MM per person-year of follow-up was 0.7%. Recognizing the limited follow-up of surviving patients at the time of this analysis, and the fact that this disease has a long clinical course (ie, patients are alive for a long period), we report the 1-, 5-, and 10-year estimates, along with the 95% confidence intervals (CIs). The estimated 1-year, 5-year, and 10-year OS rates were 96% (95% CI, 87%-100%), 76% (95% CI, 59%-97%), and 68% (95% CI, 49%-94%), respectively (Figure 2).
FIGURE 2.

Kaplan-Meier curve of overall survival of patients with idiopathic systemic capillary leak syndrome. the vertical lines in the graph denote the censors.
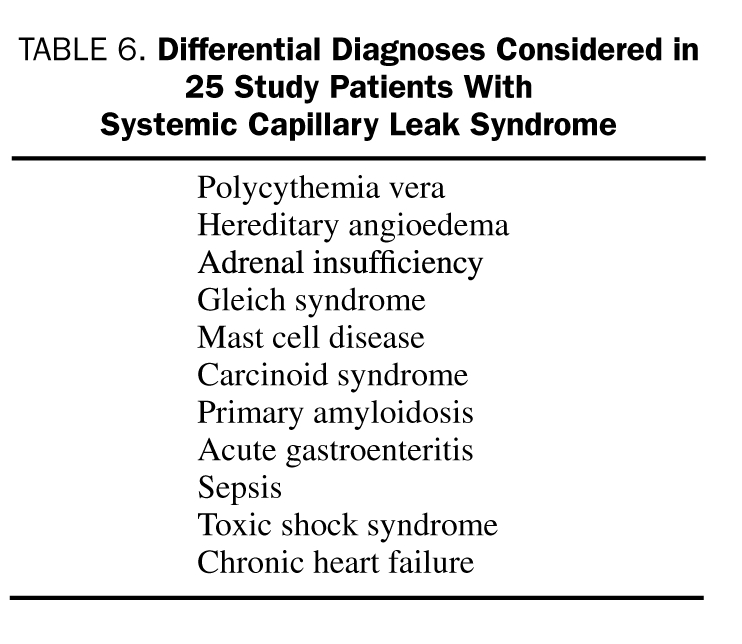
Differential Diagnosis
Visits to several subspecialists preceded the unequivocal establishment of a diagnosis of SCLS in this cohort. Due to overlap of myriad clinical and laboratory features with other diseases, SCLS has been considered a diagnosis of exclusion (Table 6). Interestingly, 12% of patients were referred for further work-up and management of misdiagnosed polycythemia vera, an important consideration in the differential diagnosis for SCLS.23 Two-thirds of these patients had undergone phlebotomy at other institutions despite normal serum erythropoietin levels.
TABLE 6.
Differential Diagnoses Considered in 25 Study Patients with Systemic Capillary Leak Syndrome
DISCUSSION
The pathogenesis of SCLS remains unclear. Cytokines, leukotrienes, vascular endothelial growth factor (VEGF), and complement have been implicated. A substantial increase in the interleukin 2 receptor–positive peripheral blood M-cell count has been noted during episodes of hyperpermeability.24 Proteins with weights up to 900 kDa can extravasate, but the exact cause of hyperpermeability remains unknown. Endothelial microvesicular body and bleb formation during attacks is suggestive of endothelial apoptosis; however, attempts to directly link M protein to endothelial damage have been unsuccessful.25,26 The appearance of this syndrome in a 17-day-old infant in our series suggests the possibility of intrauterine transfer of soluble mediators or an inherited disorder.
