

Abstract
CCN5 is one of six proteins in the CCN family. This family of proteins has been shown to play important roles in many processes, including proliferation, migration, adhesion, extracellular matrix regulation, angiogenesis, tumorigenesis, fibrosis, and implantation. In this review, we focus on the biological and putative pathophysiological roles of CCN5. This intriguing protein is structurally unique among the CCN family members, and has a unique biological activity profile as well.
Keywords: CCN proteins, Matricellular, Nuclear localization, Review
Introduction
CCN5 is a member of the CCN family proteins, named after the first 3 members of the family discovered: Cyr61, CTGF, and Nov (Chen and Lau 2009). The functional basis of the six CCN proteins appears to lie within the modular domain of their protein structure:
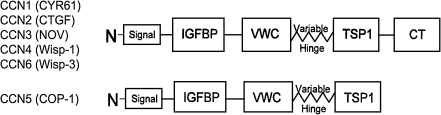 The majority of the CCN family members have four domains and an N-terminal secretion signal, or signal peptide. All six CCN proteins have conserved domains with sequence homology to insulin-like growth factor binding protein (IGFBP), von Willebrand factor type C repeat (vWC), and thrombospondin type I repeat (TSP). All CCN proteins except CCN5 have a fourth domain, a cysteine-rich carboxyl-terminal repeat (CT) (Bork 1993), although CCN5 is still a cysteine-rich protein, retaining 29 cysteines (28 in rat) within its three domains. Between the vWC and TSP domains there is a sequence of amino acids that is highly variable between CCN family members and acts as a molecular “hinge”. This flexible region is highly susceptible to proteolytic degradation by MMPs (Hashimoto et al. 2002).
The majority of the CCN family members have four domains and an N-terminal secretion signal, or signal peptide. All six CCN proteins have conserved domains with sequence homology to insulin-like growth factor binding protein (IGFBP), von Willebrand factor type C repeat (vWC), and thrombospondin type I repeat (TSP). All CCN proteins except CCN5 have a fourth domain, a cysteine-rich carboxyl-terminal repeat (CT) (Bork 1993), although CCN5 is still a cysteine-rich protein, retaining 29 cysteines (28 in rat) within its three domains. Between the vWC and TSP domains there is a sequence of amino acids that is highly variable between CCN family members and acts as a molecular “hinge”. This flexible region is highly susceptible to proteolytic degradation by MMPs (Hashimoto et al. 2002).
As noted above, a critical structural difference between CCN5 and the other members of the CCN family is that CCN5 lacks the CT domain. The potential importance of this difference has been brought into focus as the CT domain of other CCN family members has been be shown to bind to and interact in a functional manner with a number of matrix molecules, integrins, and members of important signaling pathways such as Notch 1 and LRP6 (Holbourn et al. 2009). This has led some to hypothesize that CCN5 might act as a dominant negative regulator of other CCN family members (Kubota and Takigawa 2007). Despite the attractiveness of this hypothesis, to date there is little evidence to support this idea.
CCN5 was originally cloned and published by several groups in the late 1990s. The first publication noting CCN5 was work from our laboratory (Delmolino et al. 1997). Our work on heparin as a cell-specific inhibitor of smooth muscle cell (SMC) proliferation inhibitor led us to query the genes and proteins regulated by this sulfated glycosaminoglycan using subtractive hybridization. We compared heparin-treated vascular SMC (VSMC) to chondroitin sulfate-treated VSMC, since chondroitin sulfate is a glycosaminoglycan closely related to heparin but devoid of anti-proliferative activity. This analysis revealed16 unique up-regulated clones and 25 unique down-regulated clones. DNA sequencing of one of the up-regulated clones revealed the presence of a novel transcript that was closely related to members of the CCN gene family (Delmolino et al. 2001; Lake et al. 2003) and we originally named the protein Heparin-Induced CCN-like Protein (HICP). RNAi knockdown of CCN5 in VSMC caused a 5-fold increase in the ED50 of heparin, indicating that the antiproliferative activity of heparin is strongly dependent upon CCN5 (Lake and Castellot 2003).
Independently, another group used mRNA differential display to identify a gene they named rCOP-1 as being down-regulated following transformation of rat embryo fibroblasts by inactivation of p53 and concomitant expression of a constitutively active H-ras (Zhang et al. 1998). rCop-1 (CCN5) was found to located primarily on the surface and within the cytoplasm of rat embryonic fibroblasts. Further, retroviral overexpression of rCOP-1 was found to induce apoptosis in transformed rat fibroblasts, but was unable to affect normal fibroblasts.
At about the same time, another group used selective subtractive hybridization to identify a gene they named Wnt-Inducible Secreted Protein-2 (WISP-2) because it was upregulated in the mouse mammary epithelial cell line C57MG after transformation by Wnt-1 (Pennica et al. 1998). The human WISP-2 (CCN5) gene was mapped to chromosome 20q12-20q13 and was found to have a copy number 2–4 fold higher in 23 of 25 primary human colon adenocarcinomas tested when compared to normal tissue. Paradoxically, despite the increased copy numbers of the gene in adenocarcinomas, levels of mRNA transcript for WISP-2 were down-regulated significantly in 15 of 19 human colon adenocarcinomas tested when compared to normal tissue.
A fourth group analyzed a human osteoblast cDNA library and identified an EST that contained an IGF binding domain, and based on sequence homology to the CCN family member CTGF (CCN2) named the presumed gene product CTGF-like protein or CTGF-L (Kumar et al. 1999). CTGF-L (CCN5) mRNA was found to be expressed in primary cultures of human osteoblasts, fibroblasts, ovary, testes, and heart. In situ hybridization identified CTGF-L mRNA as being highly expressed in bone-forming osteoblasts, alkaline phosphatase positive bone marrow cells, and chondrocytes. Functional studies showed the ability of endogenous CTGF-L protein to bind IGF-1 and IGF-2 and recombinant CTGF-L protein to promote the adhesion of osteoblasts, inhibit the binding of fibrinogen to purified integrin receptors and inhibit the production of osteocalcin by rat osteoblast-like Ros 17/2.8 cells.
These early studies have laid the foundation for nearly 100 publications on CCN5 and its role in cell signaling, adhesion, migration, and proliferation as well as its larger role in development and human disease. Based on the recommendations of the International CCN Society, HICP, rCOP1, WISP-2, and CTGF-L are now called CCN5, although WISP-2 is still commonly used, especially in the cancer literature.
CCN5 regulation and signaling
In the decade following the identification of CCN5 a steadily expanding literature has developed that examines possible signal transduction cascades involved in CCN5 expression and mechanisms of CCN5 gene regulation. As with many new areas of scientific study, efforts have tended to focus on well-established signaling cascades and particularly significant human disease states. In the case of CCN5, much research effort has been focused on the role of Wnt signaling [a natural expectation following the early work of Pennica et al. (1998)], estrogen signaling, p53, mitogenic growth factors, matrix molecules, and environmental stimuli.
Wnt signaling
Following the identification of CCN5 (WISP-2) as a Wnt-inducible protein, a number of investigators pursued the relationship between this signaling cascade and the function of CCN5. Wnt proteins bind to the cell surface receptors Frizzled (Fzd) and LRP5/6, thereby inhibiting glycogen synthase kinase (GSK-3β) and casein kinase 1 (CK1). Inhibition of these enzymes allows the stabilization of cytoplasmic β-catenin and its eventual translocation to the nucleus, where it interacts with TCF/LEF transcription factors and induces transcription of a select subset of proteins (Zhong et al. 2006). As mentioned above, overexpression of Wnt protein either through retroviral infection or transgenic mouse pan-overexpression was found to upregulate CCN5 at both the mRNA and protein levels (Pennica et al. 1998). Similar results were obtained by expressing a constitutively active downstream Wnt signaling molecule, s/aβ-catenin, in synovial fibroblasts, where a 2.9 fold increase in CCN5 mRNA was observed (Tanaka et al. 2005). Addition of estrogen to the s/aβ-catenin expressing fibroblasts demonstrated a synergistic effect, boosting CCN5 mRNA expression ~35-fold higher than either s/aβ-catenin or estrogen effects alone.
