

Summary
In high throughput screening (HTS) campaigns, the quality and cost of commercial reagents suitable for pilot studies often create obstacles upon scale-up to a full screen. We faced such challenges in our efforts to implement HTS for inhibitors of the phosphopantetheinyl transferase Sfp using an assay that had been validated using commercially available reagents. Here we demonstrate a facile route to the synthetic preparation of reactive tetraethylrhodamine and a quencher probes; and their application to economically produce fluorescent and quencher-modified substrates. These probes were prepared on a scale that would allow a full, quantitative HTS of more than 350,000 compounds.
Introduction
Phosphopantetheinylation is an important posttranslational modification representing an obligatory step that activates enzymes producing fatty acid, polyketide and nonribosomal peptide compounds.1 Compounds from these classes are essential to bacterial cell viability and many have been identified as small molecule virulence factors.2 The central role of this posttranslational modification to cellular maintenance and pathogenicity has not gone unnoticed as chemical actuation of this process has potential to produce antimicrobial therapeutics with a novel mode of action.3
The phosphopantetheine functionality is installed onto synthase enzymes by action of phosphopantetheinyl transferase, in conjunction with a coenzyme A (CoA) cosubstate.1 To further our development of a method to selectively isolate and identify synthase enzymes, we have sought to identify inhibitors of Sfp, a canonical representative of this enzyme class.4 To this end, we have designed a novel FRET assay platform and miniaturized it into a high-throughput screen (HTS) protocol.5 In this system, action of the enzyme assembles fluorescent tetramethylrhodamine-CoA 1 and quencher-modified acceptor peptide 2 into a non-emitting “dark” product 3 (Fig. 1).
Fig. 1.
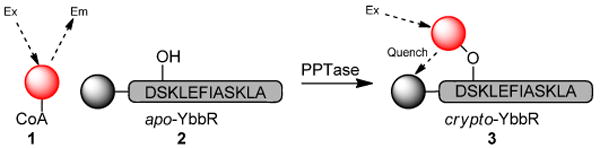
Phosphopantetheinyl transferase assay format Action of PPTase with rhodamine-CoA 1 on quencher –modified YbbR acceptor peptide 2, assembles a FRET pair 3 and decreases the rhodamine-CoA fluorescence signal.
This miniaturization study identified the first known inhibitors of this enzyme, and we now seek to further unveil new active scaffolds through the screening of large chemical libraries. Previously, pilot quantities of reagents were prepared from commercially available TAMRA maleimide 4 and Black Hole Quencher-2 (BHQ-2) carboxylic acid 5 (Fig. 2). However, in turning to interrogate large compound collections with this screen, we found the high cost of 4 and 5 prohibitive toward our ability to supply reagents. In this report, we detail our economical preparation of structurally and spectroscopically similar rhodamine and quencher probes to support large-scale HTS campaigns.
Fig. 2.
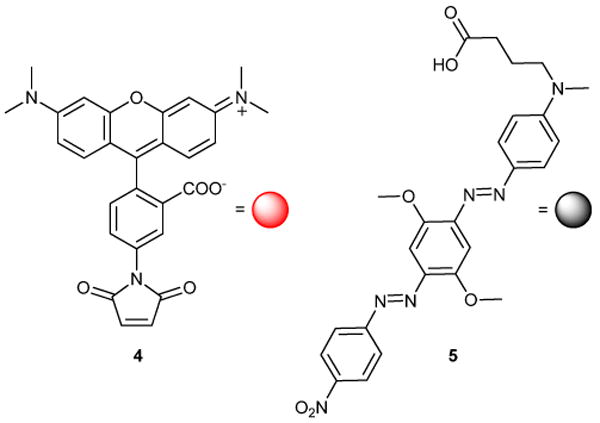
Commercially available tetramethylrhodamine maleimide 4 and Black Hole Quencher-2 carboxylic acid 5 were used to prepare pilot reagents 1 and 2.
Results and Discussion
We estimated the minimum reagent need for the quantitative HTS6 of the NIH Molecular Libraries Small Molecule Repository (311,260 compounds)7 to be 120 mg of rhodamine CoA and 400 mg of quencher-peptide. While the former was prepared from 4, no suitably inexpensive tetramethyl-precursors could be identified. We chose to evaluate a tetraethyl- analogue, Rhodamine WT 6 (Scheme 1), that is marketed for water-tracing applications and recently adapted for use by McCafferty & coworkers.8 We acquired a sample of this material (60 g) as a mixture of isomers from Pylam Dyes; available for ∼$170/lb (Pylam Dyes, Tempe, AZ, USA). This mixture may be resolved by reversed phase flash chromatography9 using methanol: 0.003% phosphoric acid as a mobile phase10 in modest yield (Scheme 1). We chose to carry forward isomer-II 7, as it more closely resembles TAMRA maleimide 4 with respect to substitution about the benzoyl ring, given that FRET characteristics depend heavily on alignment of molecular dipoles.11 The maleimide linker was assembled by mono-BOC protection of ethylene diamine 8,12 followed by conversion to maleimide 10 with N-(methoxycarbonyl)-maliemide 9 (Scheme 1).13 After purification, the BOC-group was removed with TFA/CH2Cl2 to provide TFA ammonium salt 11.
