

Abstract

This paper presents the first application of two recently developed reactions to natural product synthesis. The first method involves a 6-endo-trig cyclization to prepare a versatile chiral enaminone building block. The second is a direct C–H arylation reaction. As a showcase for the utility of these methods, (+)-antofine and (+)-ipalbidine were synthesized in only 8 steps and 24–26% overall yields.
The indolizidine alkaloids are extraordinarily prevalent in nature and are endowed with a host of biological properties that are being harnessed to treat numerous diseases.1 For this reason, methods to prepare the indolizidine core, particularly in enantiomeric form, are valuable. We have recently developed a chiral pool approach to generate mono- and bicyclic enaminones, which has enabled the synthesis of a versatile indolizidine building block from Boc-L-proline.2 The multifaceted reactivity of the enaminone scaffold lends itself to many chemoselective transformations. Having established a practical route to these molecules, we next demonstrated that cyclic enaminones were viable C–H donors in a Pd(II)-catalyzed cross-coupling reaction with aryltrifluoroborates.3 Until now, these methods have not been applied to the synthesis of more complex molecules. Herein, we show for the first time that these two methods enable an efficient synthesis of aryl indolizidine alkaloids.
Our interest in indolizidine alkaloids led us to prepare (+)-ipalbidine and (+)-antofine. These natural products were chosen not only by virtue of their prototypical structures but also by their remarkable biological profiles. (+)-Ipalbidine is a non-addictive analgesic, an oxygen-free radical scavenger, and has demonstrated inhibitory effects on the respiratory burst of leukocytes.4 (−)-Antofine, on the other hand, exhibits low nanomolar anti-proliferative activity (GI50) in drug-sensitive and multi-drug resistant cancer cell lines5 and anti-viral activity.6 This phenanthroindolizidine is the only member in its class to have established DNA and RNA binding affinities,7 which may shed light on the elusive mechanism of action of these planar natural products. We chose to synthesize the unnatural enantiomer, (+)-antofine, considering that both natural products could be synthesized in a divergent fashion from a late-stage intermediate derived from the Boc-L-proline. Although these natural products have been synthesized before,5b,6-8 this route gives access to these molecules in a remarkably concise and high-yielding fashion that will contribute to the ongoing search for their cellular target(s).
To begin our syntheses, diazoketone 1 was prepared from Boc-L-proline and was then treated with catalytic CF3CO2Ag in the presence of freshly distilled N,O-dimethylhydroxylamine (Scheme 1).9 The resulting Weinreb amide 2 was converted to ynone 3 upon addition of excess ethynylmagnesium bromide. Quenching the reaction with NaHSO4 minimized addition of the released amine into the ynone product.10 To prepare the enaminone, a modification of our one-flask, two-step procedure was used in which the Boc-protecting group was removed with formic acid. The NaI served to simultaneously activate the ynone through the formation of vinyl iodide intermediate 4, which is poised for a 6-endo-trig cyclization. This mild deprotection protocol was designed as an alternative to the previously reported1 deprotection conditions using 4N HCl/dioxane in order to mitigate racemization at the β-stereocenter.2 Racemization of this type most likely proceeds through a retro-Michael/Michael process which has been shown to be promoted by both acidic and basic conditions and is particularly problematic for β-homoproline derivatives.11 Fortunately, these deprotection/activation conditions were sufficient to suppress this undesired side-reaction, and enaminone 5 could be obtained with an e.r. of 98:212 following treatment of vinyl iodide 4 with methanolic K2CO3.
Scheme 1. Synthesis of indolizidine scaffolda.
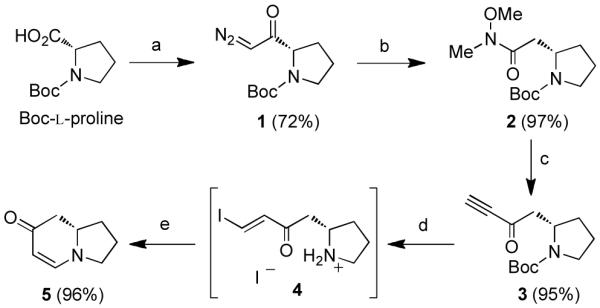
aReagents and conditions: (a) Et3N, ClCO2Et, THF; then CH2N2. (b) CF3CO2Ag (20 mol%), HN(OMe)Me, Et3N, THF. (c) ethynylmagnesium bromide (5 equiv), THF, 0 °C; then NaHSO4 (aq.). (d) NaI, HCO2H. (e) K2CO3, MeOH.
