

Abstract
We report a rhodium catalyst that exhibits high reactivity for the hydroamination of primary aminoalkenes that are unbiased toward cyclization and that possess functional groups that would not be tolerated in hydroaminations catalyzed by more electrophilic systems. This catalyst contains an unusual diaminophosphine ligand that binds to rhodium in a κ3-P,O,P mode. The reactions catalyzed by this complex typically proceed at mild temperatures (room temperature to 70 °C), occur with primary aminoalkenes lacking substituents on the alkyl chain that bias the system toward cyclization, occur with primary aminoalkenes containing chloride, ester, ether, enolizable ketone, nitrile, and unprotected alcohol functionality, and occur with primary aminoalkenes containing internal olefins. Mechanistic data imply that these reactions occur with a turnover-limiting step that is different from that of reactions catalyzed by late transition metal complexes of Pd, Pt, and Ir. This change in the turnover-limiting step and resulting high activity of the catalyst stem from favorable relative rates for protonolysis of the M-C bond to release the hydroamination product vs reversion of the aminoalkyl intermediate to regenerate the acyclic precursor. Probes for the origin of the reactivity of the rhodium complex of L1 imply that the aminophosphine groups lead to these favorable rates by effects beyond steric demands and simple electron donation to the metal center.
Introduction
The hydroamination of unactivated alkenes is a conceptually simple method for the preparation of alkylamines.1-5 The intramolecular version of this process is an attractive route to nitrogen-containing heterocycles, a common structural motif in biologically active molecules,6 from readily available aminoalkenes. Due to the utility of this reaction in academic, pharmaceutical, and industrial settings, much effort has been spent on developing catalysts to effect this transformation.
Specifically, complexes based on rare-earth,7-10 alkali-earth,11,12 and Group IV13-19 metals are known to catalyze the intramolecular hydroamination of primary and secondary aminoalkenes; however, these catalysts are often highly air and moisture sensitive and incompatible with many functional groups. For example, aminoalkenes containing acidic protons, such as unprotected alcohols, and carbonyl functionality, such as ketones and esters, have not been reported to undergo hydroamination with the highly basic rare-earth, and alkali-earth metal catalysts.7,13,20 In a similar vein, group IV metal catalysts have not catalyzed hydroaminations of aminoalkenes containing carbonyl or ether functionalities. The group IV catalyst that reacts with the broadest scope was reported recently by Schafer and is based on a tethered ureate ligand.19 Even this catalyst, however, has been shown to tolerate a limited range of functionality (an aromatic dioxolane, an N-alkyl pyrrole, and a tertiary aromatic amine), and substrates containing functional groups are all secondary amines with a bias toward cyclization. All primary aminoalkenes that react with this catalyst have lacked additional functionality, and all reported reactions of primary aminoalkenes lacking gem-diphenyl substituents with this catalyst were conducted at 145 °C.
Catalysts derived from zinc complexes21,22 and Bronsted acids23-26 have also been shown to catalyze intramolecular hydroamination with a limited range of reactivities. For example, the reaction of a secondary aminoalkene lacking gem-substitution in the presence of a zinc catalyst derived from diethylzinc and a protic additive required 21 days at 180 °C. Primary aminoalkenes containing gem-disubstitution of the alkyl linker required 120 °C, and a single example of a substrate lacking gem-disubstitution on the alkyl chain (2-amino-hex-5-ene) gave the product in only 19% conversion after 36 h.21 Hydroaminations catalyzed by Brønsted acids required high temperatures or protection of the nitrogen with an electron-withdrawing group.24-26
Complexes of late-transition metals are more stable to air and moisture and are more tolerant of polar functional groups than complexes containing early transition metals and lanthanides.27 However, intramolecular hydroaminations catalyzed by late transition metal complexes often require an N-H donor in which the nitrogen is electron-deficient and contained in an amide, carbamate or sulfonamide functionality.26,28-31 In the last five years, complexes of platinum,32,33 copper,34 rhodium,35-37 and iridium38-40 have been shown to catalyze the intermolecular hydroamination of aminoalkenes containing more basic, secondary alkylamines. In 2008, the author's group reported a cationic rhodium complex that catalyzed the cyclization of both secondary and primary aminoalkenes.35
Although data with this original catalyst35 and that with catalysts based on copper,34 rhodium,37 and iridium40,46 reported since this time demonstrated that primary aminoalkenes can undergo cyclization, reactions of such such amines catalyzed by these systems have significant limitations. First, the cyclization of primary aminoalkenes catalyzed by late-metal systems has been limited to cases that are biased toward cyclization by a Thorpe-Ingold effect; second, these reactions were limited to aminoalkenes containing terminal olefins; third, these reactions required high temperatures (>100 °C); and fourth, reactions of primary aminolkenes containing a second functional group have not been reported with any catalyst. Thus, the identification of a complex containing a late transition metal that catalyzes the cyclization of unactivated, unbiased aminoalkenes, as well as an understanding of the factors controlling the reactions of primary amines catalyzed by late transition metal complexes, is needed.26,41,42
We previously investigated the cyclization of a secondary aminoalkene catalyzed by a series of rhodium complexes containing bisphosphine ligands.35 One example of the cyclization of a secondary aminoalkene catalyzed by the combination of bis(diethylamino)xantphos ligand L1’43 (Table 1) and a cationic rhodium precursor was included in this study. Complexes of aminophosphines have been investigated rarely as components of catalysts, let alone for hydroamination.44 Thus, we investigated the structure of this catalyst and its reactivity toward a broader range of aminoalkenes.
Table 1.
Rh-Catalyzed Hydroamination of Primary Aminoalkenes: Initial Results
 | ||||||||
|---|---|---|---|---|---|---|---|---|
| entry | R | mol % [Rh] | ligand | solvent | time (h) | conversion (%)a | ratioa2:3:4:5 | Isolated yield |
| 1 | Ph | 1 | L1 | tBuOH | 2 | 100 | 18:1:1:0.7 | 80% |
| 2 | Ph | 1 | L1’ | tBuOH | 2 | 100 | 11:1:1:0.2 | |
| 3 | Ph | 1 | L1 | THF | 2 | 100 | 14:1:1:0.4 | 71% |
| 4 | H | 3 | L1 | tBuOH | 15 | 100 | 9:1:1:0.5 | 66%c |
| 5 | H | 3 | L1’ | tBuOH | 15 | 100 | 12:1:1:0.5 | |
| 6 | H | 3 | L1 | THF | 15 | 77 | 6:1:0.6:1.6 | |
Determined by 1H NMR spectroscopy.
b A trace amount of internal alkene from isomerization observed.
A 1.3 : 1 ratio of diastereomers was obtained.