Scheme 1.
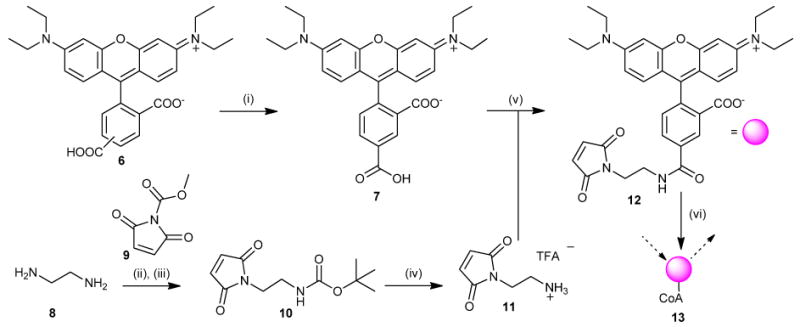
Rhodamine WT (i) C18-silica, MeOH/0.003% H3PO4, 20% (ii) (BOC)2O, DCM, 87%. (iii) 6, NaHCO3, THF, 58%. (iv) TFA, DCM quant. (v) EtiPr2N, HBTU, DMF. 76% (vi) Coenzyme A, Na-H2PO4, quant.
From here, we evaluated several activation schemes for 7. These experiments identified the HOBt ester to react readily with amine 11 in anhydrous DMF, and provided 12 in very good yield (Scheme 1). Conversion of maleimide 12 to the Coenzyme A analogue 13 proceeded quantitatively in phosphate buffer and the latter was biochemically indistinguishable from 1 (vide infra).
Finding no preparative literature concerning BHQ-2 carboxylic acid 5, we developed a facile route based on published patents (Scheme 2).14 This sequence began with diazotization of aniline 14 in 3M HCl. Dilution with hydrofluoroboric acid yielded the expected diazonium tetrafluoroborate salt. Conversion to diazoaniline 16 was accomplished by slow addition to 15 in DMF. Moving forward, the aqueous insolubility of 16 required conversion to the diazonium salt in concentrated sulfuric acid with nitrosylsulfuric acid as the oxidant. Subsequent recovery of the tetrafluoroborate salt and reaction with aniline 17 cleanly afforded alcohol 18.
Scheme 2.
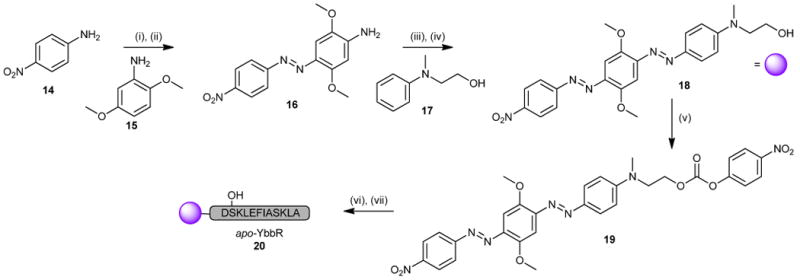
(i) 3M HCl,NaNO2; HBF4. (ii) 2,5-dimethoxyaniline, DMF, 69%. (iii) HSO3NO, H2SO4; HBF4. (iv) N-methyl-N-(2-hydroxyethyl)-aniline, DMF, 47% (2 steps). (v) p-NO2PhCOCl, DIPEA, quant. (vi) H2N-Ahx-DSKLEFIASKLA-O-[PEG]-PS resin, DIPEA. (vii) 96:2:2 TFA:TIPS:H2O.
To activate alcohol 18 for peptide coupling, we had initially planned for oxidation to the corresponding carboxylic acid. However, the compound proved unreactive or prone to decomposition with common oxidants. As such, we investigated the use of an activated carbonate to install the alcohol onto the peptide. In this respect, we found that 18 could be converted to p-nitrophenyl carbonate 19 with ease (Scheme 2).15
We prepared the final product by assembling the aminocaproate-terminated YbbR peptide (sequence: NH2-Ahx-DSKLEFIASKLA–CO2H)16 using FMOC-based solid phase peptide synthesis protocols on a 0.3 mmol scale.17 Treatment of the resin with 2 molar equivalents (630 mg) of 19 overnight gave clean conversion to peptide 20. HPLC purification of this material afforded 228 mg of product at > 98% purity (Scheme 2). The process was iterated three times to satisfy the projected amount required for HTS (> 500 mg).
Evaluation of the above-prepared reagents 13 and 20 demonstrated that they performed indistinguishably from their commercial counterparts, providing a stable and highly reproducible dose-response screen of the LOPAC1280 (Library of Pharmacologically Active Compounds, Sigma-Aldrich), performed in triplicate using a fully-automated robotic screening system (Fig. 3).18 As seen in Fig. 3A, the robotic screen was associated with consistently high Z'-factor 19 and near-constant IC50 values for an inhibitor dose-response series included on every screening plate. Furthermore, two previously noted screening hits,5 PD 404,182 and calmidazolium chloride, displayed uniform concentration-response curves which overlapped those previously obtained in with the initial reagent set (Fig. 3B). These results confirmed that the assay retained its sensitivity to inhibitors with the new reagents.
