

Introduction
1Food intake in humans and other mammals is a complex process, resulting from the integration of homeostatic systems that monitor energy stores with hedonic drive resulting from the behaviorally reinforcing properties of food (Fig. 1) (Lutter and Nestler, 2009). Under conditions of food scarcity, the hedonic and homeostatic drives cooperate to enhance the intake of highly palatable food. During times of nutrient excess, the homeostatic drive reduces food intake and increases energy expenditure in an attempt to maintain body weight. However, the hedonic drive to feed, conditioned through exposure to highly palatable food, continues to promote food consumption and excessive intake of calories. Several neuropsychiatric disorders, including Prader-Willi syndrome (PWS), bulimia nervosa, and binge eating disorder exhibit derangements in food intake resulting from perturbations in the hedonic drive to feed. In addition, genetic studies in patients with obesity are identifying mutations in genes that increase food consumption. Techniques developed in recent years now allow for a more sophisticated assessment of feeding behaviors in both laboratory animals and humans (Fig. 2). In this review, we will discuss recent advances in our understanding of food reward using a combination of laboratory animal and human research techniques.
Figure 1.
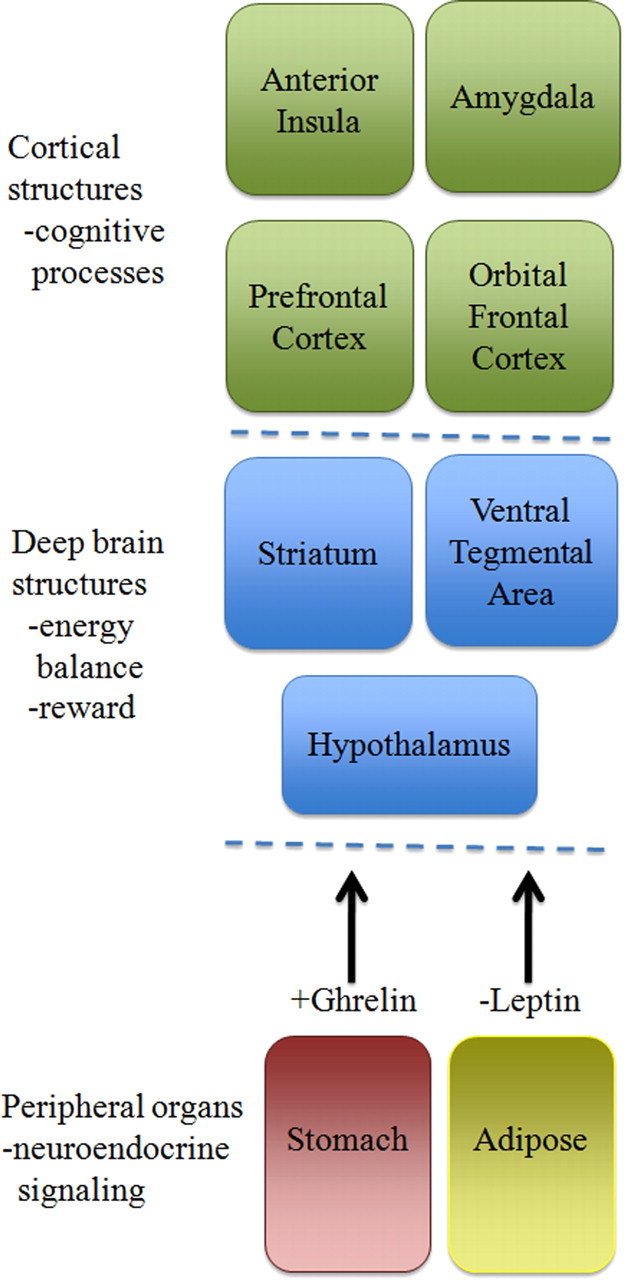
Hierarchical representation of feeding regulation. Peripheral organs like the stomach and adipose tissue produce endocrine signals, such as the hormones ghrelin and leptin, in response to changes in energy balance. Those signals are relayed primarily to structures in the deep brain, including the hypothalamus, VTA, and striatum, to regulate metabolic processes and food reward. Cortical regions then provide higher-order levels of processing of emotional responses to food.
Figure 2.

Translational neuroscience approaches to studying eating behaviors. Several techniques are available to study eating behaviors at different sites of regulation. Complementary techniques in laboratory animals and humans can be used to extend initial findings and generate new hypotheses. For instance, electrophysiological studies are of limited use in humans but readily amendable for use in laboratory animals.
Disorders associated with hyperphagia
Several disorders are associated with elevated food intake. PWS is an uncommon disorder characterized by complete or partial loss of a segment of the paternal chromosome 15 (q11-13). This neurodevelopmental disorder is characterized by marked hyperphagia and obesity, among other deficits (Goldstone, 2004). Unmonitored, individuals with PWS will eat both food and nonfood objects resulting in consumption of severalfold more calories than is required under normal conditions.
Approximately 1.5% of women and 0.5% of men suffer from bulimia nervosa, a psychiatric illness marked by repetitive episodes of binge eating in which individuals consume larger amounts of food than would be considered appropriate within a 2 h period of time (Treasure et al., 2010). Importantly, patients with bulimia nervosa feel out of control of their eating behaviors and use strategies to compensate for the increased calorie intake. These methods include self-induced vomiting (i.e., purging), laxative abuse, and excessive exercising to prevent weight gain (Treasure et al., 2010).