Here we report a full account of our findings that reveal the high reactivity of this unusual metal-ligand system for the hydroamination of primary amines, along with detailed mechanistic studies that reveal the origins of the high reactivity of this system. These new studies show that the active catalyst posseses a rhodium POP-pincer structure and is highly active for the hydroamination of primary amines. The reactions catalyzed by this complex typically proceed at mild temperatures (room temperature to 70 °C), occur with primary aminoalkenes lacking any substituents on the alkyl chain that would bias the system toward cyclization, and occur with a high degree of tolerance for polar functional groups, including aryl chlorides, esters, ethers, enolizable ketones, nitriles, and unprotected alcohols. Initial results imply that the aminophosphine groups on the ligand are involved in creating the fast rates and high selectivity, and mechanistic data show that the reactions of primary amines catalyzed by these complexes occur with a turnover-limiting step that is different from that of secondary aminoalkenes catalyzed by complexes of Pd,45 Pt,32 and Ir.46
Results and Discussion
Our prior observation that bis(diethylamino)xantphos L1' and Rh(COD)2BF4 catalyzed the cyclization of a secondary aminoalkene, the novelty of a diaminophosphine as a component for catalysis, the potential flexibility and ease of synthesis of this class of ligand, and the possibility that this catalyst combination could lead to more hydroaminations than just the cyclization of secondary amines with gem-disubstitution led us to explore the reactivity of this catalyst for aminoalkenes that have resisted cyclization with previous late transition metal catalysts. To do so, we explored the activity of the catalyst generated from L1 and cationic rhodium catalyst precursors for reactions of primary aminoalkenes possessing and lacking gem-disubstitution on the alkyl linker. We found that just 1 mol% of the complex generated from [Rh(CH3CN)2COD]BF4 and L1 catalyzed the hydroamination of primary aminoalkene 1a containing 3,3-diphenyl substituents in THF at 70 °C to form the pyrrolidine product 2a with high selectivity (14:1:1:0) for formation of the cyclic secondary amine product over the combination of imine product 3a and hydrogenation product 4a (Table 1, entry 1) Pure amine was isolated in 71% yield. The pairwise formation of the imine and hydrogenation products would occur by oxidative amination and transfer of the hydrogen to the olefin unit of the starting aminoalkene. The internal alkene 5a, resulting from isomerization of the terminal olefin in the starting material, was observed in only trace amounts. Based on the potential that proton transfer is the turnover-limiting step of the catalytic cycle (vide infra), we also studied reactions in alcohol solvents. The same reaction of 1a occurred with higher selectivity and higher isolated yield in tert-butanol (Table 1, entry 3).
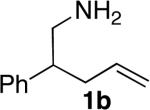
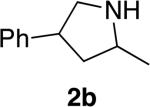
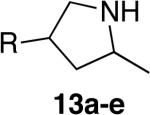
Encouraged by these initial results, we investigated reactions of primary aminoalkenes that have not undergone cyclization with previous late transition metal catalysts. Initial studies conducted on the hydroamination of aminoalkene 1b lacking gem-disubstitution on the alkyl linker in the presence of catalytic amounts of [Rh(CH3CN)2COD]BF4 and L1 showed that the rate and selectivity of reactions in ethereal solvents were significantly lower than those of the gem-disubstituted substrate 1a (Table 1, compare entries 3 and 6). However, the same reaction in tBuOH led to the first cyclization of an unbiased primary amine with a late metal catalyst with acceptable rates and high selectivity (9:1 amine:imine) for the desired amine product (entry 4). The pyrrolidine product 2b was isolated from this reaction in 66% yield as a 1.3:1 mixture of diastereomers (see also Table 2, entry 2).
Table 2.
Scope of Rh-Catalyzed Hydroamination of Primary Aminesa
| entry | mol % [Rh] | aminoalkene | product | time (h) | yieldb (dr)c | ratio (amine:imine)c | |
|---|---|---|---|---|---|---|---|
| 1 | 3 |
|
|
15 | 66% (dr = 1.3:1) | 9:1 | |
| 2d | 4 |
|
|
7 | a; n=1; 76%e | >95:5 | |
| 3d | 4 | 8 | b; n=2; 77% | >95:5 | |||
| 4f,g | 18 |
|
|
15 | 40% | >95:5 | |
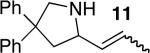
| 5f,h | 3 |
|
|
14 | 64%i | 7:1 | |
| 6j | 3 |
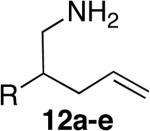
|
a; R=OTBDPS |
|
18 | 71% (dr = 1.6:1) | >95:5 |
| 7 | 3 | b; R=4-(CO2Me)C6H4 | 21 | 64% (dr = 1.6:1) | 10:1 | ||
| 8 | 5 | c; R=4-(CH2CN)C6H4 | 12 | 79% (dr = 1.5:1) | >95:5 | ||
| 9 | 5 | d; R=4-(CH3(O)C)C6H4 | 16 | 61% (dr = 1.5:1) | 10:1 | ||
| 10 | 5 | e; R=4-(CH2OH)C6H4 | 8 | 56%k (dr = 1.1:1) | 7:1 | ||
|
|
||||||
| 11 | 1 | a; R = H | a; R = H | 5 | 87% | >95:5 | |
| 12 | 2 | b; R = Cl | b; R = Cl | 6 | 84% | >95:5 | |
| 13 | 1 |
|
|
7 | 74% dr = (1.1:1) | >95:5 |
Reaction conditions: 0.5 mmol aminoalkene, [Rh(CH3CN)2COD]BF4 (1-18 mol %), and L1 (1.2-19 mol %) in 1 mL of tBuOH at 70 °C for unless otherwise specified.
Isolated yield.
Ratio determned by 1H NMR spectroscopy.
Product isolated as Boc carbamate.
NMR yield; the product was isolated in 67% yield as an 10:1:1 mixture of 7a-Boc:BocNEt2:N-Boc-1-amino-3-pentene
Reaction run at 100 °C in dioxane.
3.3:1 mixutre of E:Z isomers.
10:1 mixture of E:Z isomers.
Isolated as a 1.8:1 mixture of olefin isomers (internal:terminal).
Reaction run on a 0.25 mmol scale.
Isolated as a 97:3 mixture of alcohol:aldehyde products.