Fig. 3.

Utilization of the new reagents in a triplicate robotic screen of the LOPAC1280 library. (A) Excellent screening assay performance as evidenced by the consistently high Z' factor and the unchanging IC50 for a control inhibitor SCH-202676. (B) Switching to the new reagents did not change the assay's sensitivity to inhibitors as evidenced by the nearly identical dose responses obtained for two inhibitors of different potency, PD 404,182 (average IC50 3.2 μM) and calmidazolium chloride (average IC50 20 μM) when reagents 1 and 2 (PD 404,182, ○; calmidazolium chloride, □) or 13 and 20 (PD 404,12: ◆, ●, and *; calidazolium chloride: ■, ▲, and ▼) were used.
Conclusion
The methods presented here for the general preparation of tetraethylrhodamine and quencher reporters are economical and executable at a preparative scale. Preparation of these probes enabled the initiation of a HTS campaign where the cost and availability of commercial probes had previously limited access. These procedures should be found applicable not only for the preparation of labeled peptides as above, but also as a direct route to probes for other FRET-based assay platforms (i.e. nucleic acids).
Experimental
All reagents and chemical compounds were used as purchased from commercial sources unless noted. Pyridine was distilled from KOH. Stirring was accomplished magnetically with a teflon-coated stir bar, and all non-aqueous reactions were performed under a balloon of dry argon in septum-sealed, oven-dried glassware. When required, compounds were purified via flash chromatography20 on 230-400 mesh Silica Gel 60 (EMD Chemicals, Gibbstown, NJ, USA). Analytical TLC was performed using 250 μm silica layers on glass plates (Silica Gel 60 F254, EMD Chemicals, Gibbstown, NJ, USA) and separated compounds visualized by illumination with UV light. Analytical reversed phase TLC (rp-TLC) was performed using 200 μm layers of Partisil® KC18 on glass plates (Cat. No. 4801-425, Whatman Inc, GE Healthcare, Piscataway, NJ, USA). High pressure liquid chromatography (HPLC) separations were performed with an Agilent 1100 series instrument (Agilent Technologies, Santa Clara, CA, USA) equipped with a preparative scale autosampler (fitted with a 900 μL injection loop) and a diode array detector. Analytical separations were performed on a 4.6 × 150 mm Ultrasphere ODS® column (Cat. No. 235330, Beckman Coulter, Brea, CA, USA) with injection volumes ranging from 50 – 100 μL. Semipreparative separations were performed on a 10 × 250 mm Biotage KP-C18-HS 35/70u column (Cat. No. S1L0-1119-95050, Biotage, Charlotte, NC, USA) using an injection volume of 500 – 900 μL. Preparative reversed phase separations were performed on a Waters instrument (Waters, Milford, MA, USA) comprised of a model 680 gradient controller, model 510 pumps fitted with high flow volume (225 μL) heads, model 2487 dual wavelength absorbance detector, Hewlett Packard 3396-series II integrator (Agilent Technologies, Santa Clara, CA, USA), Pharmacia Frac-100 fraction collector (GE Healthcare, Piscataway, NJ, USA) and a 22 × 250 mm Econosphere C18 (10 μm) column (cat. no. 50195422, W.R. Grace & Co., Deerfield, IL, USA). Prior to separation, samples were prepared using Waters SepPak C18 (Waters, Milford, MA, USA) solid phase extraction columns to remove salts and nonpolar contaminants that irreversibly adsorb to reverse phase media. Characterization data and yields correspond to homogeneous materials. NMR data were collected on a 300 MHZ Varian Mercury or 400 MHz Varian Mercury Plus spectrometers operating at 300.077 MHz or 399.913 MHz for 1H-NMR and 75.462 MHz or 100.567 MHz for 13C-NMR, respectively, at the UCSD Department of Chemistry and Biochemistry NMR facility. FID files were processed using MestReNova software version 6.1.0 (MestreLab Research, Escondido, CA, USA). Chemical shifts were calibrated using the residual solvent resonance21: D6-DMSO (δ 2.50, pentet, 1H-NMR) and (δ 39.52, heptet, 13C-NMR), D1-chloroform (δ 7.26, singlet, 1H-NMR) and (δ 77.16, triplet, 13C-NMR), or D4-methanol (δ 3.31, pentet, 1H-NMR) and (δ 49.00, heptet, 13C-NMR). Resonance multiplicities are reported as s = singlet, d = doublet, t = triplet, q = quartet, p = pentet, dd = doublet of doublets, m = multiplet. 1H-NMR data are reported as follows: Chemical shift (number of protons, multiplicity, coupling constants). Mass spectrometric data for all compounds except 13 and 20 were collected by Dr. Yongxuan Su at the UCSD Department of Chemistry and Biochemistry Small Molecule Mass Spectrometry facility on Finnigan LCQDECA and ThermoFinnigan MAT900XL spectrometers. Mass spectrometric data for compounds 13 and 20 were collected by Dr. William Leister at the NIH Chemical Genomics Center using an Agilent 6210 time-of-flight mass spectrometer fitted with a 1200 series HPLC system for sample introduction (Agilent Technologies, Santa Clara, CA, USA).