Our cohort included only patients who had spontaneous attacks without a clear etiology. Another term, secondary SCLS, has been coined for similar attacks of generalized vascular hyperpermeability in which the causative factor of SCLS is known. Tumor necrosis factor, interferon-α and β, acitretin, epoprostenol, gemcitabine, interleukin 2, denileukin diftitox, and filgrastim have been implicated in secondary SCLS.27-31 Hepatitis C infection and malignancies have also been reported as etiological factors, but none of the patients in our cohort had such illnesses concurrently at the time of initial evaluation.32-35
Similarities in age and sex distribution were noted between our cohort and that by Amoura et al.19 The poor representation of African Americans was likely a reflection of referral bias. Only a few cases have been reported in the pediatric age group thus far, and our study adds another 2, suggesting the rarity of this disease in children.36-39
To increase awareness of this disorder, a report on SCLS was recently added to the National Organization of Rare Disorders database. A patient-support Web site (http://www.rareshare.org) has also included SCLS. An increase in the reported cases of SCLS in the past decade could perhaps be attributed to greater awareness among clinicians rather than an increase in its incidence.3
As underscored by many illustrative cases, delayed diagnosis and misdiagnosis increase patient morbidity and may adversely affect outcome. Protracted lag time (median, 13.5 months) between the onset of symptoms and the establishment of the correct diagnosis was a noteworthy finding in our study.
We used stringent criteria to define patients with SCLS in this series. Coexisting MGUS, warning signs, triggering factors, oliguria with onset of an attack, pulmonary edema, and polyuria in the recruitment phase followed by spontaneous resolution of symptoms are typical features but are not mandatory for making a formal diagnosis.
Although most patients were able to recognize the onset of an attack because of symptom recurrence and similar, if not identical, triggering factors, the severity of the attack was less predictable, making it important for the patient to promptly seek urgent care at the onset of an attack regardless of its severity.
Except for an isolated case of a newborn to a patient with SCLS, family history was noncontributory, a finding that compares well with previous reports. The frequency of attacks varied widely. As in a previously reported case, a subset of patients (8%) was diagnosed as having chronic hyperpermeability, characterized by stable blood pressure, persisting edema (albeit to a lesser degree between the acute flares), and, on occasion, serous effusions.40
In Clarkson's patient, a clear temporal relationship to the menstrual cycle was established. In our series, only 1 patient had menses-related crises and, unlike previous reports, no attacks were observed in the peripartum period.41
Preservation of mentation is not uncommon during an SCLS attack. In this series, only 2 patients presented with seizures during crisis, 1 of whom had cerebral edema, a well-documented finding in SCLS.42 Cerebral hypoperfusion resulted in a stroke in 1 patient. Acute renal failure, noted in nearly half of patients, is a common complication and a consequence of hypotension-related reduced kidney perfusion, acute tubular necrosis, and rhabdomyolysis-related pigmenturia.43-45 Tissue salvage (decompression) therapy was required by 3 of the 5 patients who had developed compartment syndrome, a grave complication of SCLS due to massive fluid leakage into the muscle tissue.46-48 Acute deep venous thrombosis, which occurred in 8% of patients, perhaps resulted from attack-induced activation of the components of the Virchow triad.
The detection of M protein in most patients (n=19; 76%) may be attributed to the selection bias of the patients referred to our dysproteinemia clinic. However, the results are similar to those of the French study by Amoura et al,19 wherein all 13 cases reported MGUS (11 of the κ type), and a subsequent review, in which 39 of 50 cases had MGUS (28 of the κ type).3 It is important to reiterate that the absence of an M-spike does not exclude SCLS.49,50 Unlike other PC disorders such as POEMS syndrome (polyneuropathy, organomegaly, endocrinopathy, monoclonal gammopathy, and skin changes), which is almost always associated with IgG-λ or IgA-λ, and Schnitzler syndrome (chronic urticarial rash, monoclonal gammopathy, fever, arthralgia, bone pain, and lymphadenopathy), which is associated with IgM monoclonal gammopathy, patients with Clarkson's disease predominantly have IgG-κ or IgA-κ monoclonal gammopathy.51,52 In contrast to Schnitzler and POEMS syndromes, the M component does not appear to be a major criterion required for diagnosis of SCLS.51,52
The evolution to MM, plasma cell leukemia, and, rarely, systemic amyloidosis has been reported.2,6,53 In our cohort, the rate of transformation was 0.7% per person-year of follow-up, which is comparable to 1% per year reported in MGUS.54 Surveillance for MM is therefore necessary. Recurrence (or severity) of attacks did not correlate with the size of the M-spike.