One unique characteristic of cyclic enaminones of this type is their exceptional nucleophilicity at the C3-position. Although its reactivity is attenuated in comparison to a naked enamine, the enaminone has the distinct advantage of being less susceptible to degradative processes such as hydrolysis. In this regard, we exploited this reactivity to install the aryl moiety.
In our initial studies, enaminone 5 was treated with iodine and DMAP which furnished iodoenaminone 6 (Scheme 2). The p-methoxylphenyl (PMP) substituent was then installed by way of Buchwald’s modified Suzuki-Miyaura protocol.13 Although the yields of this reaction were comparable to our previously reported microwave-assisted cross-coupling reaction,14 the Buchwald method was used because the product could be obtained in higher purity. Despite the success of this iodination/cross-coupling sequence, we pursued a more direct route involving a C–H arylation reaction. We reasoned that the nucleophilicity of the unsubstituted enaminone could be harnessed to access a palladated intermediate which could then be intercepted with an appropriate coupling partner (PMP–X). This reasoning led us to the discovery of a Pd(II)-catalyzed C-H-arylation reaction where organotrifluoroborates proved to be viable aryl donors.3 This direct-arylation was used to prepare arylindolizidine 7 (Scheme 3), from which both (+)-antofine and (+)-ipalbidine could be synthesized.
Scheme 2. Methods for arylation of indolizidine enaminonea.
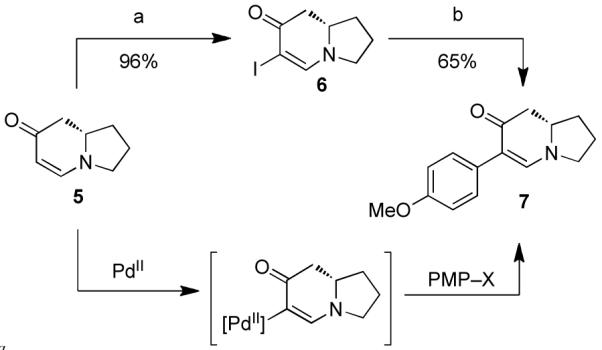
aReagents and conditions: (a) I2, DMAP, CH2Cl2. (b) Pd(OAc)2 (1.0 mol%), S-Phos (2.0 mol%), PMP–BF3K, K2CO3, MeOH, 50 °C, 5 h. PMP = p-methoxyphenyl.
Scheme 3. Synthesis of (+)-ipalbidinea.
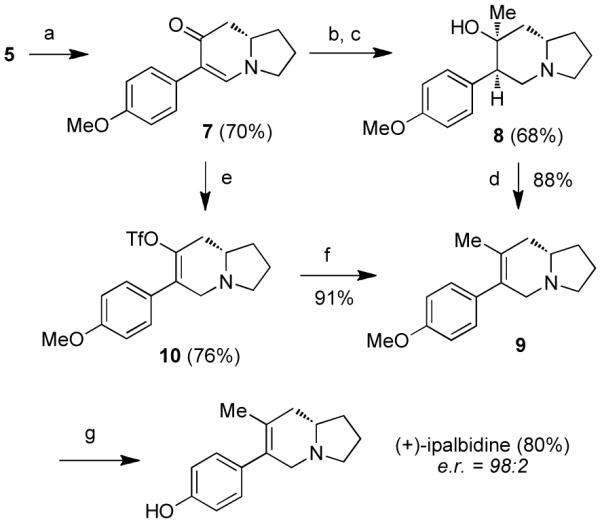
aReagents and conditions: (a) Pd(OAc)2 (30 mol%), Cu(OAc)2 (3 equiv), PMP–BF3K, K2CO3, t-BuOH/AcOH/DMSO (20:5:1), 60 °C; (b) L-Selectride, THF, −78 to 0 °C; (c) MeLi, THF, −78 °C; (d) SOCl2, pyridine, THF, −30 to −10 °C; (e) L-Selectride, THF, −78 to 0 °C; then Comins’ reagent, −78 to 0 °C; (f) Pd(PPh3)4 (10 mol%), MeZnBr, THF, 60 °C; (g) BBr3, CH2Cl2, −78 °C to rt. PMP = p-methoxyphenyl.