Separation of Rhodamine WT isomers10
Rhodamine WT 6 (1 g) was dissolved in methanol (200 mL). Dichloromethane was added (400 mL) and the solution extracted with 1M HCl (400 mL). The organic phase was concentrated in vacuo to give a burgundy solid. The solid was dissolved in methanol (50 mL) and diluted with 0.003% phosphoric acid (70 mL). This solution was loaded on a preparative C18-silica column (7 cm × 20 cm bed dimensions) equilibrated in 35:65 methanol: 0.003% phosphoric acid. The isomers were resolved by a step gradient from 35 to 60% methanol that increased in increments of 5% methanol. Fractions containing the separated isomers were pooled, diluted with an equal volume 1M HCl and extracted with dichloromethane (3 × total volume). the pooled organic phases were dried over Na2SO4 and evaporated to give rhodamine WT isomer I (355 mg, 35%) and rhodamine WT isomer II 7 (162 mg, 16%) in a ratio of approximately 2:1 (isomer I: isomer II).
Isomer I
δ H (400 MHz, cd3od) 8.44 – 8.33 (2 H, m), 7.98 (1 H, d, J 1.1), 7.12 (2 H, d, J 9.5), 7.03 (2 H, dd, J 9.5, 2.4), 6.98 (2 H, d, J 2.4), 3.67 (8 H, q, J 7.1), 1.30 (12 H, t, J 7.1). δ C (101 MHz, DMSO) 166.59, 166.53, 157.53, 155.37, 135.11, 131.56, 131.49, 131.06, 114.82, 113.10, 96.62, 45.91, 13.12. MS (ESI) m/z 487.40 ([M+]+, 100%); HRMS (ESI-FT) m/z calcd for C29H31N2O5 487.2227, found 487.2229.
isomer II 7
δ H (400 MHz, CD3OD) 8.90 (1 H, d, J 1.4), 8.41 (1 H, dd, J 7.9, 1.5), 7.53 (1 H, d, J 7.9), 7.10 (2 H, d, J 9.5), 7.03 (2 H, dd, J 9.5, 2.2), 6.97 (2 H, d, J 2.2), 3.67 (8 H, q, J 7.0), 1.30 (12 H, t, J 7.0) δ C (75 MHz, CDCl3) 170.72, 170.24, 162.60, 161.96, 159.85, 142.10, 137.20, 137.06, 136.98, 136.28, 136.11, 135.98, 135.04, 134.90, 118.31, 117.26, 100.13, 100.02, 49.65, 15.75, 15.60. MS (ESI) m/z 487.42 ([M+]+, 100%); HRMS (ESI-FT) m/z calcd for C29H31N2O5 487.2227, found 487.2233.
tert-butyl 2-aminoethylcarbamate 2212
A solution of 1,2-diaminoethane 8 (7.7 mL, 114 mmol) in chloroform (450 mL) was prepared and cooled to 0°C in an ice bath with stirring. A solution of di-tert-butyl dicarbonate (5.26mL, 22.9 mmol) in chloroform (46mL) was cooled to 0°C and added dropwise via a pressure equalizing addition funnel over 2h. The ice bath was removed and the reaction stirred overnight at room temperature to give a heterogenous solution. Solids were filtered and the filtrate evaporated. The resulting oil was dissolved in EtOAc (100 mL), washed with half-saturated brine (3 × 50 mL), dried over Na2SO4, and concentrated in vacuo to give the monoamine product (3.220 g, 20.1 mmol, 87 %). δ H (400 MHz, CDCl3) 3.15 (2 H, dd, J 11.5, 5.7), 2.77 (2 H, t, J 5.9), 1.42 (9 H, s, J 9.4). δ C (101 MHz, CDCl3) 177.14, 156.47, 79.28, 43.30, 41.63, 28.44. MS (ESI) m/z 160.95 ([M+H]+, 100%); HRMS (ESI-FT) m/z calcd for C7H17N2O2 161.1285, found 161.1286.
N-(methoxycarbonyl)-maleimide 922
To a solution of maleimide (5 g, 51.5 mmol) in ethyl acetate (250 mL) was added N-methyl morpholine (5.6 mL, 51.5 mmol) via syringe and the solution cooled on ice for 20 minutes. Methyl chloroformate (4.8 mL, 61.8 mmol) was added dropwise and the reaction stirred 30 min. Solids were collected on a Bqüchner funnel and washed with ethyl acetate (100 mL). The Filtrate and washes were combined, washed successively with water (1 × 100 mL) and brine (1 × 100 mL). The organic phase was dried over Na2SO4 and evaporated to dryness. The resulting solid was recrystallized from EtOAc/iPr2O to give methyl carbamate 9 (6.211 g, 40 mmol, 78 %). NB: DIPEA is not an acceptable substitute for NMM. δ H (400 MHz, CDCl3) 6.84 (2 H, s, J 4.6), 3.94 (2 H, s, J 0.4). δ C (101 MHz, CDCl3) 165.89, 148.32, 135.53, 54.52. MS (ESI) m/z 156.04 ([M+H]+, 100%); HRMS (ESI-FT) m/z calcd for C6H6NO4 156.0291, found 156.0293.