Control experiments showed that the tBu groups on the xanthene backbone had little effect on the reactivity or selectivity of the catalyst (Table 1, compare entries 1 and 2 and entries 4 and 5). Thus, L1 (R = tBu) or L1’ (R = H) can be used interchangeably in catalytic hydroaminations. During the latter course of this work, L1’ became available commercially, but both ligands are prepared by a similar one-pot metalation of 4,5-dibromo-2,7-di-t-butyl-9,9-dimethylxanthene or 9,9-dimethylxanthene and quenching with bis(diethylamino)chlorophosphine.47
1. Scope of the Intramolecular Hydroamination
1.1. Cyclization of unbiased aminoalkenes and internal alkenes
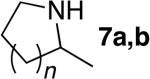
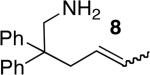
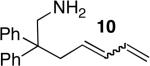
The scope of the hydroamination of primary amino alkenes that lack gem-disubstitution on the alkyl chain or that contain internal olefins or dienes catalyzed by the combination of [Rh(CH3CN)2COD]BF4 and L1 is illustrated in Table 2. As noted in the previous section, the reaction of substrate 1b containing just one substituent on the carbon beta to the amino group and alkene unit (Table 2 entry 1) gave a 9:1 ratio of amine to imine and led to isolation of the amine in 66% yield. The high activity of this catalyst system for reactions of primary aminoalkenes in tBuOH solvent was particularly exemplified by reactions with aminoalkenes lacking any substituents on the linker between the amino group and the alkene. Reactions of these aminoalkenes occurred to generate both 5- and 6-membered ring products. For example, 1-aminopentene and 1-aminohexene underwent cyclization in the presence of 4 mol % [Rh(CH3CN)2COD]BF4 and L1 to afford 2-methylpyrrolidine 7a and 2-methylpiperidine 7b (Table 2, entries 2 and 3). Internal olefins also underwent cyclization. In these cases, the solvent had little effect on the conversion or selectivity of hydroamination, and intramolecular additions to monoene 8 and the diene 10 (entries 4 and 5) were accomplished in dioxane at 100 °C. A higher catalyst loading (18 mol %) was required to achieve high conversion of the unactivated internal alkene 8 due to competitive catalyst decomposition, but these reactions constitute the first examples of cyclization of a primary amine with an internal alkene catalyzed by a complex of a late transition metal.
Free diethylamine was detected in the crude reaction mixtures by GC and 1H NMR spectroscopy, and this observation suggests that one pathway for catalyst decomposition occurs by displacement of the diethylamino groups on the phosphorus by the primary amine substrate. For substrates with slower rates of hydroamination (i.e unactivated internal olefins), the rate of catalyst decomposition becomes competitive with the rate of cyclization.
1.2. Tolerance for auxiliary functional groups
The objective of developing a late transition metal catalyst for hydroamination stems from the expectation that hydroamination with this type of catalyst will occur with a high tolerance for auxiliary functional groups. To determine if this assertion was valid, we studied cyclizations of substrates that are both unbiased for cyclization and possessing potentially reactive functional groups. Indeed, such cyclizations catalyzed by the combination of [Rh(CH3CN)2COD]BF4 and L1 occurred with aminoalkenes containing a siloxy group (entry 6), a methoxycarbonyl group (entry 7), a cyano group (entry 8), an acetyl group (entry 9) and even an unprotected hydroxyl group (entry 10). Cyclization of the aminoalkene containing the unprotected alcohol (12e) occurred with a small amount of competitive formation of an aldehyde byproduct at high conversions, but the hydroxyl-substituted product 13e was isolated in 56% yield. Cyclizations of substrates containing esters, ketones, and alcohols have not been reported with catalysts based on lanthanides or group IV metals.
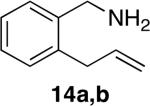
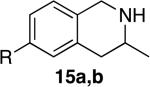
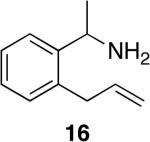
Tetrahydroisoquinolines are a common structural motif in numerous biologically active molecules.48,49 3-Methyl-tetrahydroisoquinolines were readily synthesized by hydroamination of 2-allylbenzylamines in excellent yield and selectivity (entries 11-13) with low catalyst loadings (1-2 mol %). The hydroamination of the parent 2-allylbenzylamine 14a even occurred at room temperature (10 mol % Rh, THF, 24 h) to afford the product in 77% isolated yield. Although reactions of a broad range of functionalized 2-allylbenzylamines were not explored, the aryl chloride function was tolerated, as demonstrated by the high-yielding cyclization of substrate 14b (entry 13). In addition, reactions of aminoalkenes that are branched at the α position have not occurred previously with catalysts of the late transition metals,50 but the α-branched (entry 14) aminoalkene 16 readily formed the tetrahydroisoquinoline product in the presence of catalytic amounts of [Rh(CH3CN)2COD]BF4 and L1.
1.3. Reactions of secondary aminoalkenes
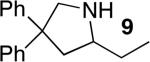
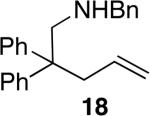
The reactivity of this catalyst was not limited to primary amines. Secondary amines also underwent hydroamination in good to excellent yields with low catalyst loadings (1-3 mol %) to furnish N-alkyl pyrrolidines and N-alkyl tetrahydroisoquinolines (Table 3). The commonly studied aminoalkene 18 containing gem-diphenyl substituents cyclized in high yield with a high ratio of amine to imine. However, secondary aminoalkene 20 containing no substituents in the linking unit also underwent cyclization with a high ratio of amine to imine to give 65% yield of isolated pyrrolidine product. Even the sterically encumbered N-iPr-substituted aminoalkene 22a readily underwent cyclization (entry 3). Finally, hydroamination with the secondary aminoalkene 22b occurred at room temperature (10 mol % Rh, THF, 24 h) to give the corresponding product and 23b in good yield (74%), further exemplifying the high reactivity of the catalyst derived from [Rh(CH3CN)2COD]BF4 and L1.
Table 3.
Rh-Catalyzed Hydroamination of Secondary Amines.a
| entry | mol% [Rh] | aminoalkene | product | time (h) | yieldb | ratio (amine:imine)c |
|---|---|---|---|---|---|---|
| 1 | 1 |
|
|
5 | 83% | >95:5 |
| 2 | 3 |
|
|
5 | 65%d | >95:5 |
|
|
|||||
| 3 | 1 | a; R = iPr | a; R = iPr | 8 | 78% | >95:5 |
| 4 | 1 | b; R = Bn | b; R = Bn | 5 | 86% | >95:5 |
Reaction conditions: 0.5 mmol aminoalkene, [Rh(CH3CN)2COD]BF (1-3 mol %), L1 (1.2-3.5 mol %) in 1 mL of tBuOH at 70 °C unless otherwise specified.
Isolated yield.
Ratio determined by 1H NMR spectroscopy.
NMR yield.