Two separate routes were developed to install the indolizidine methyl substituent for (+)-ipalbidine (Scheme 3). The first approach, which takes cues from Liu’s endgame sequence,8r involved 1,4-reduction of enaminone 7 with L-Selectride. To the resulting ketone was added MeLi, which furnished 75% of the desired tertiary alcohol 8. Although the stereochemistry of the newly formed chiral centers was not critical to this sequence, it is worth mentioning that both reactions proceeded with high diastereoselectivity (>95%).
In the penultimate step, the endo-olefin was obtained upon dehydration of alcohol 8. A number of dehydrating conditions were investigated,15 but the highest yields were obtained with use of SOCl2 and pyridine, which provided a single isomer of indolizidine 9. Upon BBr3 deprotection, (+)-ipalbidine was obtained in 80% yield.
An alternative route to (+)-ipalbidine was devised in order to shorten this synthetic sequence and improve yields. In this second approach, enaminone 7 was again reduced with L-Selectride only the resulting enolate was trapped in situ with 2-[N,N-bis(trifluoromethanesulfonyl)amino]-5-chloropyridine (Comins’ reagent).16 The methyl group was then installed by subjecting this triflate to Negishi cross-coupling conditions17 to furnish common intermediate 9 in 69% yield over two steps. As before, upon methyl deprotection, the natural product was obtained. The e.r. of synthesized (+)-ipalbidine was 98:2, showing that the stereochemical integrity had been retained over the previous three steps.
With a rising medicinal interest in the phenanthropiperidine and phenanthroquinolizidine alkaloids, we were inspired to apply our methodologies to the construction of these natural products as well. Thus, (+)-antofine was synthesized in two steps from triflate 10 (Scheme 4), whose synthesis has already been discussed. Firstly, antofine’s seco-analog was prepared using Negishi cross-coupling conditions17 to install the 3,4-dimethoxyphenyl group in 95% yield. In the final step, the phenanthrene system was established with a PhI(O2CCF3)2-mediated biaryl coupling to furnish (+)-antofine in 70% yield. The optical rotation for the natural product was in agreement with data reported in the literature.8a–k
Scheme 4. Synthesis of (+)-antofinea.
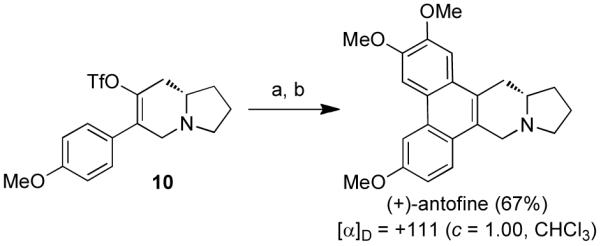
aReagents and conditions: (a) Pd(PPh3)4 (10 mol%), 3,4-dimethoxyphenylzinc bromide, THF, 60 °C; (b) PhI(O2CCF3)2, BF3·Et2O, CH2Cl2.
In summary, the syntheses of (+)-antofine and (+)-ipalbidine has been accomplished with the aid of two methodologies: a deprotection/6-endo-trig cyclization reaction of a Boc-L-proline–derived ynone and a direct Pd(II)-catalyzed C–H arylation of enaminones with an organotrifluoroborate. This is the first time that these methods have been applied to natural product synthesis. Their utility is exemplified by the fact that these natural product were prepared in only 8 steps, with overall yields of 24–26% and in up to 96% ee.
Experimental Section
(S)-2,3,8,8a-Tetrahydroindolizin-7(1H)-one (5)
Ynone 3 (3.74 g, 15.8 mmol, 1.0 equiv) was dissolved in formic acid (50 mL) solution under a N2 atmosphere and NaI (7.09 g, 47.3 mmol, 3.0 equiv) was added. The reaction was left stirring for 6 h at room temperature. The solvent was removed by passing N2 over the reaction mixture. The remaining residue was placed under vacuum for 15 minutes and then dissolved in MeOH (100 mL). A separate flask was charged with 700 mL of MeOH and K2CO3 (10.9 g, 79.0 mmol, 5.0 equiv). To this flask was added the solution of deprotected ynone over 15 minutes. The reaction was stirred for 1 h and the solvent was evaporated. At this time CH2Cl2 was added to redissolve the product (but not the inorganic salts), the slurry was suction filtered, and the filtrate concentrated. To the solid residue was added more CH2Cl2 and the precipitates were once again filtered away. This residue after removal of solvent was purified via SiO2 flash chromatography to provide the title compound as an off-white solid (2.05 g, 96% yield) after SiO2 flash chromatography (100% acetone): Spectral data of the title compound was identical to that reported in the literature2 with the exception of optical rotation. [α]D = −727 (c 1.00, CHCl3).