tert-butyl 2-(2,5-dioxo-2,5-dihydro-1H-pyrrol-1-yl)ethylcarbamate 1013
A solution of tert-butyl 2-aminoethylcarbamate (1.13 g, 7.1 mmol) in saturated sodium bicarbonate (35 mL) was prepared, the resulting solids filtered, and cooled to 0°C. Finely ground N-methoxycarbonyl maleimide 9 (1.1 g, 7.1 mmol) was added and the reaction stirred for 15 minutes at room temperature. THF (55 mL) was added and the reaction stirred 45 min. Water (50 mL) was added and the solution washed with ethyl acetate (3 × 75 mL). The organic washes were pooled, dried over Na2SO4, and concentrated to give an oil that solidified upon standing. The residue was purified by chromatography on SiO2 with a step gradient of hexane: ethyl acetate to give the title compound 10 as a white solid (1.0922 g, 4.55 mmol, 58 %). δ H (400 MHz, CDCl3) 6.68 (2 H, s), 3.67 – 3.55 (2 H, m), 3.29 (2 H, dd, J 11.1, 5.8), 1.36 (9 H, s). δ C (101 MHz, CDCl3) 171.04, 134.37, 79.67, 39.53, 38.19, 28.51. MS (ESI) m/z 263.04 ([M+Na]+, 100%); HRMS (ESI-FT) m/z calcd for C11H16N2O4Na 263.1002, found 263.1008.
2-(2,5-dioxo-2,5-dihydro-1H-pyrrol-1-yl)ethanammonium trifluoroacetate 11
tert-butyl 2-(2,5-dioxo-2,5-dihydro-1H-pyrrol-1-yl)ethylcarbamate 10 (1.000 g, 4.15 mmol) was dissolved in dichloromethane (10 mL) and cooled on ice. Trifluoroacetic acid (2 mL) was added and the solution stirred overnight. The reaction was diluted into diethylether (38 mL), cooled on ice 1 h, and filtered to provide the ammonium salt 11 (1.051 g, 4.14 mmol, 99%) as a white crystalline solid. δ H (300 MHz, CD3OD) 6.89 (2 H, s), 3.90 – 3.73 (2 H, t, J 5.6), 3.24 – 3.09 (2 H, t, J 5.6). δ C (75 MHz, CD3OD) 171.19, 162.47, 162.01, 161.55, 161.09, 134.56, 38.65, 35.05. MS (ESI) m/z 141.09 ([M+H]+, 100%); HRMS (ESI-FT) m/z calcd for C6H9N2O2 141.0658, found 141.0659.
Rhodamine-WT malemide 12
Rhodamine WT isomer II 7 (10 mg, 0.02 mmol) was dissolved in DMF (2 mL). HBTU (8.56mg, 0.02 mmol) and DIPEA (5.31 mg, 7.15 μL, 0.04) are added successively with stirring. After 30 minutes, the reaction was checked by rp-TLC (CH3OH / 0.003% H3PO4; 80/20) to ensure the formation of the HOBt ester (Rf = 0; deep purple). N-(2-aminoethyl) maleimide TFA salt 11 (10.43mg, 0.04 mmol) was added and the reaction followed to completion by rp-TLC (product Rf = 0.5).The reaction was diluted with dichloromethane (50 mL) and washed with NaH2PO4 buffer (50 mM, pH 7.4, 1 × 50 mL), brine (1 × 50 mL), dried over Na2SO4, and evaporated. The resulting purple solid was further purified by reversed phase HPLC to yield maleimide 12 (9.5 mg, 76 %). δ H (400 MHz, CD3OD) 8.69 (1 H, d, J 1.6), 8.16 (1 H, dd, J 7.9, 1.7), 7.51 (1 H, d, J 7.9), 7.12 (3 H, d, J 9.5), 7.03 (3 H, dd, J 9.5, 2.3), 6.98 (3 H, d, J 2.3), 6.84 (2 H, s), 3.87 – 3.76 (2 H, m), 3.75 – 3.60 (15 H, m), 1.30 (27 H, t, J 7.2). δ C (101 MHz, CDCl3) 171.37, 159.61, 157.95, 155.64, 136.66, 135.98, 134.40, 131.80, 131.65, 130.27, 130.20, 114.16, 113.69, 96.21, 46.11, 39.02, 37.69, 12.67. MS (ESI) m/z 609.47 ([M+]+, 100%); HRMS (ESI-FT) m/z calcd for C35H37N4O6 609.2708, found 609.2720.
Rhodamine coenzyme A 13
Coenzyme A (76 mg, 0.10 mmol) was dissolved in 20 mM NaH2PO4 pH 7.4 (100 mL) and cooled on ice. A solution of rhodamine maleimide 12 (76 mg, 0.13 mmol) in methanol (20 mL) was added in 1 mL portions. The flask was wrapped in foil and stirred for 3 h; at which point no detectable coenzyme A was present (determined by HPLC). The reaction was transferred to a separatory funnel and washed with dichloromethane (5 × 100 mL). The resulting aqueous phase was passed over a Waters SepPak solid phase extraction column equilibrated in 0.05% TFA, washed with 5 column volumes 20% acetonitrile/0.05% TFA and rhodamine CoA 13 eluted with 80% acetonitrile/0.05% TFA. Acetonitrile was removed by rotary evaporation and provided 13 as a fine purple precipitate. HRMS (ESI-TOF) m/z calcd for C56H74N11O22P3S 1377.3938, found 1377.3934.