Importantly, physiological activators of Wnt signaling have also been shown to induce CCN5 expression. Expression of the hepatitis C virus core protein in the human hepatocellular carcinoma cell line Huh-7 upregulated expression of Wnt -1 protein and subsequently induced CCN5 expression (Fukutomi et al. 2005). MC3T3-E1 osteoblast cells treated with a small molecule inhibitor of GSK-3β called GSK3βi (which causes nuclear accumulation of β-catenin and activation of canonical Wnt signaling) caused a 2-fold increase in CCN5 mRNA. When this inhibitor is combined with mechanical loading of the cells a synergistic effect is produced, increasing CCN5 mRNA expression to seven fold that of untreated control (Robinson et al. 2006). Similar results were found when MC3T3-E1 osteoblasts cells were placed under mechanical strain and treated with Wnt3A, with a 7-fold increase in CCN5 mRNA compared to controls.
Alternatively, the signaling machinery of the Wnt signaling cascade can be engaged by other signaling molecules to increase CCN5 expression. Treatment of the human osteoblast cell line Saos-2 with either parathyroid hormone (PTH) or the adenylate cyclase activator forskolin causes activation of protein kinase A (PKA), which then phosphorylates GSK-3β Ser9. This in turn causes inactivation of the protein and activation of the downstream Wnt signaling pathway, with eventual upregulation of CCN5 transcription (McManus et al. 2005; Suzuki et al. 2008). Similar increases in CCN5 mRNA have also been noted in MCF-7 cells treated with the PKA activator, CT/IBMX (Inadera 2003). The effects of PKA signaling to increase CCN5 expression have also been shown in a non-canonical pathway of signaling. Primary pigmented nodular adrenocortical disease (PPNAD) is caused by inactivating mutations in the PKA regulatory subunit type 1A, resulting in increased PKA activity upon stimulation by cAMP. These tumors are also associated with decreased expression of the microRNA, miR449. Using serial analysis of gene expression data along with bioinformatics algorithms to predict miRNA-gene target pairs, the target of miR-449 was found to be CCN5 (Iliopoulos et al. 2009). Thus, in PPNAD-derived cell lines, PKA is over activated, resulting in decreased miR449 and increased expression of CCN5. By inhibiting the activity of PKA, levels of miR449 are increased, thereby decreasing CCN5 expression. This effect is not limited to PPNAD derived cell lines, as the osteoblast cell line Saos-2 experiences a significant down-regulation of miR-449 after treatment with the PKA activator, forskolin.
Estrogen signaling
Estrogen became a focus of CCN5 regulation when the Banerjee laboratory identified it early on as an mRNA and protein that was significantly upregulated in estrogen receptor (ER) positive breast cancer cell lines following treatment with estrogen (Banerjee et al. 2003; Inadera et al. 2000; Zoubine et al. 2001). Importantly, they were able to extend this observation to the in vivo setting by showing a strong correlation between expression of CCN5 protein and estrogen receptor alpha (ER-α) positivity in human breast cancer samples (Banerjee et al. 2003). Progesterone was also shown to induce CCN5 protein in ER-positive human breast cancer cells.
Using ten autologous pairs of SMC cultures derived from human uterine leiomyoma tumors and the surrounding normal myometrium, our laboratory found that CCN5 mRNA and protein levels were highly elevated in the non-proliferating normal myometrial SMC compared to the leiomyoma SMC. The greatest differences were seen in samples harvested during the proliferative (high estrogen) phase of the menstrual cycle (Mason et al. 2004b). Similarly, we were able to demonstrate a strong correlation between expression levels of CCN5 protein in the rat uterus and the rat estrus cycle, with the high levels of CCN5 protein found during the proestrus (high estrogen) phase of the cycle and very low levels observed in the diestrus (low estrogen) phase (Mason et al. 2004a). Furthermore, treatment of ovariectomized rats with estrogen increased CCN5 protein expression in the rat uterus 5.5 fold compared to control animals. Interestingly, unlike breast cancer cells, in which CCN5 was induced by both estrogen and progesterone, CCN5 mRNA and protein expression in the rat uterus was induced by estrogen but not progesterone. This is but one of many observations in the CCN5 field indicating that the regulation and function of CCN5 is highly dependent on the pathophysiologic milieu, which is consistent with other members of the CCN family whose matricellular actions and properties are highly contextual in nature.
The estrogen positive MCF-7 cell line has been useful for establishing CCN5 as a target of estrogen signaling and for exploring the mechanism of action of CCN5 in breast cancer cells. Addition of 17β-estradiol to MCF-7 cells in culture increases expression of CCN5 mRNA and protein in a dose-dependent manner (Banerjee et al. 2003; Inadera et al. 2002). CCN5 protein levels in MCF-7 cells increase over 24 h and can reach maximum levels by 72 h (Banerjee et al. 2003; Inadera et al. 2002). Activation of the signaling cascade was found to be specific to molecules that interact with ER as a number of other hormones and steroids, including dexamethasone, tri-iodothyronine and 2,3,7,8-tetrachlorodibenzo-p-dioxin, do not affect CCN5 expression, whereas several xenoestrogens increase CCN5 expression (Inadera et al. 2002). Further, the ability of estrogen to upregulate CCN5 expression was completely blocked by the pure anti-estrogen, ICI 182,780 (Banerjee et al. 2003; Inadera et al. 2000).
The estrogen receptor alpha (ER-α) appears to be primarily responsible for estrogen’s induction of CCN5 expression, as cultured human mammary epithelial cells that lack ER-α do not respond to estrogen stimulation with an upregulation of CCN5 expression. However, stable transfection of ER-α into these cells restores the ability of estrogen to induce CCN5 expression (Banerjee et al. 2003). There is some evidence to suggest that estrogen might also function to stabilize CCN5 mRNA (Banerjee et al. 2003). ER-α has been shown to interact with the human CCN5 promoter along with the CLIM and RLIM transcription cofactors, coregulatory proteins that usually interact and regulate with the LIM-homeodomain (LIM-HD) transcription factors for which they are named. Using an siRNA approach it was shown that knockdown of CLIM decreases ER-α-mediated CCN5 mRNA transcription, similar to the CLIM effects on LIM-HD transcription factors (Johnsen et al. 2009). However, in contrast to the negative regulatory effects observed for RLIM on LIM-HD transcriptional activity, knockdown of RLIM actually decreased CCN5 mRNA transcription.
Signal cascade cross-talk within the estrogen/CCN5 signaling cascade has also been the subject of several investigations. Treatment of MCF-7 cells with 12-O-tetradecanoylphorbol-13-acetate (TPA), a protein kinase C (PKC) activator, completely blocked estrogen induced CCN5 mRNA transcription although it had no effect on another estrogen responsive gene, pS2 (Inadera 2003). Epidermal growth factor (EGF) has been shown to induce expression of CCN5 mRNA in MCF-7 cells in a dose- and time-dependent manner and can act synergistically with estrogen to raise CCN5 expression levels, possibly through the PI3K and MAPK signaling pathways (Banerjee et al. 2005). EGF signaling in MCF-7 cells appears to require an ER since the pure ER antagonist, ICI 182,780, completely blocks EGF-induced up-regulation of CCN5, and EGF was unable to effect CCN5 expression in ER negative MDA-MB-231 cells. However, this unusual effect appears to be confined to MCF-7 cells since MDA-MB-231 cells transfected with ER-α do not show similar synergistic effects. A similar set of studies was performed by the same group for IGF-1 and noted similar results to those observed for EGF. IGF-1 can induce CCN5 mRNA expression in a dose and time-dependent manner and knockdown of CCN5 abrogates the ability of IGF-1 to stimulate MCF-7 cell proliferation (Dhar et al. 2007b). This IGF-1 induction of CCN5 expression can be blocked by a pure anti-estrogen drug, but unlike EGF the signaling cross-talk appears to function through PI3K/AKT signaling rather then MAPK.