(S) - 6 - (4-Methoxyphenyl) - 2,3,8,8a - tetrahydroindolizin- 7(1H)-one (7)
Freshly purified enaminone 5 (750 mg, 5.5 mmol, 1.0 equiv), Pd(OAc)2 (370 mg, 1.7 mmol, 0.30 equiv), anhydrous Cu(OAc)2 powder (3.0 g, 17 mmol, 3.0 equiv) and granular K2CO3 (1.5 g, 11 mmol, 2.0 equiv) were combined in a 20:5:1 mixture of degassed t-BuOH/AcOH/DMSO (55 mL) under N2 and stirred for 5 min (Note: Solvents were used without purification). The reaction mixture was heated to 60 °C and the p-methoxyphenyl trifluoroborate (3.5 g, 17 mmol, 3.0 equiv) was added incrementally over 5 h as a solid. Approximately, 0.20 equivalents of trifluoroborate were added every 30 min. The reaction was stirred for an additional hour and was monitored by TLC using 100% EtOAc as the mobile phase. The reaction mixture was diluted with EtOAc and added to a separatory funnel containing brine. The product was extracted with EtOAc (3x). The combined organic layers were washed brine (2x) and sat. NaHCO3 (aq.) (2x), dried over Na2SO4 and concentrated in vacuo. The title compound was obtained as a pale yellow solid (930 mg, 70% yield) after SiO2 flash chromatography (80% EtOAc/hexanes). The spectral data was identical to that of reported in the literature.3 [α]D = −97.5 (c 1.00, CHCl3). Enantiomeric ratio was determined to be 98:2 via chiral HPLC using a Chiralcel OJ column. Conditions: i-PrOH 70% in hexanes, 30 min, 1.0 mL/min, 30 °C. (−)-Enantiomer: Rt = 20.9 min; (+)-Enantiomer: Rt = 11.1 min.
(S) - 6 - (4-Methoxyphenyl) - 1, 2, 3, 5, 8, 8a – hexahydroindolizin – 7 – yl –trifluoromethanesulfonate (10)
Enaminone 7 (590 mg, 2.4 mmol, 1.0 equiv) was dissolved in anhydrous THF (30 mL) under a N2 atmosphere. The solution was cooled to −78 °C at which point L-Selectride (0.23 mL, 0.23 mmol, 1.0 equiv, 1.0 M in THF) was added over 15 min. After stirring for 1 h, the reaction was slowly warmed to 0 °C over 2 h. The reaction mixture was once again cooled to −78 °C and 2-[N,N-bis(trifluoromethanesulfonyl)amino]-5-chloropyridine (1.1 g, 2.7 mmol, 1.1 equiv) was added all at once. The mixture was stirred for another hour at −78 °C and then slowly warmed to 0 °C over 2 h. The reaction was quenched with a saturated solution of NaHCO3 (aq.) and added to a separatory funnel. The product was extracted with EtOAc (3x). The combined organic layers were dried over Na2SO4 and concentrated in vacuo. The title compound was obtained as a colorless oil (690 mg, 76% yield) after SiO2 flash chromatography (10% EtOAc/hexanes (1% Et3N): 1H NMR (500 MHz, CDCl3) δ 1.49-1.57 (m, 1H), 1.76-1.84 (m, 1H), 1.87-1.96 (m, 1H), 2.00-2.07 (m, 1H), 2.22 (dd, J = 18.1, 9.2 Hz, 1H), 2.47-2.62 (m, 3H), 3.06 (d, J = 15.8 Hz, 1H), 3.18 (dt, J = 4.4, 2.1 Hz, 1H), 3.72-3.75 (m, 1H), 3.74 (s, 3H), 6.83 (d, J = 8.8 Hz, 2H), 7.14 (d, J = 8.8 Hz, 2H); 13C NMR (125 MHz, CDCl3) δ 22.2, 30.5, 35.2, 53.4, 55.3, 55.6, 60.5, 113.9, 118.1 (q, JCF = 320 Hz), 126.6, 129.2, 129.5, 142.0, 159.5; IR (neat) 2959, 1610, 1512, 1416, 1209, 1146 cm−1; HRMS (ESI+) m/e calc’d for [M+H]+ C16H19F3NO4S: 378.0987, found 378.0974; [α]D = +66.3 (c 1.00, CHCl3).
