

Abstract
Vascular endothelial growth factor (VEGF) is required for proper lung development and is transcriptionally regulated in alveolar epithelial cells by hypoxia inducible factor (HIF). Previous findings in a newborn mouse model of bronchopulmonary dysplasia (BPD) suggest that thioredoxin interacting protein (Txnip) is a novel regulator of VEGF expression. The present studies were designed to test the hypothesis that Txnip negatively regulates VEGF through effects on HIF-mediated gene expression. To test this hypothesis, we first examined the levels of VEGF and Txnip protein in the lungs of 1 day-old newborn and E19 embryos and detected a significant inverse correlation. To elucidate the mechanisms underlying this relationship, we studied the effects of Txnip overexpression on HIF-mediated transcription using murine lung epithelial (MLE-12) cells. Overexpression of Txnip inhibited HIF-mediated reporter activity in both hypoxia and room air. Suppression of HIF activity by Txnip appeared to be independent of the ability of Txnip to bind to thioredoxin. Thus, our studies support a model in which Txnip is a potentially critical regulator of HIF-mediated gene transcription in the murine lung. Alterations in Txnip expression could alter lung VEGF expression in prematurely born human infants and contribute to the development of BPD.
Keywords: Thioredoxin-1, thioredoxin interacting protein, vascular endothelial growth factor, hypoxia-inducible factor, bronchopulmonary dysplasia
Introduction
Each year approximately 14,000 preterm infants in the United States develop bronchopulmonary dysplasia (BPD), a disease characterized histologically by impaired alveolar and vascular growth with alveolar simplification, enlarged distal airspaces, and a reduction in the size of the pulmonary capillary bed [1]. Infants with BPD require prolonged respiratory support with mechanical ventilation and supplemental oxygen therapy and experience morbidities that often persist into adulthood [2–4]. The hypoxic fetal environment favors angiogenesis and promotes lung microvascular development and epithelial branching morphogenesis. Recent studies suggest that the dramatic shift in oxygen homeostasis that occurs with premature birth interrupts the hypoxia-mediated signaling pathways necessary for proper lung development [5–7].
Proper vascular development is necessary for normal lung development [8]. Vascular endothelial growth factor (VEGF) is produced by alveolar type 2 cells in both the developing mouse lung and in the human fetal lung [9] and plays a crucial role in angiogenesis. Inhibition of VEGF expression in animal models alters lung vascular and alveolar development resulting in a phenotype similar to human BPD [4, 10–12]. Lung VEGF expression is regulated through the activation of hypoxia-inducible transcription factors (HIFs), consisting of an α and β subunit, via a hypoxia response element (HRE) in the VEGF promoter [13, 14].
Thioredoxin interacting protein (Txnip) was originally described for its ability to inhibit the activity of thioredoxin-1 (Trx1) through the formation of a mixed disulfide between cysteine-247 (designated as C247, hereafter) of Txnip and cysteine-32 of the Trx1 active site [15, 16]. Trx1 enhances HIF-dependent VEGF production and promotes angiogenesis in solid tumors [17, 18]. It is unclear whether Trx1 has a similar role in lung development. Txnip expression is inversely correlated with VEGF expression in a number of cancer cells [19] and during normal postnatal lung development in newborn mice [20].
In the present studies, we examined the relationship between Txnip and VEGF in the lungs of embryonic day 19 and 1 day-old mice and our data confirm an inverse correlation during normal development. To elucidate the mechanisms underlying this relationship, we determined the effect of Txnip overexpression on HIF transcriptional activity in murine lung epithelial (MLE-12) cells. Our data indicate that Txnip overexpression decreases HIF-mediated transcriptional activity in alveolar epithelial cells independent of the ability to interact with Trx1 via C247.
Experimental Procedures
Animal model
Animal study protocols were approved by the Institutional Animal Care and Use Committee at The Research Institute at Nationwide Children’s Hospital. C3H/HeN mice were bred and dated as to time of pregnancy. Pregnant dams were euthanized at E19 by cervical dislocation and the pups were removed. One day-old pups were euthanized by intraperitoneal injections of 200 mg/kg of sodium pentobarbital. Lungs from both E19 and 1 day old pups were removed, snap frozen in liquid nitrogen, and stored at −80°C for subsequent analyses.
Western blot analyses
Frozen murine lung tissues were homogenized in lysis buffer as previously described [20] and protein concentrations were determined using a Bio-Rad Protein Assay kit. Protein samples (50 μg) were separated by SDS-PAGE (4–12% bis-tris gel, Invitrogen) and transferred to nitrocellulose membranes (iBlot, Invitrogen). Txnip and VEGF contents were evaluated using affinity-purified polyclonal rabbit anti-human Txnip (kindly provided by Simon Hui, San Diego State University; 1:1000 in TBS-T) and polyclonal rabbit anti-human VEGF (Santa Cruz Biotechnology sc-152; 1:500 in TBS-T) primary antibodies which cross-react with murine Txnip and VEGF protein, respectively. Peroxidase-conjugated goat anti-rabbit IgG secondary antibody (Bio-Rad; 1:12,000 in TBS-T) was then used. The membranes were developed using enhanced chemiluminescence (GE Healthcare) and the resulting bands were quantitated using ImageQuant software version 5.0 (Molecular Dynamics/GE Healthcare). To control for variances in protein loading, the band density of the protein of interest for each sample was normalized to the density of β-actin protein detected using a mouse anti-human β-actin primary antibody (Abcam; 1:5000 in TBS-T); and a goat anti-mouse secondary antibody (Bio-Rad; 1:12,000 in TBS-T). Exposure time remained uniform for each individual antibody and all data were reported as ratios of density of the band of interest to density of β-actin.