1.4. Current limitations in the scope of the cyclizations of aminoalkenes
The results in the previous three sections demonstrate that complex derived from [Rh(CH3CN)2COD]BF4 and L1 is the most active late-transition metal catalyst for intramolecular hydroaminations of unactivated primary aminoalkenes reported. However, we also identified limitations of this catalyst system. For example, substrates containing internal olefins, such as substituted styrenes and cyclohexenes (i.e. to generate bicyclic ring systems), did not undergo cyclization. Moreover, a basic amine was required; aminoalkenes that lacked a basic amine, such as N-aryl- and N-Cbz-protected aminoalkenes did not react with this catalyst under these conditions. Finally, the reaction of 2,2-diphenylhept-6-en-1-amine with catalytic amounts of [Rh(CH3CN)2COD]BF4 and L1 afforded only small amounts of the seven-membered ring product. Isomerization of the starting aminoalkene was the major reaction of the heptenylamine substrate.
2. Identification of the Structure of the Active Catalyst
To understand the origins of the high reactivity of this catalyst system for cyclization of primary aminoalkenes, we identified the structure of the active catalyst and the factors that control the reaction rates and selectivity for formation of hydroamination vs oxidative amination products. The reaction of [Rh(CH3CN)2COD]BF4 with L1 or L1’ in THF at room temperature formed [(L1)Rh(NCMe)]BF4 and [(L1’)Rh(NCMe)]BF4 in 89% and 67% yield, respectively (Eq. 1). Complexes of L1 and L1’ were characterized by 1H, 13C, and 31P NMR spectroscopy and elemental analysis, and the structure of [(L1’)Rh(NCMe)]BF4 was determined by X-ray diffraction. An ORTEP diagram of this complex is shown in Figure 1. This structure shows that L1 is bound in a κ3-fashion to the rhodium atom to generate a square-planar POP-pincer structure. The Rh-O bond distance (Rh-O = 2.129 Å) is similar to that in known cationic Rh-POP complexes reported previously.51,52 These complexes catalyzed the hydroamination of aminoalkenes with reactivity and selectivity that are similar to those of reactions catalyzed by the combination of the rhodium precursor [Rh(CH3CN)2COD]BF4 and ligand L1 or L1’. For example, cyclization of substrate 1b with 5 mol% [(L1)Rh(NCMe)]BF4 in tBuOH at 70 °C resulted in full conversion of starting material after 7 h to afford a 10:1 ratio of amine:imine products 2b:3b as determined by 1H spectroscopic analysis of the crude reaction mixture. Under otherwise identical reaction conditions, the combination of 5 mol% [Rh(CH3CN)2COD]BF4 and 6 mol % ligand L1 gave complete conversion of aminoalkene to a 9:1 ratio of 2b:3b. The tridentate, “pincer” coordination mode of the xantphos derivative is likely integral to the selectivity for hydroamination over oxidative amination by inhibiting the β-hydride elimination step that leads to the formation of the imine byproduct (Eq. 2).53 Additional discussion on the influence of the pincer structure on selectivity is presented in the mechanistic conclusions section.
 |
(Eq 1) |
 |
(Eq 2) |
Figure 1.

ORTEP drawing of catalyst [(L1’)Rh(NCMe)]BF4 (35% ellipsoids, hydrogens and BF4 counterion omitted). Selected bond distances (Å) and angles (deg): Rh1-O1 2.129; Rh1-P1 2.250; Rh1-P2 2.242; Rh1-N5 1.943; P1-Rh1-P2 168.0; N5-Rh1-O1 178.8; O1-Rh1-P1 84.1; O1-Rh1-P2 83.9; N5-Rh1-P1 96.9; N5-Rh1-P2 95.0.
3. Effect of Phosphine Ligand on Hydroamination Reactivity
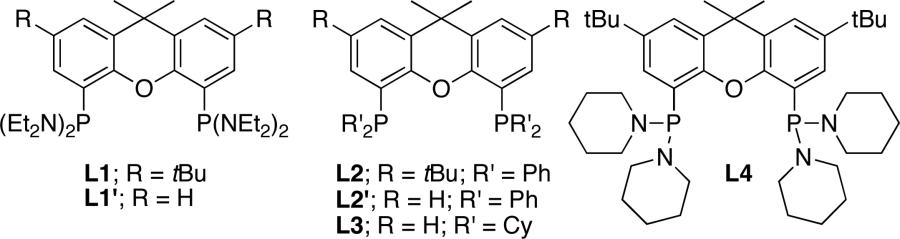
To probe the influence of the phosphine substituents on reactivity and selectivity, we investigated the reactions of aminoalkene 1a catalyzed by complexes of xantphos ligands containing diaryl- and dialkylphosphino groups. Specifically, we evaluated the parent xantphos ligands L2 and L2’, containing diphenylphosphino groups (Table 4, entries 3 and 4),54 and Cy-xantphos L3,55 containing dicyclohexylphosphino groups (entry 5). Reactions catalyzed by complexes of these ligands occurred with lower conversions and selectivity than those catalyzed by the complex of bis(diethylamino) xantphos L1 or L1’ (entries 1 and 2). Specifically, the reaction catalyzed by the combination of ligand L2 or L2’ and [Rh(CH3CN)2COD]BF4 afforded mainly the isomerized product 5a, and the reaction catalyzed by the combination of ligand L3 and the same rhodium species occurred with low conversions and poor selectivity for hydroamination versus oxidative amination (2:1 ratio of 2a:3a).
Table 4.
Evaluation of Xantphos Ligands in the Hydroamination of 1a.a
| entry | % Rh | ligand | time (h) | %convb | ratiob2a:3a:4a:5a |
|---|---|---|---|---|---|
| 1 | 1 | L1 | 2 | 100 | 18:1:1:0.7 |
| 2 | 1 | L1’ | 2 | 100 | 11:1:1:0.2 |
| 3 | 3 | L2 | 2 | 83 | 1:0.1:0:4.4 |
| 4 | 3 | L2’ | 2 | 62 | 1:0.2:0:9.1 |
| 5 | 3 | L3 | 2 | 47 | 2:1:1:0c |
| 6 | 3 | L4 | 2 | 100 | 5.7:1:1:0.4 |
| |||||
Reaction conditions: 0.2-0.3 mmol 1a, 1-3 mol % [Rh(CH3CN)2COD]BF4, and 1.2-3.5 mol % ligand in 0.4-0.6 mL of tBuOH at 70 °C.
Determined by 1H NMR spectroscopy.
Trace amount of isomerization observed.
We hypothesized that the amino groups on the phosphorus ligand could be responsible for the enhanced rate and selectivity observed in the catalytic hydroamination reaction. To distinguish between steric and electronic effects of the amino groups on this reactivity and selectivity, we prepared L4,56 an aminophosphine analogue of Cy-xantphos L3. Ligand L4 contains piperidinyl groups that are nearly isosteric with cyclohexyl groups. The cyclization of 1a catalyzed by the combination of L4 and [Rh(CH3CN)2COD]BF4 generated the amine product with higher conversion and selectivity for hydroamination (100% conversion, 6:1 ratio of 2a:3a, entry 6) than the reaction catalyzed by the complex of L3 (47% conversion, 2:1 ratio of 2a:3a, entry 4) and close to the selectivity of the reactions catalyzed by rhodium and L1 or L1’. These results suggest that amino groups on the phosphorus ligand are responsible for the enhanced rate and selectivity.