(E)-2,5-dimethoxy-4-((4-nitrophenyl)diazenyl)aniline 1614
Sigma-Aldrich supplied 2,5-dimethoxyaniline 15 as pellets of a black solid that required purification before proceeding. Aniline 15 (10 g) was dissolved in ethyl acetate (200 mL) and the persisting solids filtered to give a black solution. Activated carbon (2 g) was added and stirred 20 minutes; after which Celite® was added and the solution filtered. The resulting faint yellow filtrate could not be further decolorized by reiteration of the above treatment, and was concentrated in vacuo to give a solid. This crude material was recrystallized from boiling hexanes to give 15 as white crystalline flakes.
2,5-dimethoxyaniline 15 (0.9506 g, 6.21 mmol) was dissolved in DMF (10 mL). A solution of p-diazoniumnitrobenzene tetrafluoroborate (prepared from 14, see ref. 14) in DMF (10mL) was added slowly and evolved a deep red color. Saturated sodium bicarbonate was added every 5 min (6 × 1 mL additions). After 1h, the reaction was diluted with H2O (200 mL) and placed on ice. The dark volumous precipitate was collected by filtration, washed with H2O, and dessicated over Ca2CO3. The resulting solid is recrystallized from Hexanes:EtOAc (3:1) to yield the diazoaniline 16 (1.24 g, 4.1 mmol, 69%) as metallic green crystals. δ H (400 MHz, CDCl3) 8.30 (2 H, d, J 9.1), 7.90 (2 H, d, J 9.0), 7.40 (2 H, s), 6.35 (2 H, s), 4.61 (2 H, s), 3.98 (3 H, s), 3.90 (3 H, s). δ C (101 MHz, CDCl3) 157.56, 156.74, 146.87, 145.79, 142.18, 133.64, 124.79, 122.59, 97.69, 97.00, 56.62, 55.86, 50.08. MS (ESI) m/z 303.06 ([M+H]+, 100%); HRMS (ESI-FT) m/z calcd for C14H15N4O4 303.1088, found 303.1090.
2-((4-((E)-(2,5-dimethoxy-4-((E)-(4-nitrophenyl)diazenyl)phenyl)diazenyl)phenyl)(methyl)amino)ethanol 18
Diazoaniline 16 was dissolved in sulfuric acid (concentrated, 100 mL) at room temperature to give a thick, deep purple solution that was cooled on ice (20 minutes). Nitrosyl sulfate was prepared by dissolving NaNO2 (100 mg) in sulfuric acid (5 mL) and warming to 50°C. After complete dissolution, the solution is cooled in an ice bath and added dropwise to diazoaniline 16. After 1 hour of stirring, ice cold 1M hydrogen tetrafluoroborate (300 mL) is slowly added and the solution extracted with dichloromethane (5 × 100 mL). The organic extract is pooled, dried over Na2SO4 and evaporated to yield 0.661 g of the diazonium tetrafluoroborate salt that was dissolved in THF (200 mL) and to which a solution of N-methyl-N-(2-hydroxyethyl)-aniline 17 (0.286 g, 1.73 mmol) in THF (20 mL) was added dropwise. The reaction was stirred for 1 h and stripped of solvent. The residue was dissolved in dichloromethane (300 ml) and washed with 1M HCl (1 × 150 mL) and brine (1 × 150 mL) and dried over Na2SO4 to give 18 as a green-purple solid (0.748 g, 1.56 mmol, 47 %). δ H (400 MHz, CDCl3) 8.38 (2 H, dd, J 9.3, 2.3), 8.05 (2 H, dd, J 9.3, 2.3), 7.95 (2 H, d, J 9.1), 7.49 (2 H, d, J 13.3), 6.85 (2 H, d, J 9.1), 4.10 (3 H, s), 4.05 (3 H, s), 3.91 (2 H, t, J 5.6), 3.66 (2 H, t, J 5.7), 3.17 (3 H, s). δ C (101 MHz, CDCl3) 156.57, 153.71, 152.67, 150.98, 148.50, 146.82, 144.61, 142.19, 126.34, 124.90, 123.70, 111.74, 101.19, 100.18, 59.81, 56.87, 54.70, 50.40, 39.44. MS (ESI) m/z 465.10 ([M+H]+, 100%); HRMS (ESI-FT) m/z calcd for C23H24N6O5Na 487.1700, found 487.1702.