In addition to estrogen and IGF, there is also evidence to suggest that progesterone might also be involved in molecular cross-talk with estrogen to stimulate CCN5 expression. Treatment of MCF-7 cells with progesterone alone induces expression of CCN5 mRNA, and this increase can be blocked by the progesterone receptor antagonist, RU486 (Banerjee et al. 2003). However, when estrogen and progesterone are given simultaneously, then both the estrogen and progesterone induction of CCN5 mRNA expression are blocked, with CCN5 mRNA remaining at basal levels.
Tumor suppressors, cytokines, and transcription factors
A series of reports have implicated CCN5 as a player in tumor suppressor and oncogene regulation. Characterization of CNN5 in rat embryonic fibroblasts showed concomitant inactivation of the master tumor suppressor p53 and activation of the oncogene H-ras induced the loss of CCN5 mRNA protein expression (Zhang et al. 1998). Tissue array analysis for pancreatic adenocarcinoma showed an association between the loss of CCN5 protein expression and the overexpression of p53 (which can often occur with p53 mutational changes) (Dhar et al. 2007a). Similar results were seen in other breast carcinoma cells lines, where overexpression of mutant p53 was associated with loss of CCN5 expression (Dhar et al. 2008). Expression of p53 mutants in the MCF-7 and ZR-75-1 non-invasive breast carcinoma cells lines knocked down CCN5 mRNA expression, despite the fact these cell lines usually constitutively express CCN5. Additional evidence for the role of CCN5 in oncogene/tumor suppressor function comes from studies of the tumor suppressor Tip30, which can inhibit estrogen-induced transcription in mammary epithelial cells. Knockout of Tip30 in mammary epithelial cells resulted in increased cell proliferation and a 40-fold increase in CCN5 mRNA expression (Pecha et al. 2007). siRNA mediated knockdown of CCN5 expression in Tip30−/− mammary epithelial cells inhibited cell proliferation by over 50%. Most recently, CCN5 mRNA expression was shown to be regulated by the LATS1/2 Ser/Thr kinases, tumor suppressors found to be mutated in a number human cancers and whose knockout in mice is associated with the development of soft tissue sarcomas and ovarian tumors. Knockdown of LATS1/2 expression by siRNA was found to upregulate expression CCN5 mRNA in HELA cells (Visser and Yang 2010).
Several cytokines and other modulators of cell function have been examined for their ability to regulate CCN5 levels. For example, TGF-β treatment of murine osteoblasts increased expression of CCN5 mRNA and protein in a time specific manner, with expression peaking at 24 h (Parisi et al. 2006). Nuclear run-on assays confirmed that TGFβ actually induces CCN5 transcription in murine osteoblasts. However, in rat vascular smooth muscle, TGFβ was unable to induce CCN5 expression (Delmolino et al. 2001). PMA, a phorbol ester produced by plants has been shown to induce proliferation in ER-α and CCN5 positive breast carcinoma cell lines, but not in breast cancer cell lines that lack CCN5 expression (Sengupta et al. 2006). PMA is believed to upregulate CCN5 expression through an interaction of the PKCα-MAPK/ERK and SAPK/JNK signaling pathways. Knockdown of CCN5 abrogates the ability of PMA to induce cellular proliferation in MCF-7 cells. At the transcriptional level, when MCF-7 cells are placed under hypoxic conditions, hypoxia-inducible factor-2α (HIF-2α) and the ETS transcription factor, ELK-1, have both been found to bind to the promoter of CCN5 and induce CCN5 mRNA expression (Aprelikova et al. 2006). In support of this observation, siRNA-mediated knockdown of ELK-1 inhibited the hypoxia-induced upregulation of CCN5 mRNA.
CCN5 and proliferation, motility, invasiveness, and adhesion
Of particular interest to translational research is that CCN5 has been shown to be involved in several important human diseases or conditions that are marked by aberrant cell proliferation and migration. Both cell culture and animal studies demonstrate a potential pathophysiologic and/or pharmacologic role for CCN5 in regulating cell proliferation, motility, invasiveness, and adhesion.
Proliferation
Of all of the cell functions mentioned above, the ability of CCN5 to affect cell proliferation has been studied in the greatest depth. For example, overexpression of CCN5 protein in VSMC, or treatment of VSMC with CCN5-containing culture medium, have both been shown to strongly inhibit proliferation (Delmolino et al. 2001; Lake et al. 2003). This effect has also been observed in human uterine myometrial cells and in autologously matched uterine leiomyoma tumor cells, where overexpression of CCN5 protein inhibits serum-induced proliferation of these cells by greater than 60% (Mason et al. 2004b). In murine 3T3-L1 preadipoctyes, overexpression of CCN5 protein was also able to inhibit the proliferation of this cells by ~30% (Inadera et al. 2009). In human umbilical vein endothelial cells a peptide fragment, -TAWGPCSTTCGLGMATRV- (Accession # o760769199-216) of CCN5’s thrombospondin, type I domain, dubbed ‘wispostatin’, has been shown to inhibit serum-induced proliferation by as much as 75% (Karagiannis and Popel 2007).
Overepxression of CCN5 has been shown to inhibit the serum-induced proliferation of the highly invasive, estrogen receptor negative, breast cancer cell line MDA-MB-231 (Fritah et al. 2008). Interestingly, in the case of the poorly invasive, estrogen receptor positive MCF-7 cell line, the effect of CCN5 is not as clear. Some studies have suggested an inhibitory role of CCN5 by demonstrating that overexpression of CCN5 protein inhibits the serum-induced proliferation of MCF-7 cells, while knockdown of CCN5 expression has the opposite effect, increasing cell proliferation through the shortening of the cell doubling time (Fritah et al. 2008). Other reports have suggested a stimulatory role by showing that knockdown of CCN5 expression either inhibited serum-induced MCF-7 cell proliferation (Banerjee et al. 2003) or had no effect (Banerjee et al. 2008). More studies are needed to resolve these paradoxical findings, and the answers will undoubtedly teach us much about the mechanism of action of CCN5.
Other evidence suggests that the ability of CCN5 knockdown to inhibit MCF-7 cell proliferation might be due to interactions with distinct growth factors or cellular signaling pathways. The ability of EGF, PMA, or IGF-1 alone to induce MCF-7 cell proliferation was abrogated by CCN5 knockdown (Banerjee et al. 2005; Dhar et al. 2007b; Sengupta et al. 2006). Similarly, Tip30−/− mammary epithelial cells were shown to have reduced proliferation in response to serum after CCN5 knockdown (Pecha et al. 2007). Interestingly, knockdown of CCN5 in MCF-7 cells was found to eliminate the estrogen dependent growth requirement of these cells (Fritah et al. 2008). It is possible that the conflicting results found for the role of CCN5 in breast carcinoma cell lines proliferation might be due to differences in the cell types tested, or in the specific protocols used to stimulate and measure proliferation by different investigators.
Motility, invasiveness, and metastasis
The aberrant migration of cells, including the ability to cross tissue boundaries (invasiveness or metastasis) is a critical step that precedes proliferation in the pathogenesis of many diseases, including cancer, atherosclerosis, restenosis following vascular surgery, LAM, uterine fibroids, persistent pulmonary hypertension of the newborn, asthma, and many others. The role of CCN5 in suppression of cell motility and invasiveness has been demonstrated in a number of different cell types.