The electron-donating property of the aminophosphine was assessed by preparing the carbonyl adducts of LRh+ fragments (Eq. 3). The vCO value in the infrared spectrum of the aminophosphine complex [(L1’)Rh(CO)]+ and the Cy-xantphos complex [(L3)Rh(CO)]+ were nearly identical (1993.3 cm-1 vs 1992.7 cm-1). Thus, the aminoxantphos and Cy-xantphos ligands have similar electron-donating properties. These results, in combination with the above results comparing the difference in the reactivity of the sterically similar Cy-xantphos and piperidinyl xantphos complexes, demonstrates that the effect of the substituents on phosphorus likely stem from properties beyond steric demands and degree of electron-donation to the metal center.
 |
(Eq 3) |
4. Mechanistic Studies
4.1. Identification of Catalyst Resting States in Hydroamination Reactions of Primary Aminoalkenes
Two mechanistic pathways are typically proposed for the hydroamination of aminoalkenes catalyzed by late transition metals, one that occurs by insertion of the alkene into the metal-amide bond (path A) and one that occurs by nucleophilic addition of the amine onto a coordinated olefin (path B, Eq 4).3,57 With few exceptions, electron-deficient late transition metal catalysts, such as those based on platinum,32,33 palladium,45 and iridium,46 have been shown to react by nucleophilic attack of an amine onto a coordinated olefin. To distinguish between these two mechanisms and to determine which step of the deduced catalytic cycle is influenced by the aminophosphine ligand we determined the rhodium species present in the catalytic reaction and measured the kinetic isotope effect.
 |
(Eq 4) |
To identify catalyst resting states, we monitored reactions of aminoalkene 1b with catalytic and stoichiometric quantities of [(L1)Rh(NCMe)]BF4 by 1H and 31P NMR spectroscopy and compared the spectra to those of rhodium complexes prepared independently. Likely intermediates were readily prepared (vide infra) and were distinguishable by the magnitude of the Rh-31P coupling constant. Reactions were conducted in both THF and tBuOH to gain insight into the origin of the accelerating effect of the tBuOH solvent.
Model rhodium complexes containing alkene ([(L1’)Rh(1-hexene)]BF4), primary amine ([(L1’)Rh(4a)]BF4), and imine ligands ([(L1’)Rh(3a)]BF4) ligands were synthesized from the acetonitrile complex [(L1’)Rh(NCMe)]BF4 (Scheme 1). The imine ligand 3a was found to displace the acetonitrile ligand at room temperature in THF after 1 h to afford the corresponding imine complex [(L1’)Rh(3a)]BF4 in 75% isolated yield. The primary amine complex [(L1’)Rh(4a)]BF4 was prepared in 89% yield by heating a solution of the acetonitrile complex [(L1’)Rh(NCMe)]BF4 and 2.3 equiv. of primary amine 4a at 50 °C in THF for 14 h. Displacement of the coordinated acetonitrile with 1-hexene required a large excess of olefin (>50 equiv) to drive the equilibrium. Even after the mixture fully equilibrated, ~10% of the acetonitrile complex remained when analyzed by 31P NMR spectroscopy after 21 h in THF at room temperature. After removal of the free acetonitrile under vacuum and resubjection of the crude mixture containing this ~10:1 ratio of rhodium complexes to the reaction conditions, complete conversion of [(L1’)Rh(NCMe)]BF4 to the olefin complex [(L1’)Rh(1-hexene)]BF4 was achieved. [(L1’)Rh(1-hexene)]BF4 was obtained in 76% isolated yield. All three model rhodium complexes were characterized by elemental analysis, 1H, 13C, and 31P NMR spectroscopy.
Scheme 1.

Synthesis of model rhodium complexes for the determination of catalyst resting states.
The phosphorous-rhodium coupling constants for the acetonitrile, olefin, and amine/imine complexes are significantly different, thus enabling the identification of intermediates containing coordinated alkenes, primary amines, imines, or acetonitrile by 31P NMR spectroscopy (Scheme 1). The 31P chemical shifts and phosphorous-rhodium coupling constants for the amine and imine complexes [(L1’)Rh(4a)]BF4 and [(L1’)Rh(3a)]BF4 were found to be similar (δ 106 ppm, JRh-P = 166 and 168 Hz in tBuOH, respectively); however, a diagnostic downfield signal near ~4.5 ppm in the 1H NMR spectrum of the imine complex, assigned to the methylene protons alpha to the imine nitrogen, allowed us to distinguish between these two species.
Spectral data for these model complexes matched closely with the spectral data for species observed in the reaction of 1b catalyzed or mediated by [(L1)Rh(NCMe)]BF4. Specifically, the reaction of 1b in THF or tBuOH in the presence of stoichiometric amounts of [(L1)Rh(NCMe)]BF4 was monitored by 1H and 31P NMR spectroscopy. In THF, substrate 1b displaced the acetonitrile ligand, and a 10:1 ratio of amine complex:olefin complex (I:II) was observed at room temperature (Scheme 2). Further heating to 40 °C resulted in formation of the cyclized product. After heating at 60 °C for 25 min, aminoalkene 1b was consumed, and the catalyst reverted to the acetonitrile complex [(L1)Rh(NCMe)]BF4. Similar results were obtained for the reaction of 1b and [(L1)Rh(NCMe)]BF4 in tBuOH. However, only ~60% conversion of [(L1)Rh(NCMe)]BF4 to a 1:1 ratio of I:II was observed upon addition of aminoalkene 1b to the rhodium complex. These results demonstrate that the coordination ability of the primary amine to the rhodium center is attenuated in tBuOH and that the equilibrium ratio of amine complex to olefin complex lies more toward the olefin complex in tBuOH than it does in THF.58
Scheme 2.

Reaction of aminoalkene 1b with stoichiometric [(L1)Rh(NCMe)]BF4.
The reaction of aminoalkene 1b catalyzed by 5 mol % [(L1)Rh(NCMe)]BF4 in tBuOH at 62 °C was also monitored by 1H and 31P NMR spectroscopy. In this experiment, amine and olefin complexes I and II were identified as the catalyst resting states throughout the course of the reaction (~3:1 to 6:1 ratio I:II) (Figure 2, (a-d)). However, the imine complex was observed (Figure 2, (b-e)) as the reaction progressed and the concentration of the imine byproduct 3b increased. The imine complex was identified as the final resting state of the catalyst upon consumption of 1b (Figure 2, (e)). Although the imine product 3b is formed in minor amounts, it binds tightly to the cationic rhodium center and out competes binding of the primary amine reactant, the pyrrolidine product, and acetonitrile.