Immunohistochemistry
Lungs of 3 day old newborn C3H/HeN mice raised in room air were inflation fixed with formalin at 20 cm H2O for 5 minutes. Whole lungs were paraffin-embedded and cut into 4 μm sections. All slides were incubated for 5 minutes in a 3% hydrogen peroxide solution in water to block for endogenous peroxidase activity prior to staining. Sequential sections were incubated with either polyclonal rabbit anti-human surfactant protein C primary antibody (Santa Cruz Biotechnology, sc-13979; 1:200) or polyclonal rabbit anti-human Txnip primary antibody (Dr. Simon Hui, San Diego State University; 1:800) which cross-react with murine surfactant C and murine Txnip, respectively. Biotinylated goat anti-rabbit secondary antibody (Vector Laboratories, cat# BA-1000; 1:200) was then used and slides were developed using a Vectastain Elite kit (Vector Laboratories, cat# PK-6100). Staining was visualized with DAB chromogen and slides were counterstained with Richard Allen hematoxylin.
Cell Culture
Murine lung epithelial (MLE-12) cells, an SV-40 cell line of type 2 alveolar epithelial cell lineage, were generously provided by Dr. Jeffrey Whitsett (University of Cincinnati). Cells were cultured in HITES medium supplemented with 2% heat-inactivated fetal bovine serum, 100 U/ml penicillin, and 100 μg/ml streptomycin, and were maintained in normoxia (21% O2-5% CO2-balance nitrogen) in a humidified incubator. Because the phenotype of MLE-12 cells is passage-sensitive, the same passage number (#20) was used for all cell experiments.
Promoter activity analyses
Wild-type human Txnip and cys-247 to ser mutant (C247S) Txnip plasmids were kindly provided by Dr. Parth Patwari (Harvard University) and were generated as previously described [16]. A hypoxia response element (HRE) promoter plasmid consisting of a trimer of murine Epo 3′ enhancer fused to a luciferase reporter [21] was a gift from Dr. Paul Schumacker (Northwestern University). For all transfections, MLE-12 cells were co-transfected at 80% confluence with a total of 3 μg of plasmid DNA per well on 9.6-cm2 tissue culture plates using JetPEI (Polyplus-Transfection) according to manufacturer instructions. The DNA mixture contained 1 μg HRE-luciferase and Renilla luciferase (Promega) internal control vector plasmid DNA (50:1) and a total of 2 μg of empty vector pcDNA3.1 control and/or Txnip expression vector DNA. Twenty-four hours after transfection, cells were either maintained in normoxia (21% O2/5% CO2/balance nitrogen) or were exposed to hypoxia (1% O2/5% CO2/balance nitrogen) for 24 h. For hypoxia studies, incubations were performed in a controlled environment using ProOx 110 and ProCO2 120 controllers (Biospherix, Redfield, NY) connected to a semi-sealed culture chamber (C-chamber; Biospherix) within a humidified incubator. The ProOx and ProCO2 controllers continuously monitor conditions within the exposure chamber to maintain constant O2 and CO2 concentrations, respectively, by feedback control. Luciferase activities in cell lysates were determined using the Dual-Luciferase Reporter Assay System (Promega) and a Veritas Microplate Luminometer (Promega). Values for luciferase were normalized to Renilla luciferase.
Txnip truncation analyses
Full-length Txnip (1-391) and truncation mutants (1-300, 1-215, 1-151) were generated as described by Patwari et al [15]. MLE-12 cells were co-transfected at 80% confluence with a total of 1 μg HRE-luciferase and Renilla luciferase (Promega) internal control vector plasmid DNA (50:1) and 2 μg empty vector control or Txnip plasmid DNA as described above. Twenty-four hours after transfection cells were exposed to hypoxia (1% O2/5% CO2/balance nitrogen) for 24 h. Luciferase activities in cell lysates were determined as described above.
Statistics
Data, expressed as mean ± SEM, were obtained from 2–4 individual experiments (total n=10–22). For western blot analyses data were normalized to E19. To determine the relationship between lung VEGF and Txnip protein contents, a linear regression analysis was performed on the normalized band densities obtained on each sample by western blot analyses. Data were tested for homogeneity of variances and were log-transformed when indicated. Due to experiment-to-experiment variability in absolute firefly-luciferase/renilla-luciferase ratios, transfection data from cell culture experiments were normalized separately to exposure (room air or hypoxia) and control plasmid (pcDNA3.1) and are expressed as “percent control”. Data containing 2 groups were analyzed by Student’s t test. For multiple comparisons, data were analyzed by one-way ANOVA combined with Fisher’s Protected Least Significant Difference post hoc. All data were analyzed using SPSS Windows version 14.0 and significance was accepted at p<0.05.
