

Abstract
DNA methylation is an epigenetic mark that is critical in determining chromatin accessibility and regulating gene expression. This epigenetic mechanism has an important role in T-cell function. We used genome-wide methylation profiling to characterize the DNA methylome in primary human CD4+ T cells. We found that only 5% of CpG islands are methylated in CD4+ T cells, and that DNA methylation peak density is increased in subtelomeric chromosomal regions. We also found an inverse relationship between methylation peak density and chromosomal length. Our data indicate that DNA methylation in gene promoter regions is not always a repressive epigenetic mark. Indeed, about 27% of methylated genes are actively expressed in CD4+ T cells. We demonstrate that repressive methylation peaks are located closer to the transcription start site (TSS) compared with functionally non-repressive peaks (−893±110 bp versus −1342±218 bp (mean±s.e.m.), P-value <0.05). We also show that both a larger number and an increased CpG island density in promoter sequences predict transcriptional permissiveness of DNA methylation. TSS in the majority of genes with permissive DNA methylation peaks is in DNase I hypersensitive sites, indicating a failure of DNA methylation to induce chromatin inaccessibility in these loci.
Keywords: CD4+ T cell, DNA methylation, CpG islands, promoter methylation, methylome
Introduction
DNA methylation is an epigenetic mark that is critical in determining chromatin accessibility and regulating gene expression. DNA methylation, which refers to the addition of a methyl group to the fifth carbon in cytosine residues within CG dinucleotides, is involved in cell differentiation, imprinting, X-chromosome inactivation, and suppression of transcriptional noise and ‘parasitic’ DNA.1–4 Abnormalities in the DNA methylation pathway are associated with pathological consequences. For example, mutations in the de novo DNA methyltransferase DNMT3B result in a syndrome of Immunodeficiency, Centromeric instability and Facial anomalies (ICF syndrome).5 A complete deficiency of the DNA methyltransferase DNMT1 is incompatible with life. Furthermore, acquired abnormalities in DNA methylation are associated with disease conditions, including cancer and autoimmunity.6,7
DNA methylation has a critical role in normal T-cell function such as T helper cell differentiation and the regulation of interferon-γ, and interleukin (IL)-4 production, key cytokines produced by Th1 and Th2 cells, respectively. DNA demethylation of the FOXP3 locus is pivotal for regulatory T-cell differentiation, and demethylation of the IL-2 locus is associated with IL-2 production on T-cell activation.8 Defective T-cell DNA methylation results in T-cell autoreactivity and has an important pathogenic role in both drug-induced and idiopathic lupus, both in human disease and in animal models.7,9,10
Herein, we characterize the DNA methylome in primary human CD4+ T cells. We map DNA methylation peaks across the genome, and identify genes with promoter region methylation in CD4+ T cells using five biological replicates. We further identify distinguishing features between transcriptionally repressive and non-repressive DNA methylation in CD4+ T cells.
Results
We determined genome-wide DNA methylation peaks in primary human CD4+ T cells using DNA immunoprecipitation (IP) with an anti-5-methylcytidine antibody coupled with array hybridization. Both input and IP DNA were labeled and co-hybridized to microarray chips that included ~385 000 probes covering all UCSC-annotated CpG islands and promoter regions for all RefSeq genes (NimbleGen, Reykjavík, Iceland). The experiments were carried out using five biological replicates from five normal healthy female donors (age range from 31 to 48 years). Signal intensity data were extracted from the scanned images of each array. Scaled log2-ratios of the IP/input DNA were determined from signal intensities, and P-values for methylation enrichment were computed using the one-sided Kolmogorov–Smirnov test. Methylation peaks were determined. They represent regions with at least two probes with −log10 P-values of at least 2 within a 500 bp window, and a methylation score of at least 2. The methylation score for each peak is the average −log10 P-values from probes within that peak.