2-((4-((E)-(2,5-dimethoxy-4-((E)-(4-nitrophenyl)diazenyl)phenyl)diazenyl)phenyl)(methyl)amino)ethyl 4-nitrophenyl carbonate 19
To a solution of diazo-hydroxyethylaniline 18 (0.800 g, 1.72 mmol) in dry dichloromethane (200 mL) was added p-nitrophenylchloroformate (0.382 g, 1.89 mmol), followed by pyridine (0.416 mL, 0.408 g, 5.17 mmol), and the reaction stirred 2h. The reaction was washed (1 × 200 mL each) with 1M HCl, 50% saturated sodium carbonate, brine, and dried over Na2SO4. Silica gel (5 g) was added and the mixture stripped of solvent and dried under vacuum (<10 mmHg). A silica gel column (60 g) was dry-loaded with this material and eluted with hexanes/ethylacetate (1:1) to give p-nitrophenyl carbonate 19 (1.0551 g, 1.70 mmol, 97 %) as a purple solid. δ H (400 MHz, CDCl3) 8.38 (2 H, d, J 9.0), 8.25 (2 H, d, J 9.2), 8.05 (2 H, d, J 9.1), 7.96 (2 H, d, J 9.2), 7.49 (2 H, d, J 16.5), 7.24 (2 H, s, J 3.2), 6.85 (2 H, d, J 9.3), 4.53 (2 H, t, J 5.7), 4.10 (3 H, s), 4.06 (2 H, s), 3.87 (2 H, t, J 5.7), 3.18 (3 H, s). δ C (101 MHz, CDCl3) 177.25, 156.56, 155.47, 153.65, 152.70, 151.96, 151.17, 148.60, 146.61, 145.10, 142.47, 126.26, 125.50, 124.93, 123.75, 122.01, 111.90, 101.25, 100.29, 66.29, 56.94, 50.86, 50.65, 39.10, 29.87. MS (ESI) m/z 630.15 ([M+H]+, 100%); HRMS (ESI-FT) m/z calcd for C30H28N7O9 630.1943, found 630.1941.
Quencher-apo-YbbR peptide 20
YbbR peptide appended with a 6-aminohexanoyl residue appended to the N-terminus (sequence Fmoc-N-Ahx-DSLEFIASKLA-OH) was synthesized with a Pioneer automated peptide synthesizer (Applied Biosystems, Foster City, CA, USA) on a 0.3 mmol scale using standard 9-fluorenylmethyloxycarbonyl- (Fmoc) conditions, including 2-(1H-7-azabenzotriazol-1-yl)-1,1,3,3-tetramethyl uronium hexafluorophosphate (HATU) activation and extended coupling times (1 h per residue) in the presence of 4-fold molar excess of Fmoc-L-amino acid relative to resin capacity. After completion, the N-terminal Fmoc protecting group was left intact and the resin collected from the column after washing with dichloromethane. The resin was stored at room temperature in the dark until further use.
For quencher labeling, 1.35 g of the peptide resin (0.19 mmol/gram substitution) was swollen in an Econo-Pac disposable column (product number 732-1010, Bio-Rad, Hercules, CA, USA). A solution of piperidine was added (20 % vol/vol, 10 mL) and the resin shaken gently at room temperature. After 1 h, the resin bed was allowed to settle and the piperidine solution drained. The resin was washed with copious amounts of DMF (∼ 250 mL) and transferred to a 50 mL disposable falcon tube. To this was added a solution of p-nitrophenyl carbonate 18 (0.400 g, 0.63 mmol, 2 eq.) and diisopropylethylamine (51 μL, 0.3 mmol, 1 eq.) in DMF (30 mL) and the reaction shaken gently overnight.
In the morning, the resin was collected in an Econo-Pac column and washed with DMF until the effluent was no longer purple. The resin was transferred to a 50 mL falcon tube and 35 mL of cleavage cocktail added (2% H2O, 2 % Triisopropylsilane, 96% trifluoroacetic acid) (N.B.: reaction turns true blue). The tube was shaken intermittently for 2 h, after which the resin was filtered and washed with cleavage cocktail (15 mL). The pooled filtrate and wash was transferred to a falcon tube, evaporated to ∼5 mL under a dry stream of nitrogen, and the peptide precipitated by the addition of 40 mL of cold diethyl ether. After incubation on dry ice (1 h), the solids were collected by centrifugation (20 min × 1000 g). The pellet was titrated in cold diethyl ether (3 × 30 mL) and dried under vacuum.
The resulting black solid was dissolved in dissolution cocktail (1: 1: 1 acetic acid: acetonitrile: H2O) and purified by reverse phase HPLC to give the quencher-peptide 20 (228 mg, 42 %). The identity of the resulting peptide was confirmed by mass spectrometry. HRMS (ESI-TOF) m/z calcd for C84H11N19O25 1795.8781, found 1795.8781.
Supplementary Material
Acknowledgments
This work was funded by NIH GM086225 and R21AI090213. The authors wish to thank Drs. Yongxuan Su (UCSD Small Molecule Mass Spectrometry Facility) and William Leister (NIH Chemical Genomics Center) for performing mass spectrometric analyses; and Elizabeth Komives (UCSD) for assistance with peptide synthesis.
Footnotes
Electronic supplementary information (ESI) available: Full 1H- and 13C-NMR spectrafor compound characterization.