This disorder, particularly the unpredictability of recurrent life-threatening episodes as well as of adverse effects of treatment, took an emotional toll on most patients, some of whom also exhibited overt signs of major depression.
Much has been learned through delineating the distinct “phases” of an attack. During the initial leak phase, cautious volume expansion and central venous pressure monitoring can aid in maintaining adequate perfusion of vital organs.18 Overaggressive hydration can precipitate pulmonary edema. Corticosteroid therapy can be beneficial during an attack16,33,55 because cytokine-mediated endothelial damage has been implicated.56 In addition, plasmapheresis16,19,57,58 and intravenous γ-globulin19,50,59 have proved to be successful in the acute phase. In our study, 1 patient received intravenous immunoglobulin at an outside institution due to IgG2 deficiency before the diagnosis of SCLS was established, and only 1 underwent plasmapheresis.
To date, terbutaline and aminophylline-mediated prophylaxis17 that increases cyclic adenosine monophosphate or inhibits the intracellular Rho kinase pathway60 has had the most effect on the maintenance of quiescent periods.18 Recently, verapamil,49 leukotriene inhibitors,61,62 immunoadsorption,61 and thalidomide (which possibly acts through tumor necrosis factor α inhibition in M cells)57 have been used. Endothelial targets such as VEGF have also been exploited. Therapies involving monoclonal antibodies, such as bevacizumab (anti-VEGF) and infliximab (anti–tumor necrosis factor α), have been used with varying success.63,64 Institution of antimyeloma therapy in patients with SCLS and concomitant MM has reportedly abated subsequent SCLS attacks.19 However, this observation could not be verified in our cohort because the single patient with both SCLS and MM died shortly after the latter was diagnosed.
The 5-year OS rate of 76% in our cohort reflects an improvement in mortality when compared with previous studies,16,19 but the factor that contributes most to this improved outcome has not been definitively identified.
The constraints of studying the natural history and clinical course of a rare disorder are well known. Our study has the usual limitations of a retrospective analysis of selected patients from a tertiary care center with limited follow-up.
CONCLUSION
Although small, our series of patients with SCLS is the largest and most comprehensive series to date and has shed light on several important aspects of this disorder. First, delay in diagnosis and/or misdiagnosis led to increased patient morbidity in our series. Second, the novel finding of correlation between the degree of albumin decrement and rhabdomyolysis could potentially alert physicians to monitor patients closely for this dreaded complication. Third, the rate of progression to MM was comparable to that of MGUS patients in our study. Fourth, the prognosis of patients in the pediatric age group appears better. Fifth, to facilitate uniform reporting of objective responses to therapy in clinical trials, we have proposed novel response criteria and have successfully reported outcomes using this model. Finally, a sustained complete remission (≥2 years) was noted in nearly a quarter of the patients in our series.
Prompt institution of appropriate therapy as already outlined could reduce morbidity and prevent complications. Prophylactic therapy, although effective to a degree, remains largely empirical. A number of factors, including underrecognition in the medical community and rarity of the syndrome, have precluded analysis by rational clinical trial designs that are necessary to determine more targeted and adequate therapy, beyond the currently available symptomatic management strategies of SCLS, first described almost half a century ago.
Supplementary Material
Acknowledgments
The authors thank Ms Sue Olive for her assistance with patient correspondence.
Footnotes
An earlier version of this article appeared Online First.