In primary cultures of smooth muscle cells, the anti-motility and anti-invasive effects of CCN5 have also been well established. Overexpression of CCN5 in rat VSMC has been shown to inhibit both motility, invasiveness, and MMP-2 [a matrix metalloproteinase known to be involved in facilitating VSMC motility (Lake and Castellot 2003)] activity (Lake et al. 2003). Supporting this observation is the finding that RNAi knockdown of CCN5 within the same cell type stimulated motility and increased expression of MMP-2. Further corroboration for the anti-motility activity of CCN5 is the demonstration that ectopic expression of CCN5 in human myometrial and uterine leiomyoma cells inhibited the motility of both cell types by >50% (Mason et al. 2004b).
In the breast carcinoma cell line, MDA-MB-231, ectopic expression of CCN5 inhibited both motility and invasiveness of this aggressive breast carcinoma cell line (Fritah et al. 2008). These results were confirmed as the ability of MCF-7 conditioned media to inhibit the motility and invasiveness of MDA-MB-231 cells was abrogated by prior knockdown of CCN5 expression in the MCF-7 cells that created the conditioned media (Banerjee et al. 2008). The inhibitory effect of CCN5 on motility was also observed in MCF-7 cells where knockdown of CCN5 expression increased the IGF-1-induced motility of MCF-7 cells by 5-fold. CCN5 knockdown in MCF-7 cells also induced expression of pro-motility enzymes such as MMP-2 and MMP-9 (Banerjee et al. 2008; Fritah et al. 2008). Furthermore, MCF-7 exhibiting increased invasiveness as a result of mutant p53 overexpression were inhibited by treatment with recombinant CCN5 protein (Dhar et al. 2008).
Finally, the CCN5 thrombospondin, type I domain peptide fragment, wispostatin was found inhibit endothelial cell invasiveness by 60% (Karagiannis and Popel 2007). This is particularly interesting since the intact CCN5 protein, at least in our hands, has no effect on endothelial cell motility or proliferation in cell culture models; in fact, ectopic expression of full-length CCN5 protein in a mouse model for vascular injury strongly suppresses SMC motility and hyperplasia while permitting endothelial cell regeneration—as process that requires both motility and proliferation—to occur normally (unpublished data).
Adhesion
Unlike other members of the CCN family of genes, very little is known about the role CCN5 plays in cell adhesion. Kumar et al. observed that three different osteoblastic cell lines—primary human osteoblasts, osteosarcoma MG63, and rat osteoblast-like osteosarcoma Ros 17/2.8—attached to immobilized CCN5 in a dose dependent manner (Kumar et al. 1999). Further, possible mediators of adhesion were identified, as purified CCN5 was able to block the binding of αVβ3 integrin and αIIbβ3 integrins to fibrinogen, also in a dose-dependent manner. Recent unpublished data from our laboratory indicates that CCN5 may indeed regulate adhesion of SMC via its association with podosomes.
CCN5 and development
The relatively recent discovery of the CCN5 gene and protein coupled with the apparent early embryonic lethality of CCN5 null or transgenic mice (unpublished data) have somewhat hampered the study of CCN5’s functional roles in development. However, the fact that altering CCN5 expression in either direction leads to early embryonic death—both CCN5-null mice and transgenic mice overexpressing CCN5 die at or before the gastrulation stage and do not implant properly—strongly suggests that this protein plays a critical role in early embryonic development. Clearly, it will be necessary to generate conditional knockout and transgenic mice to study the role of CCN5 in development. Despite these difficulties, several studies have focused on CCN5 in adipocyte differentiation as well as expression patterns of CCN5 protein and mRNA both in the embryo and adult.
In a comprehensive temporal and physical mapping of embryonic gene and protein expression (see tables below), CCN5 protein was expressed in most tissues of both adult (Tables 1 and 2) and embryonic (Table 3) mice (Gray et al. 2007; Jones et al. 2007). CCN5 mRNA is expressed as early as the 4-cell embryo stage, and is quite strongly expressed by the morula, blastocyst and gastrulation stages (E3-E8; unpublished data). Other CCN proteins, notably CCN2 and CCN3, are expressed during the same time period and perhaps even earlier, at the 2-cell embryo stage, and CCN1 knockout mice have major defects in implantation and placentation (Mo et al. 2002). These observations imply a critical interplay between the CCN proteins during development that remains unexplored.
Table 1.
Summary of mouse tissue CCN5 and CCN2 expression
| Tissue | CCN2 | CCN5 |
|---|---|---|
| Heart | Cytoplasmic throughout ventricular and atrial myocardia and valves | Cystoplasmic throughout ventricular and atrial myocardia and valves |
| Lung | Cytoplasmic in all cell types; many nuclei | Cystoplasmic in all cell types; many nuclei |
| Stomach | Cytoplasmic—all cell types; nuclear-gastric pit cells | Cystoplasmic—all cell types; nuclear-gastic pit cells |
| Duodenum | Cytoplasmic—all cell types; some nuclei in villi | Cystoplasmic—all cell types; some nuclei in villi |
| Liver | Cytoplasmic in all cell types | Cystoplasmic in all cell types |
| Kidney | Cytoplasmic in all cell types; many nuclei in all cell types | Cystoplasmic in all cell types; many nuceli in all cell types |
| Ovary | Cytoplasmic in all cell types; many nuclei among all cell types | Cystoplasmic in all cell types; many nuclei among all cell types |
| Pancreas | Cytoplasmic in all cell types | Cystoplasmic in all cell types |
| Thymus | Cytoplasmic in all cell; nuclear in most cells | Cystoplasmic in all cells; nuclear in most cells |
| Olfactory | Cytoplasmic in most cell types; nuclear in some cell types | Cystoplasmic in most cell types; nuclear some cell types |
| Uterus | Cytoplasmic in all cell types; nuclear in some stroma cells | Cystoplasmic in all cell types; nuclear in some stroma cell |
| Fallopian tube | Cytoplasmic in all cell types; nuclear in some stroma cells | Cystoplasmic in all cell types; nuclear in some stroma cell |
| Skeletal muscle | Cytoplasmic in all cells | Cystoplasmic in all cells |
| Spleen | Cytoplasmic in all cells | Cystoplasmic in all cells |
Table 2.
Summary of rat CCN5 and CCN2 expression
| Tissue | CCN2 | CCN5 |
|---|---|---|
| Heart | Cytoplasmic throughout ventricular and atrial myocardia and valves | Cytoplasmic throughout ventricular and atrial myocardia and valves |
| Lung | Cytoplasmic in all cell types; many nuclei | Cytoplasmic in all cell types; many nuclei |
| Liver | Cytoplasmic in all cell types | Cytoplasmic in all cell types |
| Kidney | Cytoplasmic in all cell types; many nuclei in all cell types | Cytoplasmic in all cell types; many nuclei in all cell types |
| Ovary | Cytoplasmic in all cell types; many nuclei among all cell types | Cytoplasmic in all cell types; many nuclei among all cell types |
| Brain | Cytoplasmic in all cell types; nuclear in most cells | Cytoplasmic in all cell types; nuclear in most cells |
| Uterus | Cytoplasmic in all cell types; nuclear in some stroma cells | Cytoplasmic in all cell types; nuclear in some stroma cells |
| Fallopian tube | Cytoplasmic in all cell types; nuclear in some stroma cells | Cytoplasmic in all cell types; nuclear in some stroma cells |
| Skeletal muscle | Cytoplasmic in all cells | Cytoplasmic in all cells |
| Aorta | Cytoplasmic in all cell types | Cytoplasmic in all cell types |
| Spleen | Cytoplasmic in all cells | Cytoplasmic in all cells |
Table 3.