Several known methylated genetic loci were included in our array for quality control. These included the regions in the HOXA gene cluster, H19/IGF2/KCNQ1 gene cluster and the IGF2R locus. All were methylated in all five biological replicates used in this study. Figure 1a shows the methylation status of the H19/IGF2/KCNQ1 gene cluster in our samples. The HOXA gene cluster serves both as a positive and negative control region, as it contains both known methylated and hypomethylated regions.11 Our data confirm this methylation pattern in all five CD4+ T-cell DNA samples (Figure 1b). We further validated the methylation array data in an independent set of samples from another five normal healthy women (age range from 22 to 57 years) using bisulfite DNA sequencing of both methylated and hypomethylated regions (Figure 1).
Figure 1.


Methylation enrichment signals represented as −log10 P-value scores in CD4+ T-cell DNA from five normal healthy participants in (a) the H19/IGF2/KCNQ1 genetic locus on chromosome 11, and (b) the HOXA gene cluster on chromosome 7. Bisulfite DNA sequencing was used to validate the methylation status in methylated regions within the HOXA3 gene and the KCNQ1/TRPM5 promoter region, and hypomethylated regions in the HOXA1 and HOXA13 promoter regions in an independent set of samples.
We identified 2902±187 (mean±s.e.m., n = 5) methylation peaks in CD4+ T-cell DNA. Further, we identified 388 genes that have at least one DNA methylation peak that appears in the −5 to +1 kb region relative to the transcription start site (TSS) with its center located within the −5.5 and +1.5 kb region in all the five biological replicates tested. This stringent requirement that all genes should be identified in every sample tested has the advantage of adding confidence to the target genes identified near the methylation peaks.
We used gene expression data in normal human CD4+ T cells (10 biological replicates) available from Gene Expression Omnibus, to determine whether there is any correlation between gene expression and methylation status. Expression data were available for 202 genes with a methylated promoter region in CD4+ T cells. Only 55 genes (27.2%) had at least one transcript expressed in normal human CD4+ T cells. The majority of the methylated genes (72.8%) were not expressed. Comparatively, when all annotated genes included in the expression array experiment were analyzed, we found that 43.7% of genes (9094 out of 20 828 genes examined) were expressed in normal human CD4+ T cells (χ2 = 22.0, P<0.0001) (Figure 2a). These findings are consistent with DNA methylation being largely a repressive epigenetic mark in human CD4+ T cells. However, 27.2% of methylated genes are transcriptionally active, indicating that DNA methylation is not always associated with gene silencing in CD4+ T cells. There was a significant difference in the average distance between the center of methylation peaks and TSS of methylated genes that are expressed compared with non-expressed genes. The center of methylation peaks was on an average 449 bp further upstream from the TSS in expressed genes as compared with non-expressed genes (−1342±218 bp versus −893±110 bp (mean±s.e.m.), P-value <0.05). These data suggest that a chromatin distance of three nucleosomes (449 bp divided by 147 bp/nucleosome) is important in determining whether DNA methylation is transcriptionally repressive or permissive in a given genetic locus. There was no difference in methylation intensities (as measured by −log10 P-value methylation scores) between transcriptionally repressive and permissive methylation peaks (2.97±0.04 versus 2.98±0.03 (mean±s.e.m.), P-value = 0.85). We determined the number of CpG islands and the maximum CG dinucleotide density in CpG islands within the −5.5 to +1.5 kb region from the TSS of genes that are methylated and expressed in CD4+ T cells and genes that are methylated but non-expressed. Promoter regions of expressed genes were more likely to have a CpG island compared with non-expressed genes (83.3 versus 64.7%, odds ratio = 2.7, χ2 = 6.38, P-value = 0.012). There was a significant difference in the mean CpG island CG dinucleotide densities (as measured by maximum CG observed/expected ratio) in promoter regions of expressed compared with non-expressed genes. The mean maximum CG observed/expected ratio within CpG islands in promoter regions of methylated and expressed genes was 0.95±0.03 compared with 0.88±0.02 in methylated and non-expressed genes (mean±s.e.m., P = 0.01).
Figure 2.