REFERENCES
- 1.Takabatake T. Systemic capillary leak syndrome. Intern Med. 2002;41(11):909-910 [DOI] [PubMed] [Google Scholar]
- 2.Vigneau C, Haymann JP, Khoury N, Sraer JD, Rondeau E. An unusual evolution of the systemic capillary leak syndrome. Nephrol Dial Transplant. 2002;17(3):492-494 [DOI] [PubMed] [Google Scholar]
- 3.Dhir V, Arya V, Malav IC, Suryanarayanan BS, Gupta R, Dey AB. Idiopathic systemic capillary leak syndrome (SCLS): case report and systematic review of cases reported in the last 16 years. Intern Med. 2007;46(12):899-904 [DOI] [PubMed] [Google Scholar]
- 4.Bonadies N, Baud P, Peter HJ, Buergi U, Mueller BU. A case report of Clarkson's disease: if you don't know it, you'll miss it. Eur J Intern Med. 2006;17(5):363-365 [DOI] [PubMed] [Google Scholar]
- 5.Chihara R, Nakamoto H, Arima H, et al. Systemic capillary leak syndrome. Intern Med. 2002;41(11):953-956 [DOI] [PubMed] [Google Scholar]
- 6.Ghosh K, Madkaikar M, Iyer Y, Pathare A, Jijina F, Mohanty D. Systemic capillary leak syndrome preceding plasma cell leukaemia. Acta Haematol. 2001;106(3):118-121 [DOI] [PubMed] [Google Scholar]
- 7.Kawabe S, Saeki T, Yamazaki H, Nagai M, Aoyagi R, Miyamura S. Systemic capillary leak syndrome. Intern Med. 2002;41(3):211-215 [DOI] [PubMed] [Google Scholar]
- 8.Abdul-Ghaffar NU, Farghaly MM, Swamy AS. Acute renal failure, compartment syndrome, and systemic capillary leak syndrome complicating carbon monoxide poisoning. J Toxicol Clin Toxicol. 1996;34(6):713-719 [DOI] [PubMed] [Google Scholar]
- 9.Clarkson B, Thompson D, Horwith M, Luckey EH. Cyclical edema and shock due to increased capillary permeability. Am J Med. 1960;29:193-216 [DOI] [PubMed] [Google Scholar]
- 10.Preston GM, Rees JR, Spathis GS. A man with cyclical oedema. Guys Hosp Rep. 1962;111:69-79 [PubMed] [Google Scholar]
- 11.Gleich GJ, Schroeter AL, Marcoux JP, Sachs MI, O'Connell EJ, Kohler PF. Episodic angioedema associated with eosinophilia. N Engl J Med. 1984;310(25):1621-1626 [DOI] [PubMed] [Google Scholar]
- 12.Banerji A, Weller PF, Sheikh J. Cytokine-associated angioedema syndromes including episodic angioedema with eosinophilia (Gleich's Syndrome). Immunol Allergy Clin North Am. 2006;26(4):769-781 [DOI] [PubMed] [Google Scholar]
- 13.Weinbren I. Spontaneous periodic oedema: a new syndrome. Lancet. 1963;2(7307):544-546 [DOI] [PubMed] [Google Scholar]
- 14.Horwith M, Hagstrom JW, Riggins RC, Luckey EH. Hypovolemic shock and edema due to increased capillary permeability. JAMA. 1967;200(2):101-104 [PubMed] [Google Scholar]
- 15.Atkinson JP, Waldmann TA, Stein SF, et al. Systemic capillary leak syndrome and monoclonal IgG gammopathy; studies in a sixth patient and a review of the literature. Medicine (Baltimore). 1977;56(3):225-239 [DOI] [PubMed] [Google Scholar]
- 16.Teelucksingh S, Padfield PL, Edwards CR. Systemic capillary leak syndrome. Q J Med. 1990;75(277):515-524 [PubMed] [Google Scholar]
- 17.Droder RM, Kyle RA, Greipp PR. Control of systemic capillary leak syndrome with aminophylline and terbutaline. Am J Med. 1992;92(5):523-526 [DOI] [PubMed] [Google Scholar]