CCN2 and CCN5 expression in mammalian development
| CCN2 | CCN5 | |
|---|---|---|
| Cardiovascular system | ||
| Smooth muscle | + | + |
| Cardiac muscle | + | + |
| Endothelia | + | + |
| Nervous system | ||
| Brain | + | + |
| Meninges | + | + |
| Musculoskeletal system | ||
| Cartilage | + | + |
| Bone | + | + |
| Perichondrium | +/− | + |
| Skeletal muscle | +/− | ++ |
| Skin | ||
| Epidermis | ++ | ++ |
| Hair | + | + |
| Bronchioles | ++ | + |
| Blood vessels | +/− | + |
| Secretory tissues | ||
| Liver | + | + |
| Kidney | +/− | + |
| Reproductive organs | – | + |
| Placenta | nd | + |
| Digestive tract | ++ | + |
| Endocrine glands | +/− | + |
Moving into the early-mid stages of embryonic development of mice (E9-E11), CCN5 is expressed in tissues of ectodermal, mesodermal, and endodermal origins (Jones et al. 2007). In both early embryonic and adult tissues CCN5 was found to highly expressed in endothelium and smooth muscle of veins and arteries, as well as in the myocardium of the heart (Gray et al. 2007; Jones et al. 2007). Interestingly, in the adult mouse heart, CCN5 displayed nuclear localization, a phenomenon not found in the embryonic heart (Gray et al. 2007). In the intestines, CCN5 protein was found to be highly expressed in the tips of intestinal microvilli, while expression was absent in the intestinal crypts, further suggesting a role for CCN5 in regulating cell proliferation (Jones et al. 2007). In both embryonic and adult mice, the liver expressed some of the highest levels of CCN5 protein seen for any tissue. This expression was found in both the hepatocytes and endothelial cells. In the urogenital system, glomerular kidney endothelial cells do not express CCN5 protein while in the adult they express similar levels to those seen in other arteries and veins. In the brain of embryonic mice CCN5 protein levels were widely expressed, but this expression becomes more restricted from E12 to GD16. CCN5 protein expression was found to be highly localized to the nucleus of neurons both in embryonic and adult animals. However, in the adult animal, the proportion of neurons with nuclear localization of CCN5 varied widely according to the region of the brain.
In the human fetus, CCN5 protein is expressed throughout the myocardium, and in the endothelium and smooth muscle of coronary arteries at 4 months gestation (Jones et al. 2007). CCN5 expression in the larger blood vessels of the body was found to be somewhat less. In the lung, CCN5 is expressed at low, but uniform levels in the epithelium and mesenchymal cells at 5 months. In the developing bones of the human fetus at 5 months, CCN5 protein is detected in the nuclei of osteoclasts, but is completely absent from osteocytes. CCN5 protein expression levels in hepatocytes of the fetal liver increases from 4 to 5 months gestation, while CCN5 expression remains absent in hematopoietic stem cells. In the fetal adrenal gland, CCN5 protein was expressed in the zona fasciculata, the region responsible for the production of cortisol, while it was expressed at very low levels in the zona glomerulosa, the region responsible for aldosterone production. CCN5 expression was found to be absent from the human fetal brain at 4 months gestation, although the authors conclude that his might be due to the sections of tissue being taken from tissues cores that do not allow a full view brain.
Schutze and colleagues have focused on the role of CCN5 in adipogenesis. During the stimulated adipogenic differentiation of human bone marrow-derived mesenchymal stem cells (BMC), CCN5 mRNA expression levels decreased (Schutze et al. 2005). CCN5 mRNA was present in BMC and in the initial stage of adipocyte differentiation, but then decreased slowly as differentiation progressed, and was completely absent in differentiated adipocytes. This effect was not observed during osteogenic and chondrogenic differentiation of BMC, where CCN5 mRNA levels were the same throughout. Similar results for CCN5 mRNA in adipogenic differentiation of murine 3T3-L1 preadipocytes have also been reported (Inadera et al. 2009). However, in these studies, reduction in CCN5 mRNA was found to occur within 2 h of adipogenic differentiation and lasted for 8 h, at which point CCN5 mRNA levels had risen back to levels seen in untreated preadipocytes and continued at this level out 48 h. Furthermore, induction of the Wnt signaling cascade inhibited differentiation of preadipocytes into adipocytes while concurrently raising the levels of CCN5. Interestingly, overexpression of CCN5 alone failed to inhibit adipogenic differentiation, but did inhibit preadipocytes from becoming early adipocytes. These results suggest that while CCN5 might play a role in regulating the earliest stages of preadipocyte differentiation into adipocytes, once the cell has become an adipocyte, CCN5 is no longer involved in further differentiation of the adipocyte. The potential of CCN5 to limit the number of cells becoming full-fledged adipocytes is intriguing as it suggests the possibility of using CCN5 to control adipose tissue formation.
CCN5 and human disease
Similar to the other CCN family members, after the initial discovery of CCN5 investigators focused on a defining a role for CCN5 in human disease. Much of this effort has focused on neoplasia, leiomyoma (fibroids), pancreatic carcinoma, hepatocellular carcinoma, and skin carcinoma. Significant attention has also been focused on conditions involving hyperplasia rather than neoplasia, including arterial restenosis, atherosclerosis, macronodular adrenal hyperplasia, and rheumatoid arthritis.
Breast carcinoma
Several studies have indicated that increased levels of CCN5 expression in breast tumors correlate with severity of the disease and found little to no CCN5 expression in normal breast tissue (Banerjee et al. 2008; Banerjee et al. 2003; Davies et al. 2007). Of particular interest is the study by Davies et al. in which increased expression of CCN5 was found to be associated with a number of poor prognostic indicators including node positive tumors, higher grade, and metastases (Davies et al. 2007). However, these data are paradoxical when considered in light of the studies by Banerjee et al., who found increased levels of CCN5 in breast cancer and preneoplastic tissue when compared to normal tissue; in fact, the highest expression levels were seen in preneoplastic/neoplastic tissue (AHD and DCIS). Furthermore, the Banerjee group found that CCN5 expression decreased with tumor progression from non-invasive to invasive lesions (Banerjee et al. 2008) and did not find an association between increased CCN5 expression and node positive tumors, or the tumor grade (Banerjee et al. 2003, 2008). Finally, the poor prognostic indications found by Davies et al. is inconsistent with the findings of Banerjee et al. showing a significant association between CCN5 expression and ER-α positivity in invasive breast cancer samples (Banerjee et al. 2003). Estrogen receptor positive breast carcinoma is typically considered a good prognostic indicator since these tumors respond to selective estrogen receptor modulator (SERM) treatment. Additional studies are needed to resolve this paradox before any conclusions can be drawn with respect to the function of CCN5 in tumor progression.