Results & Discussion
Lung VEGF and Txnip protein levels are altered with birth and are inversely correlated
The fetal-to-neonatal transition from the hypoxic environment of the womb to the relatively hyperoxic extrauterine environment takes place in premature infants during a period of marked susceptibility to hyperoxia. Premature delivery has deleterious effects on the oxygen-dependent biological processes that mediate lung development; in particular, the HIF/VEGF pathway. At birth, newborn mice have lungs that are structurally similar to 28-week premature human infants. Birth of newborn mice into room air represents a hyperoxic environment compared to the environment in utero. Thus, we hypothesized that the transition from the hypoxic environment in utero to the more oxygen-rich postnatal environment would inversely alter lung Txnip and VEGF protein levels in a pattern similar to that seen with postnatal hyperoxic exposure of newborn mice [20]. To test this hypothesis, we determined the expression of Txnip and VEGF protein in whole lung homogenates prepared from embryonic day 19 (E19) and 1 day-old newborn C3H/HeN mice. Western blot analyses indicated that Txnip protein levels were 3.4 fold greater in the lungs of 1 day-old mice than in the lungs of mice sacrificed at E19 (Figure 1A). In contrast, lung VEGF protein levels in 1 day-old mice were 41% less than in E19 mice (Figure 1B). To examine the relationship between lung Txnip and VEGF protein levels, we performed a regression analysis on normalized Txnip and VEGF expression levels (Figure 1C). Our data indicated a significant inverse correlation between lung VEGF and Txnip protein levels (n=18, R=−0.85, p<0.001). These data are consistent with our working hypothesis that Txnip is a potentially novel modulator of VEGF expression in the developing murine lung.
Figure 1.
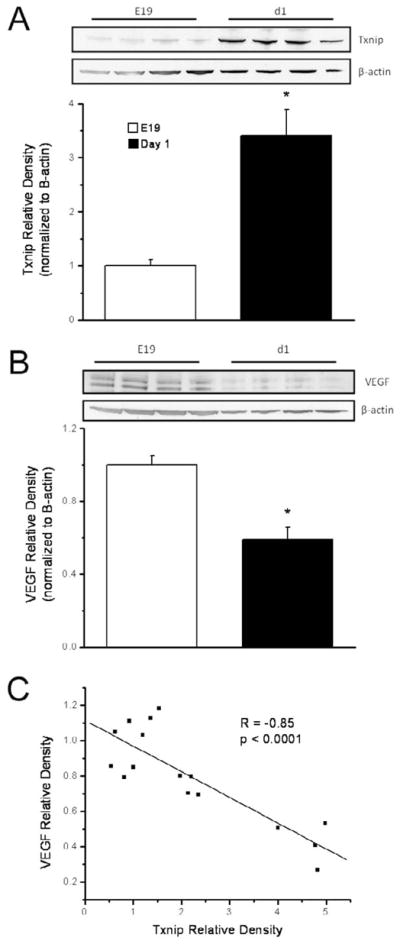
Lung Txnip (A) and VEGF (B) contents in embryonic day 19 and 1 day-old newborn C3H/HeN mice. Western blots were quantified by densitometry and were normalized to β-actin. Density data, expressed as mean ± SEM (n=8), are representative of 2 individual experiments and were analyzed by 1-way ANOVA (*p<0.001 vs E19). Double bands correspond to splice variants of VEGF. Lung Txnip protein levels in 1 day-old newborn mice were 3.4 times greater and VEGF protein levels 2.4 times less than in E19 mice. C) A regression analysis of lung Txnip and VEGF contents indicated a significant inverse correlation between lung VEGF and Txnip protein levels (n=16).
Txnip is expressed by type 2 alveolar epithelial cells in the lungs of newborn mice
Previously, Filby et al demonstrated the presence of Txnip in type 2 alveolar epithelial cells in the lungs of lambs in utero [22]. However, no data exist regarding the expression of Txnip in the newborn mouse lung. To determine if Txnip was expressed in type 2 alveolar epithelial cells in the lungs of newborn mice, we performed immunohistochemical analyses of paraffin-embedded lung sections obtained from 3 day-old newborn mice. Sequential lung sections from the same fixed lung were immunostained for Txnip and surfactant protein C (Sp-C), a marker for type 2 alveolar epithelial cells (Figure 2). Txnip immunostaining (Figure 2A) was present in cells that also stained most intensely for surfactant protein C (Figure 2B), indicating that Txnip is produced by type 2 alveolar epithelial cells.
Figure 2.

Immunohistochemical analyses for Txnip (A) and surfactant protein C (B), a marker for type 2 AECs in sequential lung sections taken from the same fixed lung of a 3 day old C3H/HeN mouse. The arrows demonstrate a cell expressing both Txnip and surfactant protein C confirming the producion of Txnip by type 2 alveolar epithelial cells.