(S) - 6 - (4 - Methoxyphenyl) - 7 - methyl - 1, 2, 3, 5, 8, 8a - hexahydroindolizine (9)
Preparation of organo zinc reagent
A flame dried round-bottom flask under N2 was charged with anhydrous THF (10.0 mL) and cooled to −78 °C. Methyl lithium (1.6 mL, 2.5 mmol, 5.0 equiv, 1.6M in Et2O) was added and the solution was allowed to sit. A separate round-bottom flask was charged with anhydrous ZnBr2 (590 mg, 2.6 mmol, 5.2 equiv). The ZnBr2 was dried by heating the round-bottom flask under a vacuum with a heat gun for 5 min. When the ZnBr2 had cooled to room temperature it was dissolved in anhydrous THF (6.0 mL) under N2. This ZnBr2 solution was slowly cannulated into the MeLi solution and the resulting mixture was stirred at −78 °C for 5 min and then allowed to warm to room temperature.
Triflate 10 (190 mg, 0.50 mmol, 1.0 equiv), dissolved in a minimal amount of THF, and Pd(PPh3)4 (29 mg, 0.025 mmol, 5.0 mol%) were added sequentially to the zinc reagent. If the reaction had not gone to completion after 1 h at room temperature, the reaction mixture was heated to 50 °C. Upon consumption of the triflate starting material (as judged by TLC) SiO2 was added to the reaction mixture and the solvent was evaporated to leave a free flowing powder. Following flash chromatography (20% EtOAc/hexanes (1% Et3N)) 110 mg (91%) of the title compound was obtained as a colorless oil: 1H NMR (400 MHz, CDCl3) δ 1.46-1.54 (m, 1H), 1.60 (s, 3H), 1.73-1.81 (m, 1H), 1.85-1.94 (m, 1H), 2.00-2.32 (m, 5H), 2.91 (d, J = 15.4 Hz, 1H), 3.22 (dd, J = 3.2, 3.2 Hz, 1H), 3.62 (d, J = 15.4 Hz, 1H), 3.82 (s, 3H), 6.86 (d, J = 8.7 Hz, 2H), 7.10 (d, J = 8.7 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 20.0, 21.4, 30.8, 38.5, 54.2, 55.2, 57.8, 60.2, 113.5, 127.9, 130.0, 130.3, 133.8, 158.1; IR (neat) 2907, 1609, 1510, 1244, 1175, 831 cm−1; HRMS (ESI+) m/e calc’d for [M+H]+ C16H22NO: 244.1701, found 244.1688; [α]D = +142 (c 1.00, CHCl3).
(+)-Secoantofine
Using 3,4-dimethoxyphenyllithium instead of MeLi, the above procedure was used for the preparation of the title compound. Following flash chromatography [40% EtOAc/hexanes (1% Et3N)] 380 mg (96%) of the (+)-secoantofine was obtained as a yellow oil: 1H NMR (400 MHz, CDCl3) δ 1.51-1.63 (m, 1H), 1.78-2.01 (m, 2H), 2.06-2.14 (m, 1H), 2.25 (dd, J = 9.0, 9.0 Hz, 1H), 2.36-2.45 (m, 2H), 2.69-2.77 (m, 1H), 3.07 (dt, J = 16.0, 3.1 Hz, 1H), 3.29 (dt, J = 4.3, 2.0 Hz, 1H), 3.54 (s, 3H), 3.72 (s, 3H), 3.81 (s, 3H), 3.86 (d, J = 15.8 Hz, 1H), 6.47 (d, J = 1.1 Hz, 1H), 6.66-6.69 (m, 4H), 6.97 (d, J = 8.8 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 21.5, 30.8, 38.6, 54.3, 55.1, 55.5, 55.7, 57.9, 60.4, 110.4, 113.1, 113.4, 120.7, 130.2, 132.6, 132.7, 133.6, 135.1, 147.1, 147.9, 158.0; IR (neat) 2955, 1607, 1511, 1245, 1030, 755 cm−1; HRMS (ESI+) m/e calc’d for [M+H]+ C23H28NO3: 366.2064, found 366.2068; [α]D = +169 (c 1.00, CHCl3).