Many of the studies in breast cancer have been extended to cell culture models where increased expression of CCN5 was found in a number of human breast carcinoma cell lines when compared to normal mammary epithelial cells (Saxena et al. 2001). Further studies showed that CCN5 expression in breast carcinoma cells lines could be induced by treatment with estrogen, progesterone, EGF and IGF-1 (Banerjee et al. 2003, 2005; Dhar et al. 2007b). In the case of EGF and IGF-1 this upregulation of CCN5 expression was dependent on the presence of a functional estrogen receptor since the pure antiestrogen ICI- 182,780 was able to block these mitogenic effects (Banerjee et al. 2005; Dhar et al. 2007b). Studies in breast carcinoma cell lines have also shown that CCN5 has a role in maintaining the non-invasive, differentiated phenotype of these cells. Poorly-invasive, low- metastasizing cell lines such as MCF-7, T47-D, ZR-75.1, and SKBr3 exhibit greatly increased expression of CCN5 compared to highly invasive and metastatic cell lines such as MDA-MB-231, MDA-MB-468, BT-20, and DU4475 (Fritah et al. 2008). As mentioned above, the effects of CCN5 on the proliferation of breast carcinoma cells have produced conflicting results. Knockdown of CCN5 in MCF-7 cells inhibits their serum-induced and IGF-1-induced proliferation (Banerjee et al. 2008; Fritah et al. 2008), and in the Tip 30−/− mouse model of ductal hyperplasia, knockdown of CCN5 in primary cultures of Tip 30−/− mammary epithelial cells inhibits their serum-induced proliferation (Pecha et al. 2007). Conversely, Fritah et al. show that overexpression of CCN5 in MCF-7 cells causes decreased DNA synthesis and that knockdown of CCN5 eliminates the estrogen requirement for MCF-7 cell proliferation in culture (Fritah et al. 2008). When addressing the role CCN5 plays in the invasive phenotype of breast carcinoma cell lines, the data are easier to interpret. Knockdown of CCN5 expression increases the motility and invasiveness of MCF-7 cells directly, while the overexpression of CCN5 within MDA-MB-231 cells inhibits the proliferation, motility, and invasiveness of these cells (Banerjee et al. 2008; Fritah et al. 2008). Finally, knockdown of CCN5 in MCF-7 cells causes a decrease in epithelial cell markers such as E-cadherin and cytokeratin 18, while simultaneously upregulating the mesenchymal marker vimentin (Banerjee et al. 2008; Fritah et al. 2008). Further, suppression of CCN5 expression upregulates the matrix metalloproteinases, MMP2 and MMP9, which are often found to be highly expressed in the invasive phenotype (Banerjee et al. 2008). Finally, CCN5 expression was found to be inversely correlated with the mutational activation of p53 in human breast carcinoma cells lines, and expression of mutated p53 in MCF-7 cells inhibits expression of CCN5 (Dhar et al. 2008). With the exception of the finding that CCN5 is required for serum-induced and IGF-1 induced proliferation of MCF-7 cells, these results suggest that CCN5 might have a role in maintaining a non-invasive epithelial phenotype in breast carcinoma cells.
Human pancreatic adenocarcinoma, hepatocellular carcinoma, and skin cancer
Similar to what is seen in breast carcinoma, pancreatic adenocarcinoma (PAC) exhibits greatly decreased levels of CCN5 expression compared to adjacent normal pancreatic tissue and areas of chronic pancreatitis (a precursor of PAC) (Dhar et al. 2007a). Also similar to the observations in breast carcinoma, CCN5 mRNA loss was associated with overexpression of p53 in PAC. Treatment of the highly aggressive PAC cell line, MIA-PaCa-2 with recombinant CCN5 protein reduced the expression of the mesenchymal cell marker vimentin and altered the gross appearance of the cells to a cobble stoned, epithelial-like phenotype. These results suggest that CCN5 might have a role in maintaining an epithelial-like phenotype in pancreatic adenocarcinoma cells and thereby decrease their invasive potential.
Overexpression of the Hepatitis C viral core protein in the human hepatocellular carcinoma derived cell line, Huh-7, caused upregulation of Wnt-1 and CCN5, a downstream target of Wnt signaling, in addition to causing increased proliferation of the cells (Fukutomi et al. 2005). Further study is required to establish a causal link for CCN5 in hepatocellular carcinoma.
CCN5 is the most highly expressed mRNA of the CCN family in normal human skin (Quan et al. 2009). Solar-simulated UV irradiation of human skin reduced the levels of CCN5 mRNA by 50% in a time-dependent manner and did not return to basal levels until 48 h post irradiation. These results suggest further study is warranted to address CCN5 levels in samples of frank skin carcinoma.
Pathologies of smooth muscle
Human uterine leiomyoma is characterized by benign neoplastic growths of uterine smooth muscle cells that form large tumors within the uterine wall. Their malignant transformation is exceedingly rare. CCN5 mRNA expression has been shown to be 6–8 fold lower in uterine leiomyoma when compared to the surrounding myometrial tissue (Mason et al. 2004b). Further, overexpression of CCN5 inhibited the proliferation and motility of human leiomyoma and myometrial cells in vitro. In a series of recent studies, we have shown that CCN5 strongly inhibits the ability of both human leiomyoma organoids and Eker rat tsc1 mutant cells to form fibroid-like tumors in immunocompromised NOD-SCID mice (unpublished data).
Arterial restenosis and atherosclerosis are both diseases that are defined by the pathological hyperplasia of VSMC in the artery wall that eventually leads to vessel occlusion and ischemia. In a rat model of arterial restenosis, CCN5 protein expression by VMSC in the artery wall is downregulated following artery injury, at which time VSMC of the artery wall are maximally proliferating. Following stenosis caused by a period of VSMC hyperplasia followed by cessation of VSMC growth, CCN5 protein expression levels returns to levels previously seen in the uninjured artery (Lake et al. 2003). This observation placed CCN5 at the right time and place in the injured artery to exert an effect on SMC proliferation. Overexpression of CCN5 protein in VSMC by introducing adenoviral vectors making CCN5 into the injured carotid artery for 25 min resulted in almost complete inhibition of proliferation, motility, and neointima formation. Importantly, the endothelial cells were not affected by CCN5 and regenerated normally. The effects of ectopic expression of CCN5 on proliferation and motility were also observed in cultured SMC from a variety of sources. Furthermore, knockdown of CCN5 in cultured VSMC causes increased motility and MMP-2 activity (Lake and Castellot 2003).
In a series of preliminary studies, we have shown that CCN5 inhibits proliferation and motility of human and mouse airway SMC. Remodeling of these cells is an important component of asthma that current therapies have not been able to address. We will soon test the intriguing possibility that CCN5 can prevent or reduce the remodeling of airway SMC that is a major problem in chronic asthma. Given the prevalence of asthma (nearly 20 million Americans have been diagnosed with it, and 5,000 individuals—mostly children—die from asthma each year), this is a potentially valuable clinical application of CCN5.
Taken together these results lend strong support to the hypothesis that CCN5 plays important physiologic roles in regulating smooth muscle proliferation and motility. The possible therapeutic use of CCN5 should be examined further using adenovirus-associated virus vectors (which appear to produce much less inflammation) and biologically active recombinant CCN5, when it becomes available.
Conclusion
Like its five family members, CCN5 is proving to be a protein with important roles in embryonic development, normal cell function, and disease. The functions of CCN5 are strongly dependent upon the particular cell and tissue context in which it is studied. Though potent viral expression vectors, highly specific antibodies, and RNAi constructs are now readily available to dissect the mechanism of action of CCN5, the availability of biologically active and stable recombinant protein is required to complete the armamentarium of tools needed to fully explore the physiologic and pathophysiologic roles of CCN5. Domain analysis of the CCN5 protein is still in the rudimentary stages, yet this approach is likely to be vital not only for understanding mechanism of action, but for the development of possible therapeutics as well.
Finally, we would note that an underexplored yet important area is the interaction between the different CCN family members. The intriguing idea that CCN5 is the “anti-CCN” that opposes the action of the other CCN proteins was clearly overly simplistic, as coordinate expression as well as opposite expression patterns are frequently observed in different tissues and disease states. Furthermore, some isoforms of CCN3 display the same biologic function as CCN5. Examining the interplay of the CCN proteins will undoubtedly lead to a more complete understanding of the biology and pathophysiology of this fascinating family of genes and proteins.