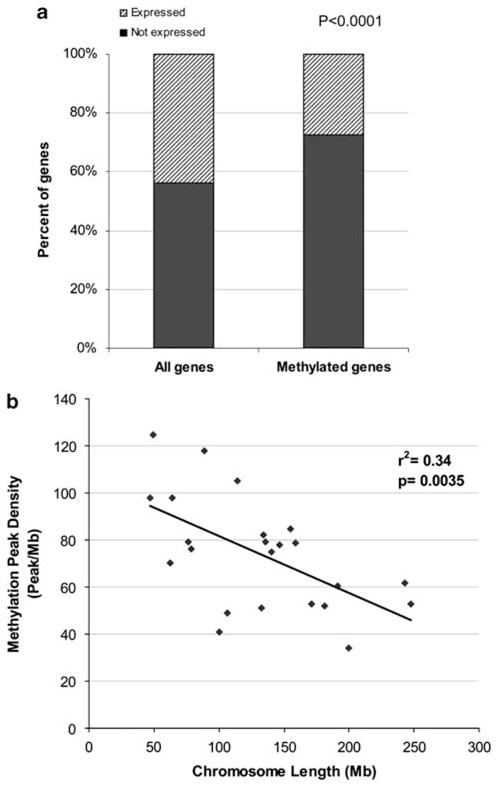
(a) DNA methylation is significantly associated with transcriptional repression in CD4+ T cells (P<0.0001). About 27% of genes with promoter region methylation escape transcriptional repression and are actively expressed. (b) Methylation peak density normalized to tiled regions (peak/Mb) negatively correlates with chromosomal length (r2 = 0.34).
We carried out functional analysis of genes that are methylated but expressed in CD4+ T cells and in genes that are methylated and non-expressed using Ingenuity Pathway Analysis software (Ingenuity Systems, Redwood City, CA, USA). Interestingly, the top functional networks identified in genes that are expressed showed involvement in basic cell functions, such as cellular signaling and interaction, and cellular growth and proliferation (Figure 3a). The non-expressed genes showed functional association with antigen presentation, cell-mediated immune response and humoral immune response (Figure 3b). This suggests that the methylation in non-expressed genes is functionally relevant and that those genes that are involved in immune functions are non-expressed in primary CD4+ T cells and can demethylate and become transcriptionally active once T cells are activated and differentiated.
Figure 3.
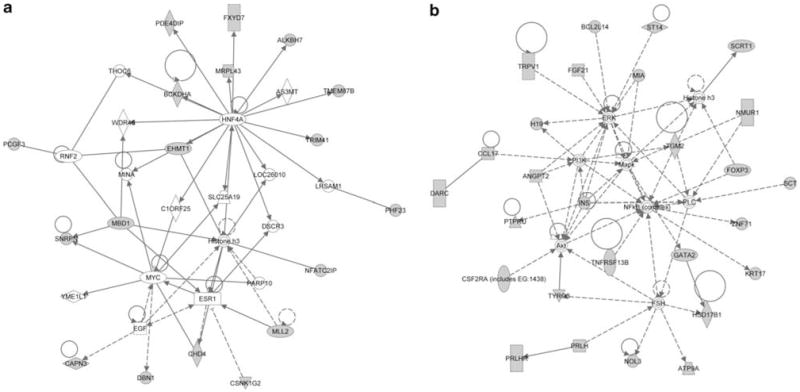
Functional network analysis of methylated genes that are expressed in primary human CD4+ T cells identified involvement in basic cell functions, including cellular signaling and interaction, and cellular growth and proliferation (a). Genes that are methylated and non-expressed are functionally associated with antigen presentation, cell-mediated immune response and humoral immune response (b). Gene key: solid lines indicate direct interaction, dotted lines indicate indirect interaction, an arrow from a to b indicates that a acts on b, a line without an arrowhead indicates binding only and a line with a small vertical line at the end from a to b indicates a inhibits b. Gray indicates genes that are methylated, white indicates genes that are not user specified but incorporated into the network through relationships with other genes. Node shapes are: square, cytokine; diamond (vertical), enzyme; diamond (horizontal), peptidase; dotted rectangular (vertical), ion channel; solid rectangular (vertical), G-protein-coupled receptor; triangle, kinase; oval (horizontal), transcription regulator; oval (vertical), transmembrane receptor; trapezoid, transporter; circle, other.