(+)-Ipalbidine
Indolizidine 9 (56 mg, 0.23 mmol, 1.0 equiv) was dissolved in CH2Cl2 (1.0 mL) and cooled to −78 °C under N2. To this solution was added BBr3 (0.23 mL, 0.23 mmol, 1.0 M in CH2Cl2). The reaction was allowed to warm to room temperature over night. The reaction was quenched with water (1.0 mL) and then 5.0 mL of a saturated solution of NaHCO3 (aq.). The product was extracted from the aqueous layer with CH2Cl2 (3x). The combined organic layers were dried with Na2SO4, concentrated and purified via flash chromatography [80% EtOAc/hexane (1% Et3N)] to provide 42 mg (80%) of (+)-ipalbidine as a white crystalline solid (mp 122.2-124.6 °C). 1H NMR (400 MHz, CDCl3) δ 1.59 (s, 3H), 1.55-1.68 (m, 1H), 1.74-1.88 (m, 1H), 1.91-2.11 (m, 2H), 2.14-2.32 (m, 3H), 2.35-2.45 (m, 1H), 3.00 (dd, J = 15.6, 2.2 Hz, 1H), 3.26 (ddd, J = 9.1, 9.1, 2.1 Hz, 1H), 3.69 (d, J = 15.6 Hz, 1H), 6.78 (d, J = 8.6 Hz, 2H), 7.00 (d, J = 8.5 Hz, 2H); 13C NMR (101 MHz, CDCl3) δ 20.0, 21.1, 30.2, 37.6, 54.1, 57.7, 60.6, 115.5, 128.3, 129.7, 129.9, 132.0, 155.9; HRMS (ESI+) m/e calc’d for [M+H]+ C15H20NO: 230.1545, found 230.1531; [a]D = +202 (c 1.00, CHCl3). [Lit. S-enantiomer: [α]D = +233.5 (c 1, CHCl3); R-enantiomer: [α]D = −237 (c 1, CHCl3), −190.5 (c 1, MeOH)].18
(+)-Antofine
Secoantofine (48 mg, 0.13 mmol, 1.0 equiv) was dissolved in CH2Cl2 (2.0 mL) and cooled to −78 °C. To this solution was sequentially added PhI(O2CCF3)2 (62 mg, 0.14 mmol, 1.1 equiv) and BF3·Et2O (16 mg, 0.14 mmol, 1.1 equiv). The solution was stirred for 4 h while being monitored by TLC. A solution of PhI(O2CCF3)2 (62 mg in 2.0 mL CH2Cl2) was added dropwise to the reaction mixture until the reaction had gone to completion. Upon consumption of starting material the reaction was quenched with 10% NaOH (aq.) and the mixture was vigorously stirred for 1 h. The product was extracted from the aqueous layer with CH2Cl2 (3x). The combined organic layers were dried with Na2SO4, concentrated and purified via flash chromatography [70% EtOAc/hexane (1% Et3N)] to provide 34 mg (70%) of (+)-antofine as a white crystalline solid: mp 226-227 °C (decomp.) 1H NMR (400 MHz, CDCl3) ™ 1.65-1.75 (m, 1H), 1.80-2.02 (m, 2H), 2.13-2.22 (m, 1H), 2.35-2.46 (m, 2H), 2.79-2.86 (m, 1H), 3.28 (ddd, J = 15.8, 3.7, 1.5 Hz, 1H), 3.38 (dt, J = 4.3, 2.1 Hz, 1H), 3.63 (d, J = 14.9 Hz, 1H), 3.95 (s, 3H), 3.99 (s, 3H), 4.04 (s, 3H), 4.63 (d, J = 14.9 Hz, 1H), 7.13 (dd, J = 9.0, 2.5 Hz, 1H), 7.25 (s, 1H), 7.75 (d, J = 9.0 Hz, 1H), 7.83 (d, J = 2.5 Hz, 1H), 7.85 (s, 1H); 13C NMR (100 MHz, CDCl3) ™ 21.6, 31.3, 33.7, 53.8, 55.0, 55.5, 55.9, 56.0, 60.3, 104.0, 1 04.7, 114.9, 123.5, 124.1, 124.2, 125.5, 126.7, 127.0, 130.2, 148.4, 149.4, 157.5; HRMS (ESI+) m/e calc’d for [M+H]+ C23H26NO3: 364.1913, found 364.1909; [α]D = +111 (c 1.00, CHCl3). [Lit. R-enantiomer: −113.4 (c 1.23, CHCl3),8c −125.2 (c 1.27, CHCl3),8d −108.2 (c 0.71, CHCl3)19].