Figure 2.

Stacked plot of 31P NMR spectra obtained at various conversions of aminoalkene 1b catalyzed by 5 mol % [(L1)Rh(NCMe)]BF4 in tBuOH at 62 °C.
The presence of the coordinated olefin intermediate II and the absence of neutral rhodium-amido and rhodium-hydride species in reactions mediated or catalyzed by [(L1)Rh(NCMe)]BF4, is consistent with a mechanism involving nucleophilic attack of the primary amine onto a coordinated olefin (Eq 4, Path B). The observation of the amine and olefin complexes I and II as the resting state suggests two possible reaction sequences. Nucleophilic attack of the primary amine onto the coordinated olefin could be irreversible and turnover-limiting. Alternatively, nucleophilic attack of the primary amine onto the coordinated olefin could be reversible, and followed by irreversible protonolysis of the Rh-C bond. In the latter case, the combination of reversible nucleophilic attack and irreversible protonolysis together would control the rates of these reactions (see Scheme 4, steps ii and iii).
Scheme 4.

Proposed catalytic cycle
4.2. Measurment of Kinetic Isotope Effects
A kinetic isotope effect (KIE) would distinguish between a pathway in which nucleophilic attack was the first irreversible (“turnover-limiting”) step and one in which proton transfer is the first irreversible step. Thus, we measured the KIE for reaction of the aminoalkene 1b and the N-deuterated substrate 1b-D2 (Eq 5). A small KIE would be expected for the pathway involving turnover-limiting nucleophilic attack because N-H(D) bond-cleavage is not involved in this step; however, a large primary KIE would be expected for the pathway involving turnover-limiting Rh-C bond cleavage by proton transfer from an amine or ammonium nitrogen. At 61 °C with 5 mol % [Rh(CH3CN)2COD]BF4 and 6 mol % L1 in tert-butanol-d9 or -d10, the value of kH/kD of aminoalkenes 1b/1b-D2 deduced from initial rates was small (1.16±0.10).47 This small value is consistent with turnover-limiting nucleophilic attack of the amine onto the alkene. This result differs from the large primary isotope effect of 3.4 measured for cyclization of an aminoalkene catalyzed by Ir and the observation of aminoalkyl-metal complexes as the resting states in reactions catalyzed by complexes of Pd and Pt. The small KIE implies that the relative rates for protonolysis of the metal-carbon bond vs reversion to the acyclic reactant (reverse of step ii) is more favorable for the rhodium aminophosphine complex than it is for other systems containing late metals.
 |
(Eq 5) |
4.3. Investigation of Relative Rates
The relative rates of reaction of the primary and secondary amines were different in separate systems and in the same system. When measured independently, the rate of hydroamination of the secondary amine 18 to form 19 was similar to the rate of hydroamination of primary amine 1a to form 2a (k1a/k18 deduced from initial rates was 1.3) However, the primary amine reacted exclusively before the secondary amine when present in the same system. This difference reflects a difference in the resting state of the catalyst in separate systems and with the two substrates together.
The resting state of reactions of secondary amines consists of a combination of the olefin-bound secondary aminoalkene 18 complex and the acetonitrile complex [(L1)Rh(NCMe)]BF4. The resting state of the catalyst in reactions of primary aminoalkenes in THF is the primary amine complex. The order of affinities of a primary amine, secondary amine, and alkene follow the trend 1° amine > alkene > 2° amine. Treatment of alkene complex [(L1’)Rh(1-hexene)]BF4 with 1.5 equiv of benzylamine in THF led to a 26:1 ratio of the primary amine complex to the alkene complex complex after 16 h, whereas treatment of this alkene complex with the secondary amine N-methylbenzylamine formed a 2.5:1 ratio of the olefin complex to the secondary amine complex. Thus, the relative energies for the resting states and transition states can be represented by those in Figure 3, and reaction of the substrates together yields the product from the primary amine over the secondary amine.
Figure 3.

Relative ground and transition state energies for reactions of primary and secondary aminoalkenes deduced from competition binding and catalytic reations.
5. Mechanistic Conclusions
Based on these experiments we propose the mechanism in Scheme 4 involving nucleophilic attack of amine onto a coordinated olefin. In this mechanism, substitution of the tethered olefin for the primary amine in complex I occurs to deliver olefin complex II. The equilibrium ratio of the amine and olefin complexes was significantly influenced by the solvent (i.e. THF vs tBuOH). In the protic solvent tBuOH the ratio of olefin to amine complex was larger than it was in the aprotic THF solvent. Increased concentrations of the olefin complex should increase the rate of nucleophilic addition, and this increased concentration is one factor that explains the faster rates of catalytic reactions performed in tBuOH than those of reactions conducted in THF. The coordinated olefin subsequently undergoes rate-limiting nucleophilic attack (step ii) to generate an ammonium alkylrhodium adduct III that could either undergo protonolysis to generate the hydroamination product or β-hydride elimination to afford the oxidative amination product.
The pincer coordination mode of the xantphos derivative likely affects the selectivity for hydroamination over oxidative amination. Previously, a dicationic PNP-palladium complex was shown to catalyze the hydroamination of amide- and carbamate-protected aminoalkenes.28 It was proposed that the tridentate PNP ligand inhibits β-hydride elimination of an alkylmetal intermediate because it occupies the remaining three coordination sites in the square-plane of the metal. β-Hydride elimination is typically faster when a coordination site is available in the square-plane (Eq. 2).53 The POP-pincer structure of the rhodium complex likely favors hydroamination over oxidative amination for related reasons. The POP pincer structure, although less tightly bound to monocationic rhodium in a κ3 structure than the PNP ligand is bound to dicationic palladium, could still discourage β-hydrogen elimination and favor formation of hydroamination over oxidative amination products. The use of pincer-type ligands in late metal-catalyzed hydroamination reactions, therefore, appears to be a general strategy for inhibiting an oxidative amination pathway and favoring a hydroamination pathway.
The complementarity of hydroaminations with basic amines catalyzed by rhodium and hydroamination with less basic amides and carbamates catalyzed by palladium28 likely results from differences in the charge of the two complexes. The monocationic rhodium complex competitively binds the alkene and the amine, but it bears a less electrophilic coordinated alkene. Thus, a more nucleophilic amine is needed for cyclization to occur by nucleophilic attack on the coordinated alkene. On the other hand, the dicationic palladium complex bears a more electrophilic coordinated alkene, and less nucleophilic amides will attack this coordinated alkene. Hydroamination with more basic nitrogen nucleophiles, such as alkylamines, do not occur with this palladium system, perhaps because these nucleophiles bind tightly to the highly electrophilic Pd center and prevent the alkene binding that precedes C-N bond formation.