- 18.Tahirkheli NK, Greipp PR. Treatment of the systemic capillary leak syndrome with terbutaline and theophylline: a case series. Ann Intern Med. 1999;130(11):905-909 [DOI] [PubMed] [Google Scholar]
- 19.Amoura Z, Papo T, Ninet J, et al. Systemic capillary leak syndrome: report on 13 patients with special focus on course and treatment. Am J Med. 1997;103(6):514-519 [DOI] [PubMed] [Google Scholar]
- 20.Bouhaja B, Somrani N, Thabet H, Zhioua M, Yacoub M. Adult respiratory distress syndrome complicating a systemic capillary leak syndrome. Intensive Care Med. 1994;20(4):307-308 [DOI] [PubMed] [Google Scholar]
- 21.Bracho FA. Hereditary angioedema. Curr Opin Hematol. 2005;12(6):493-498 [DOI] [PubMed] [Google Scholar]
- 22.Lofdahl CG, Solvell L, Laurell AB, Johansson BR. Systemic capillary leak syndrome with monoclonal IgG and complement alterations: a case report on an episodic syndrome. Acta Med Scand. 1979;206(5):405-412 [DOI] [PubMed] [Google Scholar]
- 23.Doubek M, Brychtova Y, Tomiska M, Mayer J. Idiopathic systemic capillary leak syndrome misdiagnosed and treated as polycythemia vera. Acta Haematol. 2005;113(2):150-151 [DOI] [PubMed] [Google Scholar]
- 24.Cicardi M, Gardinali M, Bisiani G, Rosti A, Allavena P, Agostoni A. The systemic capillary leak syndrome: appearance of interleukin-2-receptor-positive cells during attacks. Ann Intern Med. 1990;113(6):475-477 [DOI] [PubMed] [Google Scholar]
- 25.Assaly R, Olson D, Hammersley J, et al. Initial evidence of endothelial cell apoptosis as a mechanism of systemic capillary leak syndrome. Chest. 2001;120(4):1301-1308 [DOI] [PubMed] [Google Scholar]
- 26.Zhang W, Ewan PW, Lachmann PJ. The paraproteins in systemic capillary leak syndrome. Clin Exp Immunol. 1993;93(3):424-429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rosenstein M, Ettinghausen SE, Rosenberg SA. Extravasation of intravascular fluid mediated by the systemic administration of recombinant interleukin 2. J Immunol. 1986;137(5):1735-1742 [PubMed] [Google Scholar]
- 28.Fellows IW, Powell RJ, Toghill PJ, Williams TJ, Cohen GF. Epoprostenol in systemic capillary leak syndrome [letter]. Lancet. 1988;2(8620):1143 [DOI] [PubMed] [Google Scholar]
- 29.Estival JL, Dupin M, Kanitakis J, Combemale P. Capillary leak syndrome induced by acitretin. Br J Dermatol. 2004;150(1):150-152 [DOI] [PubMed] [Google Scholar]
- 30.Rechner I, Brito-Babapulle F, Fielden J. Systemic capillary leak syndrome after granulocyte colony-stimulating factor (G-CSF). Hematol J. 2003;4(1):54-56 [DOI] [PubMed] [Google Scholar]
- 31.Talpur R, Apisarnthanarax N, Ward S, Duvic M. Treatment of refractory peripheral T-cell lymphoma with denileukin diftitox (ONTAK). Leuk Lymphoma. 2002;43(1):121-126 [DOI] [PubMed] [Google Scholar]
- 32.Yamamoto K, Mizuno M, Tsuji T, Amano T. Capillary leak syndrome after interferon treatment for chronic hepatitis C. Arch Intern Med. 2002;162(4):481-482 [DOI] [PubMed] [Google Scholar]
- 33.Kao NL, Richmond GW, Luskin AT. Systemic capillary leak syndrome. Chest. 1993;104(5):1637-1638 [DOI] [PubMed] [Google Scholar]