Supplementary Material
Acknowledgment
This work was supported by National Institutes of Health Grants CA90602, GM069663, GM076302 and GM081267, the Kansas Masonic Cancer Research Institute, the University of Minnesota through the Vince and McKnight Endowed Chairs. and an ACS Division of Medicinal Chemistry Predoctoral Fellowship (to M.J.N). We thank Xin Wang for some initial efforts on the synthesis of (+)-ipalbidine,
Footnotes
Supporting Information Available. Full experimental details and copies of NMR spectral data for all new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- (1).Michael JP. Nat. Prod. Rep. 2008;25:139–165. doi: 10.1039/b612166g. [DOI] [PubMed] [Google Scholar]
- (2).(a) Turunen BJ, Georg GI. J. Am. Chem. Soc. 2006;128:8702–8703. doi: 10.1021/ja0609046. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Niphakis MJ, Turunen BJ, Georg GI. J. Org. Chem. doi: 10.1021/jo100907u. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Ge H, Niphakis MJ, Georg GI. J. Am. Chem. Soc. 2008;130:3708–3709. doi: 10.1021/ja710221c. [DOI] [PubMed] [Google Scholar]
- (4).(a) Chen X, Chu Y, Han G. Zhongguo Yaolixue Tongbao. 1998;14:167–169. [Google Scholar]; (b) Chen X, Chu Y. Zhongguo Yaolixue Tongbao. 1998;14:243–244. [Google Scholar]; (c) Wang L, Chu Y. Yaoxue Xuebao. 1996;31:806–811. [Google Scholar]
- (5).(a) Gao W, Chen APC, Leung CH, Gullen EA, Fürstner A, Shi Q, Wei L, Lee KH, Cheng YC. Bioorg. Med. Chem. Lett. 2008;18:704–709. doi: 10.1016/j.bmcl.2007.11.054. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Fu Y, Lee Sang K, Min HY, Lee T, Lee J, Cheng M, Kim S. Bioorg. Med. Chem. Lett. 2007;17:97–100. doi: 10.1016/j.bmcl.2006.09.080. [DOI] [PubMed] [Google Scholar]; (c) Lee SK, Nam K-A, Heo Y-H. Planta Med. 2003;69:21–25. doi: 10.1055/s-2003-37021. [DOI] [PubMed] [Google Scholar]
- (6).Wang K, Su B, Wang Z, Wu M, Li Z, Hu Y, Fan Z, Mi N, Wang Q. J. Agric. Food Chem. 2009;58:2703–2709. doi: 10.1021/jf902543r. [DOI] [PubMed] [Google Scholar]
- (7).(a) Xi Z, Zhang R, Yu Z, Ouyang D, Huang R. Bioorg. Med. Chem. Lett. 2005;15:2673–2677. doi: 10.1016/j.bmcl.2005.02.022. [DOI] [PubMed] [Google Scholar]; (b) Xi Z, Zhang R, Yu Z, Ouyang D. Bioorg. Med. Chem. Lett. 2006;16:4300–4304. doi: 10.1016/j.bmcl.2006.05.059. [DOI] [PubMed] [Google Scholar]
- (8).For recent syntheses of antofine, see: Yang X, Shi Q, Bastow KF, Lee KH. Org. Lett. 2010;12:1416–1419. doi: 10.1021/ol902819j. Ambrosini LM, Cernak TA, Lambert TH. Tetrahedron. 2010;66:4882–4887. Wang Z, Li Z, Wang K, Wang Q. Eur. J. Org. Chem. 2010:292–299. Yamashita S, Kurono N, Senboku H, Tokuda M, Orito K. Eur. J. Org. Chem. 2009:1173–1180. doi: 10.1021/jo900311g. Su CR, Damu AG, Chiang PC, Bastow KF, Morris-Natschke SL, Lee KH, Wu TS. Bioorg. Med. Chem. 2008;16:6233–6241. doi: 10.1016/j.bmc.2008.04.032. Wang KL, Lue MY, Wang QM, Huang RQ. Tetrahedron. 