Our measured kinetic isotope effect and the observed catalyst resting states imply that the series of proton transfer steps (step iii), including cleavage of the rhodium-carbon bond to generate the cyclized product, occurs faster than reversion to the reactant by C-N bond cleavage. Cleavage of the rhodium-carbon bond could occur by either direct protonation of the sigma bond or by a mechanism involving initial formation of a RhIII-H intermediate, followed by reductive elimination to release the product.59 We propose that the aminophosphine ligand affects these relative rates by promoting proton transfer, perhaps by assistance by the electron pair. We are currently investigating this hypothesis further. As the reaction progresses, and the concentration of the imine increases, the imine complex IVb becomes the resting state. In contrast to platinum-, palladium-, and iridium-catalyzed hydroamination of secondary amines, protonation of the metal-carbon bond is not the turnover-limiting step for the hydroamination of primary amines catalyzed by [(L1)Rh(NCMe)]BF4.
Summary
We have developed a highly active catalyst for the intramolecular hydroamination of unprotected, primary aminoalkenes based on an unusual diaminophosphine ligand. For the first time with a late metal catalyst, primary aminoalkenes that do not contain gem-disubstitution on the alkyl chain undergo cyclization in high yields at mild temperatures. The substrate scope includes aminoalkenes containing various reactive functional groups that would not otherwise be tolerated with previously reported catalysts for hydroamination of primary amines. We have also demonstrated initial reactivity of this catalyst with internal alkenes.
The diaminophosphine ligand appears to be important for achieving high rates and selectivity in the intramolecular hydroamination of primary amines. A detailed mechanistic study of this catalytic reaction has been conducted. Our data support a mechanism occurring by turnover-limiting nucleophilic attack of amine onto a coordinated olefin, followed by cleavage of the metal-carbon bond. We suggest that the aminophosphine groups on the ligand create favorable relative rates for the proton transfer steps.
We have also identified the primary pathway for catalyst decomposition that will allow us to design future catalysts with improved stability. We are currently seeking to use these conclusions to develop even more active catalysts for intermolecular hydroaminations of unactivated olefins and to develop asymmetric hydroaminations with catalysts bound by ligands containing chiral diamino groups.37
Supplementary Material
Scheme 3.

Competition experiment between primary and secondary aminoalkenes.
ACKNOWLEDGMENT
We thank NIH for financial support of this work (GM-55382 to J.F.H. and GM87062 to L.D.J) and Johnson-Matthey for rhodium chloride. We also thank Shira Halperin for assisting in the synthesis of substrates and Danielle Gray for X-ray crystallography.
Footnotes
KEY WORDS (Word Style “BG_Keywords”). If you are submitting your paper to a journal that requires keywords, provide significant keywords to aid the reader in literature retrieval.
Supporting Information Available. Experimental procedures, compound characterization data, kinetic data, and details for crystal structure of [(L1’)Rh(NCMe)]BF4. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- 1.Hultzsch KC. Adv. Synth. Catal. 2005;347:367. [Google Scholar]
- 2.Mueller TE, Beller M. Chem. Rev. 1998;98:675. doi: 10.1021/cr960433d. [DOI] [PubMed] [Google Scholar]
- 3.Mueller TE, Hultzsch KC, Yus M, Foubelo F, Tada M. Chem. Rev. 2008;108:3795. doi: 10.1021/cr0306788. [DOI] [PubMed] [Google Scholar]
- 4.Mueller TE. In: Encyclopedia of Catalysis. Hovath IT, editor. Vol. 3. Wiley-Interscience; Hoboken: 2003. pp. 518–541. [Google Scholar]
- 5.Brunet J-J, Neibecker D. In: Catalytic Heterofunctionalization. Togni A, Grutzmacher H, editors. Wiley-VCH; Weinheim: 2001. pp. 91–141. [Google Scholar]
- 6.O'Hagan D. Nat. Prod. Rep. 2000;17:435. doi: 10.1039/a707613d.. See also refs 48 and 49 for examples of biologically active tetrahydroisoquinolines heterocycles.
- 7.Hong S, Marks TJ. Acc. Chem. Res. 2004;37:673. doi: 10.1021/ar040051r. [DOI] [PubMed] [Google Scholar]
- 8.Stanlake LJE, Schafer LL. Organometallics. 2009;28:3990. [Google Scholar]
- 9.Datta S, Gamer MT, Roesky PW. Organometallics. 2008;27:1207. [Google Scholar]
- 10.Kim YK, Livinghouse T. Angew. Chem. Int. Ed. 2002;41:3645. doi: 10.1002/1521-3773(20021004)41:19<3645::AID-ANIE3645>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 11.Datta S, Roesky PW, Blechert S. Organometallics. 2007;26:4392. [Google Scholar]
- 12.Crimmin MR, Casely IJ, Hill MS. J. Am. Chem. Soc. 2005;127:2042. doi: 10.1021/ja043576n. [DOI] [PubMed] [Google Scholar]
- 13.Bytschkov I, Doye S. Eur. J. Org. Chem. 2003:935. [Google Scholar]
- 14.Reznichenko AL, Hultzsch KC. Organometallics. 2010;29:24–27. [Google Scholar]
- 15.Haak E, Bytschkov I, Doye S. Angew. Chem. Int. Ed. 1999;38:3389. [PubMed] [Google Scholar]
- 16.Kim H, Lee PH, Livinghouse T. Chem. Commun. 2005:5205. doi: 10.1039/b505738h. [DOI] [PubMed] [Google Scholar]
- 17.Bexrud JA, Beard JD, Leitch DC, Schafer LL. Org. Lett. 2005;7:1959. doi: 10.1021/ol0503992. [DOI] [PubMed] [Google Scholar]
- 18.Thomson RK, Bexrud JA, Schafer LL. Organometallics. 2006;25:4069. [Google Scholar]
- 19.Leitch DC, Payne PR, Dunbar CR, Schafer LL. J. Am. Chem. Soc. 2009;131:18246. doi: 10.1021/ja906955b. [DOI] [PubMed] [Google Scholar]
- 20.Barrett AGM, Crimmin MR, Hill MS, Procopiou PA. Proc. R. Soc. 2010;466:927. [Google Scholar]
- 21.Zulys A, Dochnahl M, Hollmann D, Lohnwitz K, Herrmann J-S, Roesky PW, Blechert S. Angew. Chem. Int. Ed. 2005;44:7794. doi: 10.1002/anie.200502006. [DOI] [PubMed] [Google Scholar]
- 22.Pissarek J-W, Schlesiger D, Roesky PW, Blechert S. Adv. Synth. Catal. 2009;351:2081. [Google Scholar]
- 23.Rosenfeld DC, Shekhar S, Takemiya A, Utsunomiya M, Hartwig JF. Org. Lett. 2006;8:4179. doi: 10.1021/ol061174+. [DOI] [PubMed] [Google Scholar]
- 24.Ackermann L, Kaspar LT, Althammer A. Org. Biomol. Chem. 2007;5:1975. doi: 10.1039/b706301f. [DOI] [PubMed] [Google Scholar]
- 25.Schlummer B, Hartwig JF. Org. Lett. 2002;4:1471. doi: 10.1021/ol025659j. [DOI] [PubMed] [Google Scholar]
- 26.Zhang Z, Yang C-G, He C. J. Am. Chem. Soc. 2006;128:1798. doi: 10.1021/ja053864z. [DOI] [PubMed] [Google Scholar]
- 27.Ru-catalyzed olefin metathesis exemplifies the broad functional group tolerance exhibited by late transition metal catalysts. See: Toste FD, Chatterjee AK, Grubbs RH. Pure Appl. Chem. 2002;74:7.