- 34.Garces S, Araujo F, Rego F, Soares JL, Carlos AG. Capillary leakage syndrome: a case report and a review. Allerg Immunol (Paris). 2002;34(10):361-364 [PubMed] [Google Scholar]
- 35.Dereure O, Portales P, Clot J, Guilhou JJ. Biclonal Sezary syndrome with capillary leak syndrome. Dermatology. 1994;188(2):152-156 [DOI] [PubMed] [Google Scholar]
- 36.Stiller B, Sonntag J, Dahnert I, et al. Capillary leak syndrome in children who undergo cardiopulmonary bypass: clinical outcome in comparison with complement activation and C1 inhibitor. Intensive Care Med. 2001;27(1):193-200 [DOI] [PubMed] [Google Scholar]
- 37.Foeldvari I, Waida E, Junker AK. Systemic capillary leak syndrome in a child. J Pediatr. 1995;127(5):739-741 [DOI] [PubMed] [Google Scholar]
- 38.Karatzios C, Gauvin F, Egerszegi EP, et al. Systemic capillary leak syndrome presenting as recurrent shock. Pediatr Crit Care Med. 2006;7(4):377-379 [DOI] [PubMed] [Google Scholar]
- 39.Onal H, Aktuglu-Zeybek C, Altun G, Ozyilmaz I, Alhaj S, Aydin A. Capillary leak syndrome in a 5-month-old infant associated with intractable diarrhoea. Ann Trop Paediatr. 2007;27(1):81-86 [DOI] [PubMed] [Google Scholar]
- 40.Airaghi L, Montori D, Santambrogio L, Miadonna A, Tedeschi A. Chronic systemic capillary leak syndrome: report of a case and review of the literature. J Intern Med. 2000;247(6):731-735 [DOI] [PubMed] [Google Scholar]
- 41.Kanda K, Sari A, Nagai K, Matayoshi Y. Postpartum capillary leak syndrome: a case report. Crit Care Med. 1980;8(11):661-662 [DOI] [PubMed] [Google Scholar]
- 42.Bertorini TE, Gelfand MS, O'Brien TF. Encephalopathy due to capillary leak syndrome. South Med J. 1997;90(10):1060-1062 [DOI] [PubMed] [Google Scholar]
- 43.Dolberg-Stolik OC, Putterman C, Rubinow A, Rivkind AI, Sprung CL. Idiopathic capillary leak syndrome complicated by massive rhabdomyolysis. Chest. 1993;104(1):123-126 [DOI] [PubMed] [Google Scholar]
- 44.Biswas S, Nik S, Corrie PG. Severe gemcitabine-induced capillary-leak syndrome mimicking cardiac failure in a patient with advanced pancreatic cancer and high-risk cardiovascular disease. Clin Oncol (R Coll Radiol). 2004;16(8):577-579 [DOI] [PubMed] [Google Scholar]
- 45.Milner CS, Wagstaff MJ, Rose GK. Compartment syndrome of multiple limbs: an unusual presentation. J Plast Reconstr Aesthet Surg. 2006;59(11):1251-1252 [DOI] [PubMed] [Google Scholar]
- 46.Sanghavi R, Aneman A, Parr M, Dunlop L, Champion D. Systemic capillary leak syndrome associated with compartment syndrome and rhabdomyolysis. Anaesth Intensive Care. 2006;34(3):388-391 [DOI] [PubMed] [Google Scholar]
- 47.Prieto Valderrey F, Burillo Putze G, Martinez Azario J, Santana Ramos M. Systemic capillary leak syndrome associated with rhabdomyolysis and compartment syndrome. Am J Emerg Med. 1999;17(7):743-744 [DOI] [PubMed] [Google Scholar]
- 48.Guidet B, Guerin B, Maury E, Offenstadt G, Amstutz P. Capillary leakage complicated by compartment syndrome necessitating surgery. Intensive Care Med. 1990;16(5):332-333 [DOI] [PubMed] [Google Scholar]
- 49.Kang PM, Lawrence C, Khan GA, Hays RM. Fulminating systemic capillary leak syndrome with lymphocytosis and hypogammaglobulinemia. Ren Fail. 1995;17(5):615-617 [DOI] [PubMed] [Google Scholar]