Txnip overexpression decreases hypoxia-response element-driven promoter activity
Appropriate HIF/VEGF expression is required for normal lung development [4, 10–12, 23, 24]. Decreasing HIF-α degradation by prolonging the half-life of HIFs [8, 25–27] or by restoring VEGF expression directly [4, 28, 29] improves alveolarization and enhances lung vascular growth in animal models of BPD. VEGF production by type 2 alveolar epithelial cells is regulated by HIF via a HRE in the VEGF promoter region. While Txnip was identified as a possible modulator of lung development in the fetal lamb [22] and newborn mouse [20], the underlying mechanisms have not been clearly defined. To elucidate the mechanisms responsible for the inverse relationship between Txnip and VEGF, we tested the hypothesis that Txnip negatively regulates HIF activity. MLE-12 cells, an SV-40 transformed alveolar epithelial cell line of type 2 lineage, were co-transfected with a HRE-driven luciferase reporter together with various amounts of Txnip expression vector. The effect of Txnip expression on reporter transcription was assessed by luciferase assay. Though fetal oxygen tension is reported to be between 3 and 5% [27], 1% O2 was used for studies in vivo to promote maximal activation of the HRE-driven luciferase reporter. No abnormalities in cell function, as indicated by cell growth, were observed in hypoxia-exposed cells. As expected, exposure to 1% O2 increased HRE-driven luciferase reporter activity 9.7 fold (Figure 3A). Overexpression of Txnip caused a dose-dependent decrease in HRE-luciferase activity in cells exposed to room air for 24 h (Figure 3B). Transfection of 0.5, 1, and 2 μg of Txnip plasmid DNA per well decreased HRE-luciferase activity by 17%, 23%, and 51%, respectively, compared to cells transfected with an empty vector. Txnip not only inhibited the HRE-driven reporter expression in room air, but also significantly attenuated the reporter transcription under hypoxic conditions. Transfection with 0.5, 1, or 2 μg of Txnip expression vector resulted in a 9%, 12%, and 35% decrease in reporter activity, respectively (Figure 3C). Our data clearly indicate that Txnip overexpression decreases HIF promoter activity in alveolar epithelial cells. The finding that Txnip suppresses HIF function in both hypoxia and room air suggests that residual HIF activity under normoxic conditions is also regulated by Txnip. Though we were unable to statistically analyze the differences, the proportional effect of Txnip overexpression on HRE-driven reporter activity appears to be greater in room air than in hypoxia. We speculate that the proportional effect of Txnip overexpression on HIF transcriptional activity is less in hypoxia because HIF-1α protein levels are known to increase as oxygen tension decreases [27].
Figure 3.

HRE-luciferase activity is increased by exposure to 1% oxygen and is decreased in a dose-dependent manner by overexpression of Txnip. Data are expressed as mean ± SEM and are representative of 2–4 individual experiments. A) HRE-luciferase activity is 9.7 times greater in MLE-12 cells exposed to 1% oxygen for 24 h than in cells cultured in room air (n=22; *p<0.001 vs room air). B) HRE-luciferase activity was measured as described in Methods in room air-exposed MLE-12 cells following transient overexpression of Txnip for 24 h. HRE-luciferase activity was decreased by 17%, 23%, and 51% in cells transfected with 0.5, 1, and 2 μg Txnip plasmid DNA/well, respectively, compared to control-transfected cells (n=10–22; *p<0.001 vs control; #p<0.001 vs 0.5 and 1 μg). C) HRE-luciferase activity was measured in MLE-12 cells exposed to 1% oxygen following transient overexpression of Txnip for 24 h. HRE-luciferase activity was decreased by 9%, 12%, and 35% in cells transfected with 0.5, 1, and 2 μg Txnip plasmid DNA/well, respectively, compared to control-transfected cells (n=16–22; *p<0.01 vs control; ¥p<0.005 vs control; #p<0.001 vs 0.5 and 1 μg/well).
Txnip-dependent inhibition of HIF activity does not depend on Cysteine-247
It has been proposed that Txnip inhibits Trx1 function through a thiol-disulfide exchange reaction between C247 of Txnip and the catalytic cysteine of the Trx1 active site resulting in the formation of a mixed disulfide between Txnip and Trx1 [16]. Given that Trx1 promotes HIF function [17, 18], we hypothesized that the effect of overexpression of Txnip on HRE-luciferase activity in MLE-12 cells was mediated through the binding of Txnip to Trx1. To test this hypothesis, we transfected MLE-12 cells with empty vector control, wild-type Txnip or a C247S mutant Txnip which is incapable of binding to the catalytic active site cysteine of Trx1 [15, 16]. These expression vectors were co-transfected into MLE-12 cells with HRE-luciferase reporter and were exposed to room air or 1% O2 for 24 h. Our data indicated that both wild-type and C247S mutant Txnip suppressed HRE-luciferase activities in MLE-12 cells in a comparable manner (Figure 4). In room air, transfection of wild-type Txnip resulted in a 43% decrease in reporter activity, while transfection of the C247S mutant led to a 39% decrease in reporter activity (Figure 4A). Likewise, under hypoxic conditions transfection with wild-type and C247S mutant Txnip resulted in a 30% and a 25% decrease in HRE-luciferase activity, respectively (Figure 4B). Thus, Txnip decreases HIF activity in MLE-12 cells in a manner that is independent of C247. Given these data, it is unlikely that the effect of Txnip on HIF activity is mediated via a thiol-disulfide reaction with the catalytic active site cysteine of Trx1. These data do not exclude the possibility that Txnip interacts with Trx1 to suppress HIF activity via a mechanism that has yet to be identified.
Figure 4.