References
- Aprelikova O, Wood M, Tackett S, Chandramouli GV, Barrett JC. Role of ETS transcription factors in the hypoxia-inducible factor-2 target gene selection. Cancer Res. 2006;66:5641–5647. doi: 10.1158/0008-5472.CAN-05-3345. [DOI] [PubMed] [Google Scholar]
- Banerjee S, Saxena N, Sengupta K, Tawfik O, Mayo MS, Banerjee SK. WISP-2 gene in human breast cancer: estrogen and progesterone inducible expression and regulation of tumor cell proliferation. Neoplasia. 2003;5:63–73. doi: 10.1016/s1476-5586(03)80018-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee S, Sengupta K, Saxena NK, Dhar K, Banerjee SK. Epidermal growth factor induces WISP-2/CCN5 expression in estrogen receptor-alpha-positive breast tumor cells through multiple molecular cross-talks. Mol Cancer Res. 2005;3:151–162. doi: 10.1158/1541-7786.MCR-04-0130. [DOI] [PubMed] [Google Scholar]
- Banerjee S, Dhar G, Haque I, Kambhampati S, Mehta S, Sengupta K, Tawfik O, Phillips TA, Banerjee SK. CCN5/WISP-2 expression in breast adenocarcinoma is associated with less frequent progression of the disease and suppresses the invasive phenotypes of tumor cells. Cancer Res. 2008;68:7606–7612. doi: 10.1158/0008-5472.CAN-08-1461. [DOI] [PubMed] [Google Scholar]
- Bork P. The modular architecture of a new family of growth regulators related to connective tissue growth factor. FEBS Lett. 1993;327:125–130. doi: 10.1016/0014-5793(93)80155-N. [DOI] [PubMed] [Google Scholar]
- Chen CC, Lau LF. Functions and mechanisms of action of CCN matricellular proteins. Int J Biochem Cell Biol. 2009;41:771–783. doi: 10.1016/j.biocel.2008.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies SR, Watkins G, Mansel RE, Jiang WG. Differential expression and prognostic implications of the CCN family members WISP-1, WISP-2, and WISP-3 in human breast cancer. Ann Surg Oncol. 2007;14:1909–1918. doi: 10.1245/s10434-007-9376-x. [DOI] [PubMed] [Google Scholar]
- Delmolino LM, Stearns NA, Castellot JJ., JrHeparin induces a member of the CCN family which has characteristics of a growth arrest gene Mol Biol Cell 19978287a9190208 [Google Scholar]
- Delmolino LM, Stearns NA, Castellot JJ., Jr COP-1, a member of the CCN family, is a heparin-induced growth arrest specific gene in vascular smooth muscle cells. J Cell Physiol. 2001;188:45–55. doi: 10.1002/jcp.1100. [DOI] [PubMed] [Google Scholar]
- Dhar G, Mehta S, Banerjee S, Gardner A, McCarty BM, Mathur SC, Campbell DR, Kambhampati S, Banerjee SK. Loss of WISP-2/CCN5 signaling in human pancreatic cancer: a potential mechanism for epithelial-mesenchymal-transition. Cancer Lett. 2007;254:63–70. doi: 10.1016/j.canlet.2007.02.012. [DOI] [PubMed] [Google Scholar]
- Dhar K, Banerjee S, Dhar G, Sengupta K, Banerjee SK. Insulin-like growth factor-1 (IGF-1) induces WISP-2/CCN5 via multiple molecular cross-talks and is essential for mitogenic switch by IGF-1 axis in estrogen receptor-positive breast tumor cells. Cancer Res. 2007;67:1520–1526. doi: 10.1158/0008-5472.CAN-06-3753. [DOI] [PubMed] [Google Scholar]
- Dhar G, Banerjee S, Dhar K, Tawfik O, Mayo MS, Vanveldhuizen PJ, Banerjee SK. Gain of oncogenic function of p53 mutants induces invasive phenotypes in human breast cancer cells by silencing CCN5/WISP-2. Cancer Res. 2008;68:4580–4587. doi: 10.1158/0008-5472.CAN-08-0316. [DOI] [PubMed] [Google Scholar]
- Fritah A, Saucier C, Wever O, Bracke M, Bieche I, Lidereau R, Gespach C, Drouot S, Redeuilh G, Sabbah M. Role of WISP-2/CCN5 in the maintenance of a differentiated and noninvasive phenotype in human breast cancer cells. Mol Cell Biol. 2008;28:1114–1123. doi: 10.1128/MCB.01335-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukutomi T, Zhou Y, Kawai S, Eguchi H, Wands JR, Li J. Hepatitis C virus core protein stimulates hepatocyte growth: correlation with upregulation of wnt-1 expression. Hepatology. 2005;41:1096–1105. doi: 10.1002/hep.20668. [DOI] [PubMed] [Google Scholar]
- Gray MR, Malmquist JA, Sullivan M, Blea M, Castellot JJ., Jr CCN5 Expression in mammals. II. Adult rodent tissues. J Cell Commun Signal. 2007;1:145–158. doi: 10.1007/s12079-007-0013-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto G, Inoki I, Fujii Y, Aoki T, Ikeda E, Okada Y. Matrix metalloproteinases cleave connective tissue growth factor and reactivate angiogenic activity of vascular endothelial growth factor 165. J Biol Chem. 2002;277:36288–36295. doi: 10.1074/jbc.M201674200. [DOI] [PubMed] [Google Scholar]
- Holbourn KP, Perbal B, Ravi Acharya K. Proteins on the catwalk: modelling the structural domains of the CCN family of proteins. J Cell Commun Signal. 2009;3:25–41. doi: 10.1007/s12079-009-0048-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iliopoulos D, Bimpaki EI, Nesterova M, Stratakis CA (2009) MicroRNA signature of primary pigmented nodular adrenocortical disease: clinical correlations and regulation of Wnt signaling. Cancer Res 69(8):3278–3782 [DOI] [PMC free article] [PubMed]
- Inadera H. Estrogen-induced genes, WISP-2 and pS2, respond divergently to protein kinase pathway. Biochem Biophys Res Commun. 2003;309:272–278. doi: 10.1016/j.bbrc.2003.07.001. [DOI] [PubMed] [Google Scholar]
- Inadera H, Hashimoto S, Dong HY, Suzuki T, Nagai S, Yamashita T, Toyoda N, Matsushima K. WISP-2 as a novel estrogen-responsive gene in human breast cancer cells. Biochem Biophys Res Commun. 2000;275:108–114. doi: 10.1006/bbrc.2000.3276. [DOI] [PubMed] [Google Scholar]
- Inadera H, Dong HY, Matsushima K. WISP-2 is a secreted protein and can be a marker of estrogen exposure in MCF-7 cells. Biochem Biophys Res Commun. 2002;294:602–608. doi: 10.1016/S0006-291X(02)00530-2. [DOI] [PubMed] [Google Scholar]
- Inadera H, Shimomura A, Tachibana S. Effect of Wnt-1 inducible signaling pathway protein-2 (WISP-2/CCN5), a downstream protein of Wnt signaling, on adipocyte differentiation. Biochem Biophys Res Commun. 2009;379:969–974. doi: 10.1016/j.bbrc.2008.12.185. [DOI] [PubMed] [Google Scholar]
- Johnsen SA, Gungor C, Prenzel T, Riethdorf S, Riethdorf L, Taniguchi-Ishigaki N, Rau T, Tursun B, Furlow JD, Sauter G, Scheffner M, Pantel K, Gannon F, Bach I. Regulation of estrogen-dependent transcription by the LIM cofactors CLIM and RLIM in breast cancer. Cancer Res. 2009;69:128–136. doi: 10.1158/0008-5472.CAN-08-1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones JA, Gray MR, Oliveira BE, Koch M, Castellot JJ., Jr CCN5 expression in mammals: I. Embryonic and fetal tissues of mouse and human. J Cell Commun Signal. 2007;1:127–143. doi: 10.1007/s12079-007-0012-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karagiannis ED, Popel AS. Peptides derived from type I thrombospondin repeat-containing proteins of the CCN family inhibit proliferation and migration of endothelial cells. Int J Biochem Cell Biol. 2007;39:2314–2323. doi: 10.1016/j.biocel.2007.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubota S, Takigawa M. CCN family proteins and angiogenesis: from embryo to adulthood. Angiogenesis. 2007;10:1–11. doi: 10.1007/s10456-006-9058-5. [DOI] [PubMed] [Google Scholar]