We next analyzed DNase I hypersensitive (HS) sites in primary human CD4+ T cells, as a marker for chromatin accessibility. This revealed that TSS is located within a DNase I HS site in the majority of methylated genes that are expressed but not in non-expressed genes (85.5 versus 27.9%, χ2 = 52.45, P-value <0.0001). This further indicates that DNA methylation is functionally relevant in inducing chromatin inaccessibility at the TSS and transcriptional repression in the latter but not former group of methylated genes.
Out of all the 27 458 CpG islands examined within the 22 autosomal chromosomes and the X chromosome, we found that only 1375±65 (mean±s.e.m.) CpG islands (5% of islands) include methylation peaks in our samples. This indicates that the vast majority of CpG islands are unmethylated in CD4+ T cells. This is consistent with recent studies in other cell types.11,12
The regions in the HOXA gene cluster (Chr7: 26 924 046–27 424 045), the H19/IGF2/KCNQ1 cluster (Chr11: 1 699 992–3 143 916) and the IGF2R gene region (Chr6: 160 309 320–160 447 571) were entirely tiled in our arrays. Data from these regions (~2 Mb titled region) allowed unbiased examination of the DNA methylation pattern both in CpG islands and in surrounding genetic regions included within these sequences. We found that most methylation peaks are located outside the CpG islands. Indeed, in genetic loci close to CpG islands, methylation peaks tend to occur at CpG ‘shores’, just outside the boundaries of CpG islands. This observation has been recently reported in other human tissues.13
We found a higher density of methylation peaks in the subtelomeric regions compared with the rest of the chromosomes in CD4+ T cells. The mean methylation peak density in the tiled region within the subtelomeric regions (7 Mb regions from the telomeres) was significantly higher compared with the non-subtelomeric regions in all chromosomes combined (P<0.0001). This enrichment toward the telomeres is not explained by tiling as methylation peaks densities were normalized for the size of the region tiled, and this phenomenon has been recently reported by others.11,14 Enrichment of methylation peaks in the subtelomeric regions was evident and statistically significant in all chromosomes except for chromosome 19 (Table 1). Unexpectedly, we also found a negative correlation between chromosomal lengths and the density of methylation peaks observed within the tiled regions in our samples (r2 = 0.34, P = 0.0035) (Figure 2b). This is not explained by gene density as there is a positive correlation between the number of genes on each chromosome and chromosomal length (r2 = 0.35, P = 0.003).
Table 1.
Methylation peak density (peak/Mb) in subtelomeric (up to 7 Mb from each telomere) and non-subtelomeric regions in each chromosome
| Chromosome | Methylation peak density (peak/Mb) |
P-value | |||
|---|---|---|---|---|---|
| Subtelomeric |
Non-subtelomeric |
||||
| Mean | s.e.m. | Mean | s.e.m. | ||
| 1 | 101.5 | 6.2 | 44.3 | 3.4 | 1.75E-06 |
| 2 | 175.1 | 8.3 | 45.6 | 3.4 | 6.83E-09 |
| 3 | 62.1 | 9.0 | 31.4 | 1.6 | 0.002106 |
| 4 | 142.3 | 4.4 | 25.3 | 2.7 | 8.37E-11 |
| 5 | 120.4 | 5.8 | 28.8 | 2.5 | 6.89E-09 |
| 6 | 146.3 | 10.0 | 36.1 | 4.1 | 2.04E-07 |
| 7 | 188.9 | 9.2 | 46.7 | 4.0 | 8.62E-09 |
| 8 | 167.1 | 9.1 | 35.5 | 0.8 | 6.84E-09 |
| 9 | 101.2 | 7.1 | 63.9 | 5.1 | 0.000393 |
| 10 | 189.9 | 10.0 | 50.2 | 5.4 | 3.39E-08 |
| 11 | 121.6 | 10.2 | 39.2 | 2.9 | 2.55E-06 |
| 12 | 116.7 | 7.6 | 35.5 | 2.4 | 2.06E-07 |
| 13 | 270.6 | 15.2 | 39.2 | 3.7 | 5.51E-09 |
| 14 | 87.1 | 6.5 | 38.6 | 2.8 | 7.93E-06 |
| 15 | 65.9 | 4.4 | 38.2 | 2.4 | 4.86E-05 |
| 16 | 149.0 | 6.6 | 92.0 | 6.8 | 2.47E-05 |
| 17 | 113.1 | 10.3 | 55.6 | 3.3 | 6.66E-05 |
| 18 | 127.3 | 11.8 | 52.0 | 5.3 | 3.10E-05 |
| 19 | 111.7 | 8.7 | 89.8 | 9.7 | 0.066 |
| 20 | 106.1 | 7.4 | 43.0 | 4.1 | 3.81E-06 |
| 21 | 164.6 | 6.5 | 74.6 | 10.0 | 3.35E-06 |
| 22 | 160.5 | 12.5 | 76.4 | 4.1 | 1.45E-05 |
| X | 132.8 | 9.9 | 69.0 | 7.3 | 8.47E-05 |
Discussion