Wild-type and C247S mutant Txnip equivalently suppress HRE-luciferase promoter activity in MLE-12 cells. Data are expressed as mean ± SEM and are representative of 2–3 individual experiments. A) HRE-luciferase activity was measured as described in Methods in room air-exposed MLE-12 cells following transient overexpression of wild-type or C247S Txnip for 24 h and was decreased by 43% and 39%, respectively, compared to control-transfected cells (n=10–13; *p<0.001 vs control). B) HRE-luciferase activity was measured in MLE-12 cells exposed to 1% oxygen following transient overexpression of Txnip for 24 h and was decreased by 30% and 25%, respectively, in cells transfected with wild-type or C247S Txnip plasmid DNA/well compared to control-transfected cells (n=16; *p<0.001 vs control).
Suppression of HRE-luciferase activity by Txnip requires the C-terminal α-arrestin domain
The arrestin family of proteins (consisting of the α-arrestins, the β-arrestins, and the visual arrestins) regulate receptor signaling through many families of receptors. Txnip is the only known member of the mammalian α-arrestin family that binds to Trx1. All α-arrestins have two arrestin domains and a C-terminal tail domain [30]. Txnip is an important regulator of metabolic function and glucose homeostasis [31–34]; functions that appear to be intrinsic to arrestin domains themselves and independent of an interaction between Txnip and Trx1 [15, 33]. Patwari et al. demonstrated the necessity of the C-terminal α-arrestin domain of Txnip, but not the C-terminal tail region, for suppression of lactate release from 293T cells [15]. We hypothesized that the mechanisms responsible for the metabolic effects of Txnip might also govern the effects of Txnip on HIF activity. To test this hypothesis, we transfected MLE-12 cells with empty vector control or an expression vector containing one of the following: full-length Txnip (1-391), a Txnip mutant with truncation of the C-terminal tail (1-300), or one of two Txnip mutants with a truncation within (1-215) or of (1-151) the C-terminal α-arrestin domain. Cells were co-transfected with a HRE-driven luciferase reporter and renilla-luciferase and were exposed to 1% oxygen for 24 h (Figure 5). Both full-length Txnip and the Txnip truncation mutant lacking the C-terminal tail (Txnip 1-300) suppressed HRE-luciferase promoter activity (empty vector: 1±0.01; Txnip 1-391: 0.58±0.02; Txnip 1-300: 0.61±0.01). The effect of Txnip overexpression on HRE-luciferase promoter activity was absent in cells expressing the Txnip mutants with truncations into the C-terminal α-arrestin domain (Txnip 1-215: 1.17±0.02; Txnip 1-151: 0.93±0.01). Though the significance of these findings are unclear, HIF activity was slightly greater in cells transfected with the 1-215 truncation mutant and was slightly lower in cells transfected with the 1-151 truncation mutant than in control-transfected cells. Nonetheless, our results conclusively demonstrate that the C-terminal α-arrestin domain of Txnip is required for suppression of HIF promoter activity. These data suggest the possibility that common mechanisms regulate the metabolic and HIF-suppressive effects of Txnip and that these effects might be an intrinsic property of α-arrestins.
Figure 5.

A) Schematic representation of Txnip protein sequence highlighting the N-terminal (arrestin domain #1) and C-terminal (arrestin domain #2) arrestin domains, the pVHL-binding region, the leucine-rich CRM1 nuclear export consensus sequence (NES) [19], and the cysteine that is proposed to be responsible for the inhibition of Trx1 activity (cysteine-247) [16]. B) Truncation of the C-terminal tail (1-300) of Txnip does not affect Txnip-mediated suppression of HRE-luciferase activity. Conversely, truncation within the C-terminal α-arrestin domain (1-215) and of the C-terminal α-arrestin domain (1-151) abolishes suppression of HRE-luciferase activity by Txnip. Data are expressed as mean ± SEM and are representative of 4 individual experiments. HRE-luciferase activity was measured in lysates from MLE-12 cells overexpressing full-length Txnip (1-391) or one of three C-terminal truncation mutants (1-300, 1-215, and 1-151) and exposed to 1% oxygen as described in Methods. HRE-luciferase activity was decreased by 43% and 39%, respectively, in cells expressing Txnip-1-391 and Txnip-1-300 compared to control-transfected cells. HRE-luciferase activity was 117% of control in cells expressing Txnip-1-215 and was decreased by 7% in cells expressing Txnip-1-151 compared to control-transfected cells (n=24; *p<0.001 vs empty vector; ¥p<0.05 vs Txnip-1-215).