- Kumar S, Hand AT, Connor JR, Dodds RA, Ryan PJ, Trill JJ, Fisher SM, Nuttall ME, Lipshutz DB, Zou C, Hwang SM, Votta BJ, James IE, Rieman DJ, Gowen M, Lee JC. Identification and cloning of a connective tissue growth factor-like cDNA from human osteoblasts encoding a novel regulator of osteoblast functions. J Biol Chem. 1999;274:17123–17131. doi: 10.1074/jbc.274.24.17123. [DOI] [PubMed] [Google Scholar]
- Lake AC, Castellot JJ., Jr CCN5 modulates the antiproliferative effect of heparin and regulates cell motility in vascular smooth muscle cells. Cell Commun Signal. 2003;1:5. doi: 10.1186/1478-811X-1-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lake AC, Bialik A, Walsh K, Castellot JJ., Jr CCN5 is a growth arrest-specific gene that regulates smooth muscle cell proliferation and motility. Am J Pathol. 2003;162:219–231. doi: 10.1016/S0002-9440(10)63813-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mason HR, Grove-Strawser D, Rubin BS, Nowak RA, Castellot JJ., Jr Estrogen induces CCN5 expression in the rat uterus in vivo. Endocrinology. 2004;145:976–982. doi: 10.1210/en.2003-0823. [DOI] [PubMed] [Google Scholar]
- Mason HR, Lake AC, Wubben JE, Nowak RA, Castellot JJ., Jr The growth arrest-specific gene CCN5 is deficient in human leiomyomas and inhibits the proliferation and motility of cultured human uterine smooth muscle cells. Mol Hum Reprod. 2004;10:181–187. doi: 10.1093/molehr/gah028. [DOI] [PubMed] [Google Scholar]
- McManus EJ, Sakamoto K, Armit LJ, Ronaldson L, Shpiro N, Marquez R, Alessi DR. Role that phosphorylation of GSK3 plays in insulin and Wnt signalling defined by knockin analysis. EMBO J. 2005;24:1571–1583. doi: 10.1038/sj.emboj.7600633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mo FE, Muntean AG, Chen CC, Stolz DB, Watkins SC, Lau LF. CYR61 (CCN1) is essential for placental development and vascular integrity. Mol Cell Biol. 2002;22:8709–8720. doi: 10.1128/MCB.22.24.8709-8720.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parisi MS, Gazzerro E, Rydziel S, Canalis E. Expression and regulation of CCN genes in murine osteoblasts. Bone. 2006;38:671–677. doi: 10.1016/j.bone.2005.10.005. [DOI] [PubMed] [Google Scholar]
- Pecha J, Ankrapp D, Jiang C, Tang W, Hoshino I, Bruck K, Wagner KU, Xiao H. Deletion of Tip30 leads to rapid immortalization of murine mammary epithelial cells and ductal hyperplasia in the mammary gland. Oncogene. 2007;26:7423–7431. doi: 10.1038/sj.onc.1210548. [DOI] [PubMed] [Google Scholar]
- Pennica D, Swanson TA, Welsh JW, Roy MA, Lawrence DA, Lee J, Brush J, Taneyhill LA, Deuel B, Lew M, Watanabe C, Cohen RL, Melhem MF, Finley GG, Quirke P, Goddard AD, Hillan KJ, Gurney AL, Botstein D, Levine AJ. WISP genes are members of the connective tissue growth factor family that are up-regulated in wnt-1-transformed cells and aberrantly expressed in human colon tumors. Proc Natl Acad Sci USA. 1998;95:14717–14722. doi: 10.1073/pnas.95.25.14717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quan T, Shin S, Qin Z, Fisher GJ. Expression of CCN family of genes in human skin in vivo and alterations by solar-simulated ultraviolet irradiation. J Cell Commun Signal. 2009;3:19–23. doi: 10.1007/s12079-009-0044-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson JA, Chatterjee-Kishore M, Yaworsky PJ, Cullen DM, Zhao W, Li C, Kharode Y, Sauter L, Babij P, Brown EL, Hill AA, Akhter MP, Johnson ML, Recker RR, Komm BS, Bex FJ. Wnt/beta-catenin signaling is a normal physiological response to mechanical loading in bone. J Biol Chem. 2006;281:31720–31728. doi: 10.1074/jbc.M602308200. [DOI] [PubMed] [Google Scholar]
- Saxena N, Banerjee S, Sengupta K, Zoubine MN, Banerjee SK. Differential expression of WISP-1 and WISP-2 genes in normal and transformed human breast cell lines. Mol Cell Biochem. 2001;228:99–104. doi: 10.1023/A:1013338912642. [DOI] [PubMed] [Google Scholar]
- Schutze N, Noth U, Schneidereit J, Hendrich C, Jakob F. Differential expression of CCN-family members in primary human bone marrow-derived mesenchymal stem cells during osteogenic, chondrogenic and adipogenic differentiation. Cell Commun Signal. 2005;3:5. doi: 10.1186/1478-811X-3-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sengupta K, Banerjee S, Dhar K, Saxena NK, Mehta S, Campbell DR, Banerjee SK. WISP-2/CCN5 is involved as a novel signaling intermediate in phorbol ester-protein kinase Calpha-mediated breast tumor cell proliferation. Biochemistry. 2006;45:10698–10709. doi: 10.1021/bi060888p. [DOI] [PubMed] [Google Scholar]
- Suzuki A, Ozono K, Kubota T, Kondou H, Tachikawa K, Michigami T. PTH/cAMP/PKA signaling facilitates canonical Wnt signaling via inactivation of glycogen synthase kinase-3beta in osteoblastic Saos-2 cells. J Cell Biochem. 2008;104:304–317. doi: 10.1002/jcb.21626. [DOI] [PubMed] [Google Scholar]
- Tanaka I, Morikawa M, Okuse T, Shirakawa M, Imai K (2005) Expression and regulation of WISP2 in rheumatoid arthritic synovium. Biochem Biophys Res Commun 334(4):973–997 [DOI] [PubMed]
- Visser S, Yang X. Identification of LATS transcriptional targets in HeLa cells using whole human genome oligonucleotide microarray. Gene. 2010;449:22–29. doi: 10.1016/j.gene.2009.09.008. [DOI] [PubMed] [Google Scholar]
- Zhang R, Averboukh L, Zhu W, Zhang H, Jo H, Dempsey PJ, Coffey RJ, Pardee AB, Liang P. Identification of rCop-1, a new member of the CCN protein family, as a negative regulator for cell transformation. Mol Cell Biol. 1998;18:6131–6141. doi: 10.1128/mcb.18.10.6131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong N, Gersch RP, Hadjiargyrou M. Wnt signaling activation during bone regeneration and the role of dishevelled in chondrocyte proliferation and differentiation. Bone. 2006;39:5–16. doi: 10.1016/j.bone.2005.12.008. [DOI] [PubMed] [Google Scholar]
- Zoubine MN, Banerjee S, Saxena NK, Campbell DR, Banerjee SK. WISP-2: a serum-inducible gene differentially expressed in human normal breast epithelial cells and in MCF-7 breast tumor cells. Biochem Biophys Res Commun. 2001;282:421–425. doi: 10.1006/bbrc.2001.4584. [DOI] [PubMed] [Google Scholar]