DNA methylation is largely a transcriptionally repressive epigenetic mark that induces gene silencing and chromatin inaccessibility.15,16 DNA methylation induces chromatin inaccessibility and transcriptional repression by several mechanisms. These include the recruitment of members of the methylcytosine binding domain-containing proteins, such as MECP2, which is turn recruit histone deacetylases that result in chromatin condensation.15,16 In addition, the bulky methyl group on methylcytosine residues can prevent the binding of transcription factors to the promoter sequences of methylated genes.17 Using DNA methylation profiling in primary human CD4+ T cells, we shed light on transcriptionally permissive DNA methylation marks and find that ~27% of methylated genes in primary human CD4+ T cells are expressed. We find that most CpG islands are not methylated in CD4+ T cells, consistent with published work in a number of genetic regions in multiple cell types.18,19
We report two characteristics that distinguish transcriptionally repressive from permissive DNA methylation peaks in promoter gene sequences. A distance between TSS and the center of methylation peaks of an average of 449 bp further upstream from the TSS prevents DNA methylation from inducing chromatin inaccessibility and transcriptional silencing. Our data suggest that methylation peaks that are transcriptionally repressive are on average about six nucleosomes away from the TSS. Methylation peaks located on average about nine nucleosomes away were not functionally repressive. In addition, CpG islands in the promoter regions of genes that harbor a permissive DNA methylation peak are characterized by an increased maximal CG dinucleotide density compared with repressive peaks (Figure 4). Furthermore, we demonstrate that the majority of genes with a TSS located downstream of permissive DNA methylation peaks are sensitive to DNase I digestion at the TSS, as indicated by the presence of a DNase I HS site, indicating that chromatin is accessible and available for transcription machinery binding.
Figure 4.

A schematic representation showing distinguishing features between repressive and permissive DNA methylation. (a) Repressive methylation peaks are on average 893±110 bp upstream of the transcription start site of target genes, which are characterized by a relatively lower maximum CpG island density. Chromatin is closed at the transcription start site as indicated by the absence of DNase I HS sites. (b) Permissive methylation peaks are further upstream from transcription start site compared with repressive peaks, and are characterized by higher maximum CpG island densities within promoter sequences of target genes. These methylation peaks fail to maintain a closed chromatin configuration. This results in accessible chromatin at the transcription start sites of target genes as evidenced by the presence of DNase I HS sites and gene expression. It is possible that these methylation peaks fail to efficiently recruit transcriptional repressor complexes, such as the MECP2–SIN3A–HDAC complex. MECP2, methyl-CpG-binding protein 2; SIN3A, SIN3 homolog A; HDAC, histone deacetylase.
We also observe that transcriptionally permissive DNA methylation peaks correspond to genes involved in cell signaling, growth and proliferation, whereas repressive peaks are more closely associated with genes associated with immune response and T-cell differentiation. Modulation of repressive DNA methylation patterns at key regulatory loci has a critical role in lineage differentiation of certain T helper subsets. For example, hypomethylation of a single region in the FOXP3 locus reveals Treg cell lineage commitment and is closely related to the longevity of suppressor function.20,21
Our data indicate that proximity of methylation is directly related to the potential for methylation-dependent nucleosome remodeling and repression. We suggest a model in which failure of DNA methylation to induce inaccessible chromatin configuration is related to both the distance of the methylation peaks from TSS and perhaps reduced efficiency to recruit transcription repressor complexes, such as MECP2-SIN3A-HDAC, as a result of high CpG island density, leading to an open chromatin configuration and availability for transcription.