HIF-1α is posttranslationally regulated by oxygen-dependent prolyl hydroxylases that facilitate the binding of von Hippel-Lindau protein (pVHL) to HIF-α to target it for proteasomal degradation [13, 19]. In 293T cells, Shin et al. demonstrated that Txnip (via amino acids 148-235) binds to pVHL and enhances the interaction of HIF-1α with pVHL in the nucleus [19]. Furthermore, a CRM-1 nuclear export sequence was identified in the C-terminal region of Txnip that was shown to mediate the nuclear export of the pVHL/HIF-1α complex to the cytoplasm. Mutations in either the pVHL binding region or the CRM-1 consensus sequence of Txnip were associated with a decrease in HIF-1α nuclear translocation and an increase in HIF promoter activity. The Txnip mutant used in the present studies with a truncation at the start of the C-terminal tail (1-300) possesses both the pVHL binding region and the CRM-1 nuclear export sequence identified by Shin et al (Figure 5A). Conversely, neither of the Txnip mutants with truncations in the C-terminal α-arrestin domain (1-215 or 1-151) contains the pVHL binding region or the CRM-1 nuclear export sequence. Our finding that Txnip truncated at start of the C-terminal tail (1-300) suppressed HIF promoter activity equivalently to wild-type Txnip supports the model of Txnip-dependent regulation of HIF degradation proposed by Shin et al. Our data indicating that the Txnip mutants lacking complete pVHL or CRM-1 sequences (1-215 and 1-151) failed to suppress HIF activity further support the concept that regulation of HIF-dependent genes by Txnip is mediated through regulation of HIF degradation.
Summary/Conclusions
We speculate that Txnip is a critical regulator of VEGF expression and lung development. Suppression of basal HIF activity by Txnip underlies its fundamental importance in regulating HIF activation. Contrary to our hypothesis, however, the effect of Txnip on HIF activity appears to be independent of C247 and, therefore, is likely to be independent of binding to the Trx active site. These findings do not preclude the possibility that effect of Txnip on HIF activity is mediated through mechanism(s) of interaction with Trx that have yet to be identified. The multifunctional nature of Txnip, including its identification as the only Trx1-binding member of the α-arrestin family of proteins, makes the determination of the specific mechanism(s) by which Txnip regulates HIF activation complicated. We conclude that neither C247 nor the C-terminal tail of Txnip is required for suppression of HIF activation. Conversely, the C-terminal α-arrestin domain of Txnip is required for suppression of HIF activity, though it is unclear if this finding is unique to Txnip alone or if it is a fundamental property of all α-arrestins. Finally, our data support a model in which the effects of Txnip on HIF function are regulated through effects on HIF degradation. One must be cautious about generalizing the findings in this study to both HIF-α proteins because both HIF-1α and HIF-2α can regulate VEGF expression [14]. We speculate that the effects of Txnip on HIF transcriptional activity and VEGF expression are more likely to be mediated through alterations in HIF-1α given the oxygen concentrations used in the present studies. Ongoing studies will identify the mechanism by which Txnip regulates HIF-mediated VEGF expression as well as the precise role of Txnip in lung vascular and alveolar development.
Acknowledgments
The authors would like to thank Cynthia Hill, Morgan Locy, and Xiaomei Meng for their excellent technical assistance. This work was supported by the National Institutes of Health (K12 HD043372) and The Research Institute at Nationwide Children’s Hospital.
List of Abbreviations
- BPD
Bronchopulmonary dysplasia
- Txnip
thioredoxin interacting protein
- Trx1
thioredoxin-1
- HIF
hypoxia-inducible factor
- HRE
hypoxia response element
- pVHL
von Hippel-Lindau protein
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Van Marter LJ. Epidemiology of bronchopulmonary dysplasia. Semin Fetal Neonatal Med. 2009;14:358–366. doi: 10.1016/j.siny.2009.08.007. [DOI] [PubMed] [Google Scholar]
- 2.McLeod A, Ross P, Mitchell S, Tay D, Hunter L, Hall A, Paton J, Mutch L. Respiratory health in a total very low birthweight cohort and their classroom controls. Arch Dis Child. 1996;74:188–194. doi: 10.1136/adc.74.3.188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Thebaud B, Abman SH. Bronchopulmonary dysplasia: where have all the vessels gone? Roles of angiogenic growth factors in chronic lung disease. Am J Respir Crit Care Med. 2007;175:978–985. doi: 10.1164/rccm.200611-1660PP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Thebaud B, Ladha F, Michelakis ED, Sawicka M, Thurston G, Eaton F, Hashimoto K, Harry G, Haromy A, Korbutt G, Archer SL. Vascular endothelial growth factor gene therapy increases survival, promotes lung angiogenesis, and prevents alveolar damage in hyperoxia-induced lung injury: evidence that angiogenesis participates in alveolarization. Circulation. 2005;112:2477–2486. doi: 10.1161/CIRCULATIONAHA.105.541524. [DOI] [PubMed] [Google Scholar]