Notes and references
- 1.Lambalot RH, Gehring AM, Flugel RS, Zuber P, LaCelle M, Marahiel MA, Reid R, Khosla C, Walsh CT. Chemistry & Biology. 1996;3:923–936. doi: 10.1016/s1074-5521(96)90181-7. [DOI] [PubMed] [Google Scholar]
- 2.Ferreras JA, Stirrett KL, Lu XQ, Ryu JS, Soll CE, Tan DS, Quadri LEN. Chemistry & Biology. 2008;15:51–61. doi: 10.1016/j.chembiol.2007.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]; Horbach R, Graf A, Weihmann F, Antelo L, Mathea S, Liermann JC, Opatz T, Thines E, Aguirre J, Deising HB. The Plant cell. 2009;21:3379–3396. doi: 10.1105/tpc.108.064188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chu M, Mierzwa R, Xu L, Yang SW, He L, Patel M, Stafford J, Macinga D, Black T, Chan TM, Gullo V. Bioorg Med Chem Lett. 2003;13:3827–3829. doi: 10.1016/j.bmcl.2003.07.011. [DOI] [PubMed] [Google Scholar]; Joseph-McCarthy D, Parris K, Huang A, Failli A, Quagliato D, Dushin EG, Novikova E, Severina E, Tuckman M, Petersen PJ, Dean C, Fritz C, Meshulam T, DeCenzo M, Dick L, McFadyen IJ, Somers WS, Lovering F, Gilbert AM. J Med Chem. 2005;48:7960–7969. doi: 10.1021/jm050523n. [DOI] [PubMed] [Google Scholar]
- 4.La Clair JJ, Foley TL, Schegg TR, Regan CM, Burkart MD. Chemistry & Biology. 2004;11:195–201. doi: 10.1016/j.chembiol.2004.02.010. [DOI] [PubMed] [Google Scholar]; Foley TL, Burkart MD. Anal Biochem. 2009;394:39–47. doi: 10.1016/j.ab.2009.06.037. [DOI] [PMC free article] [PubMed] [Google Scholar]; Foley TL, Young BS, Burkart MD. The FEBS journal. 2009 doi: 10.1111/j.1742-4658.2009.07425.x. [DOI] [PubMed] [Google Scholar]; Meier JL, Niessen S, Hoover HS, Foley TL, Cravatt BF, Burkart MD. Acs Chem Biol. 2009;4:948–957. doi: 10.1021/cb9002128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yasgar A, Foley TL, Jadhav A, Inglese J, Burkart MD, Simeonov A. Mol Biosyst. 2010;6:365–375. doi: 10.1039/b913291k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Inglese J, Auld DS, Jadhav A, Johnson RL, Simeonov A, Yasgar A, Zheng W, Austin CP. Proc Nat Acad Sci USA. 2006;103:11473–11478. doi: 10.1073/pnas.0604348103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Molecular Libraries Small Molecule Repository. http://mlsmr.glpg.com/MLSMR_HomePage/project.html.
- 8.Kruger RG, Dostal P, McCafferty DG. Chem Commun. 2002:2092–2093. doi: 10.1039/b206303d. [DOI] [PubMed] [Google Scholar]
- 9.Pedersen DS, Rosenbohm C. Synthesis. 2001:2431–2434. [Google Scholar]
- 10.Vasudevan D, Fimmen RL, Francisco AB. Environ Sci Technol. 2001;35:4089–4096. doi: 10.1021/es010880x. [DOI] [PubMed] [Google Scholar]
- 11.Lakowicz JR. Principles of fluorescence spectroscopy. Kluwer Academic/Plenum; New York: 1999. [Google Scholar]
- 12.Dardonville C, Fernandez-Fernandez C, Gibbons SL, Ryan GJ, Jagerovic N, Gabilondo AM, Meana JJ, Callado LF. Bioorg Med Chem. 2006;14:6570–6580. doi: 10.1016/j.bmc.2006.06.007. [DOI] [PubMed] [Google Scholar]
- 13.U. S. Pat., 5,595,741. 1997
- 14.U. S. Pat., 7,109,312. 2005; U. S. Pat., 7205347 B2. 2007
- 15.Vatele JM. Tetrahedron. 2004;60:4251–4260. [Google Scholar]
- 16.Yin J, Straight PD, McLoughlin SM, Zhou Z, Lin AJ, Golan DE, Kelleher NL, Kolter R, Walsh CT. Proc Nat Acad Sci USA. 2005;102:15815–15820. doi: 10.1073/pnas.0507705102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Glover KJ, Martini PM, Vold RR, Komives EA. Anal Biochem. 1999;272:270–274. doi: 10.1006/abio.1999.4182. [DOI] [PubMed] [Google Scholar]
- 18.Michael S, Auld D, Klumpp C, Jadhav A, Zheng W, Thorne N, Austin C, Inglese J, Simeonov A. Assay Drug Dev Technol. 2008;6:637–657. doi: 10.1089/adt.2008.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang JH, Chung TDY, Oldenburg KR. J Biomol Screen. 1999;4:67–73. doi: 10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]
- 20.Still WC, Kahn M, Mitra A. J Org Chem. 1978;43:2923–2925. [Google Scholar]
- 21.Gottlieb HE, Kotlyar V, Nudelman A. J Org Chem. 1997;62:7512–7515. doi: 10.1021/jo971176v. [DOI] [PubMed] [Google Scholar]
- 22.Keller O, Rudinger J. Helv Chim Acta. 1975;58:531–541. doi: 10.1002/hlca.19750580224. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.