Materials and methods
CD4+ T cell isolation and DNA extraction
Peripheral blood mononuclear cells were isolated from normal healthy donor blood samples by density gradient centrifugation (Amersham Biosciences, Uppsala, Sweden). CD4+ T cells were then isolated through magnetic bead separation using direct labeling (Miltenyi Biotec, Auburn, CA, USA) following the manufacturer’s protocol. DNA was extracted using the DNeasy Kit (Qiagen, Valencia, CA, USA). The DNA concentration was then determined using a NanoDrop spectrophotometer (Thermo Sci., Wilmington, DE, USA). Our studies were carried out using ten biological replicates from ten normal healthy women. All participants signed an informed consent. Our studies are approved by our institutional review boards.
DNA IP and array hybridization
Genomic CD4+ T-cell DNA was digested with MseI and purified using Qiagen Quick PCR Purification kit. Next, we diluted 1.25 μg of MseI-digested DNA to a final volume of 300 μl in TE buffer (pH7.5), heat denatured for 10 min at 95 °C and then immediately cooled on ice for 5 min. An aliquot of 60 μl was then removed and stored at −20 °C as the input DNA. IP buffer (60 μl at 5 ×) was then added to the remaining 240 μl of the digested and denatured DNA samples. Anti-5-methylcytidine antibody (Abcam, Cambridge, MA, USA) was added and the samples were incubated overnight with agitation at 4 °C. Pre-washed protein A agarose beads were then added and the samples were incubated at 4 °C with agitation for 3.5–4 h to obtain DNA–antibody–bead complex. This reaction mix was then spun down at 6000 r.p.m. for 2 min at 4 °C and the pellet washed three times with 1 ml 1 × IP buffer with 5 min incubations with agitation in between centrifugation steps. The beads were then resuspended in 250 μl of digestion buffer and 7 μl of proteinase K (10 mg ml−1) was added and incubated overnight at 55 °C with agitation. IP DNA was then purified using ethanol precipitation. We then performed whole genome amplification (WGA2 kit, Sigma-Aldrich, St Louis, MO, USA) of input and IP DNA. Input and IP DNA from each participant were then labeled with Cy3 and Cy5, respectively, pooled, denatured and then co-hybridized to 385K methylation arrays with tiling that covers all UCSC-annotated CpG islands and promoter regions for all RefSeq genes (NimbleGen).
Bisulfite DNA sequencing
CD4+ T-cell DNA from an independent set of normal healthy controls was isolated and treated with sodium bisulfite using the EZ DNA Methylation-Gold kit (Zymo Research, Orange, CA, USA). Sodium bisulfite treatment will convert unmethylated cytosine residues to thymine, whereas methylated cytosine residues will remain as cytosines. Sodium bisulfite treated DNA was amplified and directly sequenced (primer sequences are available upon request). Percent methylation on each CG site was quantified using the Epigenetic Sequencing Methylation analysis software (ESME).22
Statistical analysis and methylation peaks identification
Signal intensity data were obtained and analyzed from each scanned array by NimbleGen. The ratio of IP versus input DNA signals in each probe from each co-hybridized sample is determined and the log2-ratio is computed and scaled to center the ratio data around zero. Scaling is carried out by subtracting the bi-weight mean for the log2-ratio values for all features on the array from each log2-ratio value (complete scaled log2-ratio data for all probes and all samples are available in online Supplementary file 1). The −log10 P-values are calculated for each probe by placing a fixed-length 750 bp window around each consecutive probe, and the one-sided Kolmogorov–Smirnov test is used to determine whether the probes are drawn from a significantly more positive distribution of intensity log2-ratios compared to those in the rest of the array. The resulting score for each probe is the −log10 P-value from the windowed Kolmogorov–Smirnov test around that probe. Methylation peaks are defined as regions with at least two probes with −log10 P-values of at least 2 within 500 bp window, and a methylation score of at least 2. Peaks that are within 500 bp apart are merged. The methylation score for each peak represents the average −log10 P-value for the probes within that methylation peak. Complete methylation peaks data for the autosomal chromosomes and chromosome X are presented in online Supplementary file 2.