- 50.Lassoued K, Clauvel JP, Similowski T, Autran B, Bengoufa D, Oksenhendler E. Pulmonary infections associated with systemic capillary leak syndrome attacks in a patient with hypogammaglobulinemia. Intensive Care Med. 1998;24(9):981-983 [DOI] [PubMed] [Google Scholar]
- 51.Dispenzieri A, Kyle RA, Lacy MQ, et al. POEMS syndrome: definitions and long-term outcome. Blood. 2003;101(7):2496-2506 [DOI] [PubMed] [Google Scholar]
- 52.de Koning HD, Bodar EJ, van der Meer JW, Simon A. Schnitzler syndrome: beyond the case reports: review and follow-up of 94 patients with an emphasis on prognosis and treatment. Semin Arthritis Rheum. 2007;37(3):137-148 [DOI] [PubMed] [Google Scholar]
- 53.Beermann W, Horstrup KA, Will R. Systemic capillary leak syndrome [letter]. Am J Med. 1998;105(6):554 [DOI] [PubMed] [Google Scholar]
- 54.Kyle RA, Therneau TM, Rajkumar SV, et al. A long-term study of prognosis in monoclonal gammopathy of undetermined significance. N Engl J Med. 2002;346(8):564-569 [DOI] [PubMed] [Google Scholar]
- 55.Fardet L, Kerob D, Rybojad M, et al. Idiopathic systemic capillary leak syndrome: cutaneous involvement can be misleading. Dermatology. 2004;209(4):291-295 [DOI] [PubMed] [Google Scholar]
- 56.Cicardi M, Berti E, Caputo V, Radice F, Gardinali M, Agostoni A. Idiopathic capillary leak syndrome: evidence of CD8-positive lymphocytes surrounding damaged endothelial cells. J Allergy Clin Immunol. 1997;99(3):417-419 [DOI] [PubMed] [Google Scholar]
- 57.Staak JO, Glossmann JP, Esser JM, Diehl V, Mietz H, Josting A. Thalidomide for systemic capillary leak syndrome. Am J Med. 2003;115(4):332-334 [DOI] [PubMed] [Google Scholar]
- 58.Treatment of systemic capillary leak syndrome [letter]. Lancet. 1988;2(8626-8627):1496 [PubMed] [Google Scholar]
- 59.Lambert M, Launay D, Hachulla E, et al. High-dose intravenous immunoglobulins dramatically reverse systemic capillary leak syndrome. Crit Care Med. 2008;36(7):2184-2187 [DOI] [PubMed] [Google Scholar]
- 60.van Nieuw Amerongen GP, van Hinsburgh VW. Targets for pharmacological intervention of endothelial hyperpermeability and barrier function. Vascul Pharmacol. 2002;39(4-5):257-272 [DOI] [PubMed] [Google Scholar]
- 61.Walinder O, Einarsson P, Lindberger K. A case report of systemic capillary leak syndrome. Effective treatment with immunoadsorption and leukotriene antagonist [in Swedish]. Lakartidningen. 2004;101(38):2880-2882 [PubMed] [Google Scholar]
- 62.Lilly CM, Silverman ES, Sheffer AL. Systemic capillary leak syndrome, leukotrienes, and anaphylaxis. J Intensive Care Med. 2002;17(4):189-194 [Google Scholar]
- 63.Dowden AM, Rullo OJ, Aziz N, Fasano MB, Chatila T, Ballas ZK. Idiopathic systemic capillary leak syndrome: novel therapy for acute attacks. J Allergy Clin Immunol. 2009;124(5):1111-1113 [DOI] [PubMed] [Google Scholar]
- 64.Lesterhuis WJ, Rennings AJ, Leenders WP, et al. Vascular endothelial growth factor in systemic capillary leak syndrome. Am J Med. 2009;122(6):e5-e7 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.