- 28.Michael FE, Cochran BM. J. Am. Chem. Soc. 2006;128:4246. doi: 10.1021/ja060126h. [DOI] [PubMed] [Google Scholar]
- 29.Komeyama K, Morimoto T, Takaki K. Angew. Chem. Int. Ed. 2006;45:2938. doi: 10.1002/anie.200503789. [DOI] [PubMed] [Google Scholar]
- 30.Bender CF, Widenhoefer RA. Org. Lett. 2006;8:5303. doi: 10.1021/ol062107i. [DOI] [PubMed] [Google Scholar]
- 31.Han X, Widenhoefer RA. Angew. Chem. Int. Ed. 2006;45:1747. doi: 10.1002/anie.200600052. [DOI] [PubMed] [Google Scholar]
- 32.Bender CF, Widenhoefer RA. J. Am. Chem. Soc. 2005;127:1070. doi: 10.1021/ja043278q. [DOI] [PubMed] [Google Scholar]
- 33.Bender CF, Hudson WB, Widenhoefer RA. Organometallics. 2008;27:2356. [Google Scholar]
- 34.Ohmiya H, Moriya T, Sawamura M. Org. Lett. 2009;11:2145. doi: 10.1021/ol9007712. [DOI] [PubMed] [Google Scholar]
- 35.Liu Z, Hartwig JF. J. Am. Chem. Soc. 2008;130:1570. doi: 10.1021/ja710126x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bauer EB, Andavan GTS, Hollis TK, Rubio RJ, Cho J, Kuchenbeiser GR, Helgert TR, Letko CS, Tham FS. Org. Lett. 2008;10:1175. doi: 10.1021/ol8000766. [DOI] [PubMed] [Google Scholar]
- 37.There has been only one report of late metal-catalyzed asymmetric intramolecular hydroamination of unactivated olefins: Shen X, Buchwald SL. Angew. Chem. Int. Ed. 2010;122:574.
- 38.Hesp KD, McDonald R, Stradiotto M. Can. J. Chem. 2010;88:1–9. [Google Scholar]
- 39.Hesp KD, Stradiotto M. Org. Lett. 2009;11:1449. doi: 10.1021/ol900174f. [DOI] [PubMed] [Google Scholar]
- 40.Kashiwame Y, Kuwata S, Ikariya T. Chem. Eur. J. 2010;16:766. doi: 10.1002/chem.200902827. [DOI] [PubMed] [Google Scholar]
- 41.Brunet J-J, Chu NC, Diallo O. Organometallics. 2005;24:3104. [Google Scholar]
- 42.Zhang Z, Lee SD, Widenhoefer RA. J. Am. Chem. Soc. 2009;131:5372. doi: 10.1021/ja9001162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Goertz W, Kamer PCJ, van Leeuwen PWNM, Vogt D. Chem. Eur. J. 2001;7:1614. doi: 10.1002/1521-3765(20010417)7:8<1614::aid-chem16140>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- 44.Gopalakrishnan J. Appl. Organometal. Chem. 2009;23:291. [Google Scholar]
- 45.Cochran BM, Michael FE. J. Am. Chem. Soc. 2008;130:2786. doi: 10.1021/ja0734997. [DOI] [PubMed] [Google Scholar]
- 46.Hesp KD, Tobisch S, Stradiotto M. J. Am. Chem. Soc. 2010;132:413. doi: 10.1021/ja908316n. [DOI] [PubMed] [Google Scholar]
- 47.See Supporting Information for experimental details
- 48.Scott JD, Williams RM. Chem. Rev. 2002;102:1169. doi: 10.1021/cr010212u. [DOI] [PubMed] [Google Scholar]
- 49.Chrzanowaska M, Rozwadowska MD. Chem. Rev. 2004;104:3341. doi: 10.1021/cr030692k. [DOI] [PubMed] [Google Scholar]
- 50.One example of an alpha-branched aminoalkene (2-amino-hex-5-ene) was reported to undergo cyclization in the presence of a zinc catalyst to give the product in low conversion (19%). See ref 21.
- 51.Alcck NW, Brown JM, Jeffery JC. J. Chem. Soc. Dalton Trans. 1976:583. [Google Scholar]
- 52.Sandee AJ, van der Veen LA, Reek JNH, Kamer PCJ, Lutz M, Spek AL, van Leeuwen PWNM. Angew. Chem. Int. Ed. 1999;38:3231. [PubMed] [Google Scholar]
- 53.Hartwig JF. Organotransition metal chemistry: from bonding to catalysis. University Science Books; Sausalito: 2010. pp. 398–400. [Google Scholar]
- 54.Kranenburg M, van der Burgt YEM, Kamer PCJ, van Leeuwen PWNM. Organometallics. 1995;14:3081. [Google Scholar]
- 55.Kuwano R, Kusano H. Chem. Lett. 2007;36:528. [Google Scholar]
- 56.L4 was synthesized from 4,5-dibromo-2,7-di-t-butyl-9,9-dimethylxanthene and known bis(piperidinyl)chlorophosphine: Amigues EJ, Hardacre C, Keane G, Miguad ME. Green Chemistry. 2008;10:660.. See supporting information for experimental details.
- 57.Hartwig JF. Organotransition metal chemistry: from bonding to catalysis. University Science Books; Sausalito: 2010. pp. 713–716. [Google Scholar]
- 58.Fu X, Wayland BB. J. Am. Chem. Soc. 2005;127:16460. doi: 10.1021/ja054548n. [DOI] [PubMed] [Google Scholar]
- 59.DFT calculations showed that reductive elimination from a neutral Ir(III)-hydride intermediate is a lower energy pathway than direct protonolysis of the Ir-C bond in the hydroamination of asecondary aminoalkene catalyzed by [Ir(COD)Cl]2. See ref 46.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.