Microarray CD4+ T-cell expression and DNase I hypersensitivity data
Gene expression profile for human CD4+ T cells obtained from 10 normal healthy participants was extracted from Gene Expression Omnibus (GEO accession: GSE4588). Expression profiling was performed using the Human Genome U133 Plus 2.0 Array (Affymetrix, Santa Clara, CA, USA). Genes were considered expressed if the mean normalized signal value in all 10 samples for at least 1 transcript was above 64. DNase I HS sites in normal human CD4+ T cells were extracted from the UCSC Genome Browser and Tables.23
Bioinformatic analysis
The number and density of CpG islands were determined algorithmically using the NCBI Build 36.1 of the human genome (HG18). CpG islands were defined as a stretch of DNA of at least 200 bp with a C + G content of at least 50% and an observed/expected CG dinucleotide frequency of at least 0.6.
Acknowledgments
This work was supported by the National Institute of Health Grant number P20-RR015577 from the National Center for Research Resources, Grant number R03AI076729 from the National Institute of Allergy and Infectious Diseases, the Oklahoma Rheumatic Disease Research Core Centers; the Department of Veterans Affairs; the University of Oklahoma Health Sciences Center; the Oklahoma Medical Research Foundation. The authors thank Dr J Donald Capra, MD, for his critical review of this paper.
Footnotes
Conflict of interest
The authors declare no conflict of interest.
Supplementary Information accompanies the paper on Genes and Immunity website (http://www.nature.com/gene)
References
- 1.Reik W, Dean W, Walter J. Epigenetic reprogramming in mammalian development. Science (New York, NY) 2001;293:1089–1093. doi: 10.1126/science.1063443. [DOI] [PubMed] [Google Scholar]
- 2.Li E, Beard C, Jaenisch R. Role for DNA methylation in genomic imprinting. Nature. 1993;366:362–365. doi: 10.1038/366362a0. [DOI] [PubMed] [Google Scholar]
- 3.Mohandas T, Sparkes RS, Shapiro LJ. Reactivation of an inactive human X chromosome: evidence for X inactivation by DNA methylation. Science (New York, NY) 1981;211:393–396. doi: 10.1126/science.6164095. [DOI] [PubMed] [Google Scholar]
- 4.Bird AP. Functions for DNA methylation in vertebrates. Cold Spring Harb Symp Quant Biol. 1993;58:281–285. doi: 10.1101/sqb.1993.058.01.033. [DOI] [PubMed] [Google Scholar]
- 5.Hansen RS, Wijmenga C, Luo P, Stanek AM, Canfield TK, Weemaes CM, et al. The DNMT3B DNA methyltransferase gene is mutated in the ICF immunodeficiency syndrome. Proc Natl Acad Sci USA. 1999;96:14412–14417. doi: 10.1073/pnas.96.25.14412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ballestar E, Esteller M. SnapShot: the human DNA methylome in health and disease. Cell. 2008;135:1144–1144. e1141. doi: 10.1016/j.cell.2008.11.040. [DOI] [PubMed] [Google Scholar]
- 7.Pan Y, Sawalha AH. Epigenetic regulation and the pathogenesis of systemic lupus erythematosus. Transl Res. 2009;153:4–10. doi: 10.1016/j.trsl.2008.10.007. [DOI] [PubMed] [Google Scholar]
- 8.Sawalha AH. Epigenetics and T-cell immunity. Autoimmunity. 2008;41:245–252. doi: 10.1080/08916930802024145. [DOI] [PubMed] [Google Scholar]
- 9.Sawalha AH, Jeffries M, Webb R, Lu Q, Gorelik G, Ray D, et al. Defective T-cell ERK signaling induces interferon-regulated gene expression and overexpression of methylation-sensitive genes similar to lupus patients. Genes Immun. 2008;9:368–378. doi: 10.1038/gene.2008.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sawalha AH, Jeffries M. Defective DNA methylation and CD70 overexpression in CD4+ T cells in MRL/lpr lupusprone mice. Eur J Immunol. 2007;37:1407–1413. doi: 10.1002/eji.200636872. [DOI] [PubMed] [Google Scholar]