- 5.van Tuyl M, Liu J, Wang J, Kuliszewski M, Tibboel D, Post M. Role of oxygen and vascular development in epithelial branching morphogenesis of the developing mouse lung. Am J Physiol Lung Cell Mol Physiol. 2005;288:L167–178. doi: 10.1152/ajplung.00185.2004. [DOI] [PubMed] [Google Scholar]
- 6.Choi KS, Bae MK, Jeong JW, Moon HE, Kim KW. Hypoxia-induced angiogenesis during carcinogenesis. J Biochem Mol Biol. 2003;36:120–127. doi: 10.5483/bmbrep.2003.36.1.120. [DOI] [PubMed] [Google Scholar]
- 7.Shweiki D, Itin A, Soffer D, Keshet E. Vascular endothelial growth factor induced by hypoxia may mediate hypoxia-initiated angiogenesis. Nature. 1992;359:843–845. doi: 10.1038/359843a0. [DOI] [PubMed] [Google Scholar]
- 8.Asikainen TM, Chang LY, Coalson JJ, Schneider BK, Waleh NS, Ikegami M, Shannon JM, Winter VT, Grubb P, Clyman RI, Yoder BA, Crapo JD, White CW. Improved lung growth and function through hypoxia-inducible factor in primate chronic lung disease of prematurity. FASEB J. 2006;20:1698–1700. doi: 10.1096/fj.06-5887fje. [DOI] [PubMed] [Google Scholar]
- 9.Shifren JL, Doldi N, Ferrara N, Mesiano S, Jaffe RB. In the human fetus, vascular endothelial growth factor is expressed in epithelial cells and myocytes, but not vascular endothelium: implications for mode of action. J Clin Endocrinol Metab. 1994;79:316–322. doi: 10.1210/jcem.79.1.8027247. [DOI] [PubMed] [Google Scholar]
- 10.Jakkula M, Le Cras TD, Gebb S, Hirth KP, Tuder RM, Voelkel NF, Abman SH. Inhibition of angiogenesis decreases alveolarization in the developing rat lung. Am J Physiol Lung Cell Mol Physiol. 2000;279:L600–607. doi: 10.1152/ajplung.2000.279.3.L600. [DOI] [PubMed] [Google Scholar]
- 11.Galambos C, Ng YS, Ali A, Noguchi A, Lovejoy S, D’Amore PA, DeMello DE. Defective pulmonary development in the absence of heparin-binding vascular endothelial growth factor isoforms. Am J Respir Cell Mol Biol. 2002;27:194–203. doi: 10.1165/ajrcmb.27.2.4703. [DOI] [PubMed] [Google Scholar]
- 12.Gerber HP, Hillan KJ, Ryan AM, Kowalski J, Keller GA, Rangell L, Wright BD, Radtke F, Aguet M, Ferrara N. VEGF is required for growth and survival in neonatal mice. Development. 1999;126:1149–1159. doi: 10.1242/dev.126.6.1149. [DOI] [PubMed] [Google Scholar]
- 13.Rajatapiti P, van der Horst IW, de Rooij JD, Tran MG, Maxwell PH, Tibboel D, Rottier R, de Krijger RR. Expression of hypoxia-inducible factors in normal human lung development. Pediatr Dev Pathol. 2008;11:193–199. doi: 10.2350/07-04-0257.1. [DOI] [PubMed] [Google Scholar]
- 14.Compernolle V, Brusselmans K, Acker T, Hoet P, Tjwa M, Beck H, Plaisance S, Dor Y, Keshet E, Lupu F, Nemery B, Dewerchin M, Van Veldhoven P, Plate K, Moons L, Collen D, Carmeliet P. Loss of HIF-2alpha and inhibition of VEGF impair fetal lung maturation, whereas treatment with VEGF prevents fatal respiratory distress in premature mice. Nat Med. 2002;8:702–710. doi: 10.1038/nm721. [DOI] [PubMed] [Google Scholar]
- 15.Patwari P, Chutkow WA, Cummings K, Verstraeten VL, Lammerding J, Schreiter ER, Lee RT. Thioredoxin-independent regulation of metabolism by the alpha-arrestin proteins. J Biol Chem. 2009;284:24996–25003. doi: 10.1074/jbc.M109.018093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Patwari P, Higgins LJ, Chutkow WA, Yoshioka J, Lee RT. The interaction of thioredoxin with Txnip. Evidence for formation of a mixed disulfide by disulfide exchange. J Biol Chem. 2006;281:21884–21891. doi: 10.1074/jbc.M600427200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Welsh SJ, Bellamy WT, Briehl MM, Powis G. The redox protein thioredoxin-1 (Trx-1) increases hypoxia-inducible factor 1alpha protein expression: Trx-1 overexpression results in increased vascular endothelial growth factor production and enhanced tumor angiogenesis. Cancer Res. 2002;62:5089–5095. [PubMed] [Google Scholar]
- 18.Ema M, Hirota K, Mimura J, Abe H, Yodoi J, Sogawa K, Poellinger L, Fujii-Kuriyama Y. Molecular mechanisms of transcription activation by HLF and HIF1alpha in response to hypoxia: their stabilization and redox signal-induced interaction with CBP/p300. EMBO J. 1999;18:1905–1914. doi: 10.1093/emboj/18.7.1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shin D, Jeon JH, Jeong M, Suh HW, Kim S, Kim HC, Moon OS, Kim YS, Chung JW, Yoon SR, Kim WH, Choi I. VDUP1 mediates nuclear export of HIF1alpha via CRM1-dependent pathway. Biochim Biophys Acta. 2008;1783:838–848. doi: 10.1016/j.bbamcr.2007.10.012. [DOI] [PubMed] [Google Scholar]