2008;64:7504–7510. Kim S, Lee YM, Lee J, Lee T, Fu Y, Song Y, Cho J, Kim D. J. Org. Chem. 2007;72:4886–4891. doi: 10.1021/jo070668x. Camacho-Davila A, Herndon JW. J. Org. Chem. 2006;71:6682–6685. doi: 10.1021/jo061053n. Fürstner A, Kennedy JWJ. Chem. Eur. J. 2006;12:7398–7410. doi: 10.1002/chem.200600592. Kim S, Lee T, Lee E, Lee J, Fan GJ, Lee SK, Kim D. J. Org. Chem. 2004;69:3144–3149. doi: 10.1021/jo049820a. Fan GJ, Lee SK, Kim D. J. Org. Chem. 2004;69(9):3144–3149. doi: 10.1021/jo049820a. For select syntheses of ipalbidine, see: Honda T, Namiki H, Nagase H, Mizutani H. Arkivoc. 2003:188–198. Honda T, Namiki H, Nagase H, Mizutani H. Tetrahedron Lett. 2003;44:3035–3038. Ikeda M, Shikaura J, Maekawa N, Daibuzono K, Teranishi H, Teraoka Y, Oda N, Ishibashi H. Heterocycles. 1999;50:31–34. Sheehan SM, Padwa A. J. Org. Chem. 1997;62:438–439. doi: 10.1021/jo961690l. Danishefsky SJ, Vogel C. J. Org. Chem. 1986;51:3915–3916. Jefford CW, Kubota T, Zaslona A. Helv. Chim. Acta. 1986;69:2048–2061. Liu Z, Lu R, Chen Q, Hong H. Huaxue Xuebao. 1985;43:992–995. Stevens RV, Luh Y. Tetrahedron Lett. 1977:979–982. Wick AE, Bartlett PA, Dolphin D. Helv. Chim. Acta. 1971;54:513–522. Govindachari TR, Sidhaye AR, Viswanathan N. Tetrahedron. 1970;26:3829–3831.
- (9).We attempted to generate HN(OMe)Me in situ from commercially available HN(OMe)Me·HCl using excess Et3N, but the yields were considerably lower.
- (10).Pirc S, Bevk D, Golobic A, Stanovnik B, Svete J. Helv. Chim. Acta. 2006;89:30–44. [Google Scholar]
- (11).Kimball FS, Turunen BJ, Ellis KC, Himes RH, Georg GI. Biorg. Med. Chem. 2008;16:4367–4377. doi: 10.1016/j.bmc.2008.02.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).The enantiomeric ratio of enaminone 5 was determined by chiral HPLC following arylation to indolizidine 7.
- (13).Barder TE, Buchwald SL. Org. Lett. 2004;6:2649–2652. doi: 10.1021/ol0491686. [DOI] [PubMed] [Google Scholar]
- (14).Wang X, Turunen BJ, Leighty MW, Georg GI. Tetrahedron Lett. 2007;48:8811–8814. doi: 10.1016/j.tetlet.2007.10.078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).See Supporting Information.
- (16).Comins DL, Deghani A. Tetrahedron Lett. 1992;33:6299–6302. [Google Scholar]
- (17).Comins DL, Chen X, Morgan LA. J. Org. Chem. 1997;62:7435–7438. doi: 10.1021/jo9711495. [DOI] [PubMed] [Google Scholar]
- (18).Wick AE, Bartlett PA, Dolphin D. Helv. Chim. Acta. 1971;54:513–22. [Google Scholar]
- (19).Kim S, Lee J, Lee T, Park H. g., Kim D. Org. Lett. 2003;5:2703–2706. doi: 10.1021/ol0349007. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.