- 11.Rauch TA, Wu X, Zhong X, Riggs AD, Pfeifer GP. A human B cell methylome at 100-base pair resolution. Proc Natl Acad Sci USA. 2009;106:671–678. doi: 10.1073/pnas.0812399106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shen L, Kondo Y, Guo Y, Zhang J, Zhang L, Ahmed S, et al. Genome-wide profiling of DNA methylation reveals a class of normally methylated CpG island promoters. PLoS Genet. 2007;3:2023–2036. doi: 10.1371/journal.pgen.0030181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Irizarry RA, Ladd-Acosta C, Wen B, Wu Z, Montano C, Onyango P, et al. The human colon cancer methylome shows similar hypo- and hypermethylation at conserved tissue-specific CpG island shores. Nat Genet. 2009;41:178–186. doi: 10.1038/ng.298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lister R, Pelizzola M, Dowen RH, Hawkins RD, Hon G, Tonti-Filippini J, et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature. 2009;462:315–322. doi: 10.1038/nature08514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nan X, Ng HH, Johnson CA, Laherty CD, Turner BM, Eisenman RN, et al. Transcriptional repression by the methyl-CpG-binding protein MeCP2 involves a histone deacetylase complex. Nature. 1998;393:386–389. doi: 10.1038/30764. [DOI] [PubMed] [Google Scholar]
- 16.Jones PL, Veenstra GJ, Wade PA, Vermaak D, Kass SU, Landsberger N, et al. Methylated DNA and MeCP2 recruit histone deacetylase to repress transcription. Nat Genet. 1998;19:187–191. doi: 10.1038/561. [DOI] [PubMed] [Google Scholar]
- 17.Bird AP, Wolffe AP. Methylation-induced repression—belts, braces, and chromatin. Cell. 1999;99:451–454. doi: 10.1016/s0092-8674(00)81532-9. [DOI] [PubMed] [Google Scholar]
- 18.Eckhardt F, Lewin J, Cortese R, Rakyan VK, Attwood J, Burger M, et al. DNA methylation profiling of human chromosomes 6, 20 and 22. Nat Genet. 2006;38:1378–1385. doi: 10.1038/ng1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Weber M, Hellmann I, Stadler MB, Ramos L, Paabo S, Rebhan M, et al. Distribution, silencing potential and evolutionary impact of promoter DNA methylation in the human genome. Nat Genet. 2007;39:457–466. doi: 10.1038/ng1990. [DOI] [PubMed] [Google Scholar]
- 20.Floess S, Freyer J, Siewert C, Baron U, Olek S, Polansky J, et al. Epigenetic control of the foxp3 locus in regulatory T cells. PLoS Biol. 2007;5:e38. doi: 10.1371/journal.pbio.0050038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Janson PC, Winerdal ME, Marits P, Thorn M, Ohlsson R, Winqvist O. FOXP3 promoter demethylation reveals the committed Treg population in humans. PLoS One. 2008;3:e1612. doi: 10.1371/journal.pone.0001612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lewin J, Schmitt AO, Adorjan P, Hildmann T, Piepenbrock C. Quantitative DNA methylation analysis based on four-dye trace data from direct sequencing of PCR amplificates. Bioinformatics. 2004;20:3005–3012. doi: 10.1093/bioinformatics/bth346. [DOI] [PubMed] [Google Scholar]
- 23.Boyle AP, Davis S, Shulha HP, Meltzer P, Margulies EH, Weng Z, et al. High-resolution mapping and characterization of open chromatin across the genome. Cell. 2008;132:311–322. doi: 10.1016/j.cell.2007.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]