- 20.Tipple TE, Welty SE, Nelin LD, Hansen JM, Rogers LK. Alterations of the thioredoxin system by hyperoxia: implications for alveolar development. Am J Respir Cell Mol Biol. 2009;41:612–619. doi: 10.1165/rcmb.2008-0224OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chandel NS, McClintock DS, Feliciano CE, Wood TM, Melendez JA, Rodriguez AM, Schumacker PT. Reactive oxygen species generated at mitochondrial complex III stabilize hypoxia-inducible factor-1alpha during hypoxia: a mechanism of O2 sensing. J Biol Chem. 2000;275:25130–25138. doi: 10.1074/jbc.M001914200. [DOI] [PubMed] [Google Scholar]
- 22.Filby CE, Hooper SB, Sozo F, Zahra VA, Flecknoe SJ, Wallace MJ. VDUP1: a potential mediator of expansion-induced lung growth and epithelial cell differentiation in the ovine fetus. Am J Physiol Lung Cell Mol Physiol. 2006;290:L250–258. doi: 10.1152/ajplung.00244.2005. [DOI] [PubMed] [Google Scholar]
- 23.Maniscalco WM, Watkins RH, D’Angio CT, Ryan RM. Hyperoxic injury decreases alveolar epithelial cell expression of vascular endothelial growth factor (VEGF) in neonatal rabbit lung. Am J Respir Cell Mol Biol. 1997;16:557–567. doi: 10.1165/ajrcmb.16.5.9160838. [DOI] [PubMed] [Google Scholar]
- 24.Maniscalco WM, Watkins RH, Finkelstein JN, Campbell MH. Vascular endothelial growth factor mRNA increases in alveolar epithelial cells during recovery from oxygen injury. Am J Respir Cell Mol Biol. 1995;13:377–386. doi: 10.1165/ajrcmb.13.4.7546767. [DOI] [PubMed] [Google Scholar]
- 25.Asikainen TM, Schneider BK, Waleh NS, Clyman RI, Ho WB, Flippin LA, Gunzler V, White CW. Activation of hypoxia-inducible factors in hyperoxia through prolyl 4-hydroxylase blockade in cells and explants of primate lung. Proc Natl Acad Sci U S A. 2005;102:10212–10217. doi: 10.1073/pnas.0504520102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Asikainen TM, Waleh NS, Schneider BK, Clyman RI, White CW. Enhancement of angiogenic effectors through hypoxia-inducible factor in preterm primate lung in vivo. Am J Physiol Lung Cell Mol Physiol. 2006;291:L588–595. doi: 10.1152/ajplung.00098.2006. [DOI] [PubMed] [Google Scholar]
- 27.Asikainen TM, Ahmad A, Schneider BK, Ho WB, Arend M, Brenner M, Gunzler V, White CW. Stimulation of HIF-1alpha, HIF-2alpha, and VEGF by prolyl 4-hydroxylase inhibition in human lung endothelial and epithelial cells. Free Radic Biol Med. 2005;38:1002–1013. doi: 10.1016/j.freeradbiomed.2004.12.004. [DOI] [PubMed] [Google Scholar]
- 28.Kunig AM, Balasubramaniam V, Markham NE, Morgan D, Montgomery G, Grover TR, Abman SH. Recombinant human VEGF treatment enhances alveolarization after hyperoxic lung injury in neonatal rats. Am J Physiol Lung Cell Mol Physiol. 2005;289:L529–535. doi: 10.1152/ajplung.00336.2004. [DOI] [PubMed] [Google Scholar]
- 29.Kunig AM, Balasubramaniam V, Markham NE, Seedorf G, Gien J, Abman SH. Recombinant human VEGF treatment transiently increases lung edema but enhances lung structure after neonatal hyperoxia. Am J Physiol Lung Cell Mol Physiol. 2006;291:L1068–1078. doi: 10.1152/ajplung.00093.2006. [DOI] [PubMed] [Google Scholar]
- 30.Alvarez CE. On the origins of arrestin and rhodopsin. BMC Evol Biol. 2008;8:222. doi: 10.1186/1471-2148-8-222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hui ST, Andres AM, Miller AK, Spann NJ, Potter DW, Post NM, Chen AZ, Sachithanantham S, Jung DY, Kim JK, Davis RA. Txnip balances metabolic and growth signaling via PTEN disulfide reduction. Proc Natl Acad Sci U S A. 2008;105:3921–3926. doi: 10.1073/pnas.0800293105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yoshioka J, Imahashi K, Gabel SA, Chutkow WA, Burds AA, Gannon J, Schulze PC, MacGillivray C, London RE, Murphy E, Lee RT. Targeted deletion of thioredoxin-interacting protein regulates cardiac dysfunction in response to pressure overload. Circ Res. 2007;101:1328–1338. doi: 10.1161/CIRCRESAHA.106.160515. [DOI] [PubMed] [Google Scholar]
- 33.Chutkow WA, Patwari P, Yoshioka J, Lee RT. Thioredoxin-interacting protein (Txnip) is a critical regulator of hepatic glucose production. J Biol Chem. 2008;283:2397–2406. doi: 10.1074/jbc.M708169200. [DOI] [PubMed] [Google Scholar]
- 34.Mogami H, Yura S, Tatsumi K, Fujii T, Fujita K, Kakui K, Kondoh E, Inoue T, Fujii S, Yodoi J, Konishi I. Thioredoxin binding protein-2 inhibits excessive fetal hypoglycemia during maternal starvation by suppressing insulin secretion in mice. Pediatr Res. 67:138–143. doi: 10.1203/PDR.0b013e3181c2f4cc. [DOI] [PubMed] [Google Scholar]