The Diagnostic and Statistical Manual of Mental Disorders, fourth edition, also includes binge eating disorder as a research diagnosis (American Psychiatric Association, 1994). In contrast to bulimia nervosa, patients who suffer from binge eating disorder engage in bouts of binge eating but do not use compensatory measures such as purging or exercise. Current estimates indicate that 3.5% of women and 2.0% of men would meet criteria for binge eating disorder, if it becomes an official diagnosis, which would make it the most common eating disorder in the general population (Treasure et al., 2010).
It should be noted that binge eating disorder is not synonymous with obesity. Although obesity is common among patients with binge eating disorder, obesity is best viewed as a heterogeneous illness with many factors, including food motivation, satiety, and energy expenditure, contributing to its development. Human genetic studies have begun to identify subsets of patients with genetic mutations that promote food intake leading to obesity. Two prominent examples of this approach are the melanocortin 4 receptor (MC4R) and fat mass and obesity associated (FTO) genes. Mutations in the MC4R gene have been reported in ∼5% of individuals with severe obesity (Yeo et al., 1998; Farooqi et al., 2003b). Recently, genetic variants in the FTO gene were identified as a risk factor for the development of severe and early onset obesity (Dina et al., 2007; Frayling et al., 2007). Approximately 16% of individuals of European ancestry are homozygous carriers of this FTO variant, which confers a 1.67-fold increase in the odds of developing obesity.
Methodological review
Human methods
A variety of methods are available for measuring the hedonic value of food in humans. Traditionally, food diaries and test meals consumed under laboratory settings have been used to monitor consumption of palatable foods (de Castro, 2000). In recent years, these techniques have been supplemented by self-report questionnaires such as the Child Eating Behavior Questionnaire (Wardle et al., 2001) and the Power of Food Scale (Lowe et al., 2009) that measure certain aspects of the appetite for highly palatable foods. More recently, computer-based programs have been developed for use in humans that measure the behaviorally reinforcing quality of food using operant responding tasks (Nasser et al., 2008). A similar program seeks to distinguish “liking” or hedonic value of a food from “wanting” or incentive salience of a rewarding food (Finlayson et al., 2007). Together, these studies provide methods for the initial characterization of the components of food reward.
Noninvasive neuroimaging techniques, such as functional magnetic resonance imaging (fMRI), have recently been adapted to study food reward. fMRI is a specialized MRI scan that measures changes in oxygen consumption and blood flow caused by changes of neural activities in the brain (Ogawa et al., 1990). This technique has helped to identify brain regions that are activated by food-related cues such as images or smell (Pelchat et al., 2004; Bragulat et al., 2010). In addition, fMRI imaging has identified regions activated by appetite-regulating hormones, including leptin, PYY3-36, and ghrelin (Batterham et al., 2007; Farooqi et al., 2007; Malik et al., 2008). A powerful use of functional imaging has been to reveal regions of the brain differentially regulated between patients with eating disorders and normal individuals. Together, these studies provide new insight on how the human brain responds to food-related information, identify neural circuits involved in food-related reward behavior, and suggest how dysregulation of these pathways may contribute to eating disorders.
Rodent methods
Most current studies analyze food intake in rodents by allowing animals ad libitum access to food and then measuring the amount of food consumed within a set period of time. Allowing rodents ad libitum access to food, however, eliminates the requirement of effortful responding and instead largely measures satiety (i.e., meal cessation or how “full” or “satisfied” an animal is). To study the reinforcing properties of food, many approaches originally developed to study drug addiction have been applied to study food reward in both humans and rodents. Diet preference tests allow animals to choose between specially selected research diets composed of different macronutrient content (such as high fat vs high carbohydrate). The amount of food consumed for each diet is recorded and relative preference for each diet is determined. Test animals typically consume more of the diet that they find more palatable. This test aims to measure the “liking” component of the diet and has helped to identify signaling pathways that are involved in the relative reward valuation of different macronutrients, such as opiate and dopamine signaling (Naleid et al., 2007; Johnson and Kenny, 2010).
Operant responding tests allow animals to establish an association between a particular effortful behavior (such as lever pressing) and obtainment of a reward (such as the availability to a palatable food). Test animals are willing to expend more effort in the particular behavior when they find a food to be more rewarding. The reinforcing value is determined by measuring how much effort the experimental animal is willing to give to acquire the food reward (i.e., “wanting”). In contrast, in the food conditioned place preference (CPP) task, animals learn to associate environmental cues (such as visual patterns of the wall and the floor texture) with certain stimuli, such as palatable food, during training sessions. The reward value is determined by measuring the preference an animal displays for the environmental cues associated with the rewarding stimuli. In general, the CPP test measures various aspects of motivated behaviors, including the liking and associative learning components of the reward pathways while operant responding assesses wanting. These paradigms have been used to evaluate the consequences of certain pharmacological or genetic perturbations on food reward. For instance, operant responding tasks have been used to identify a role for many signaling pathways in food reward, including the glutamatergic, opiate, ΔFosB, and serotonin signaling (Herzig et al., 2005; Olausson et al., 2006; Yonghui et al., 2006; Jarosz et al., 2007; Sanders et al., 2007).
Neuroendocrine systems involved in food reward
Prader-Willi syndrome
The hyperphagia of PWS likely involves aberrations in neuronal pathways involved in the control of hedonic feeding, because changes in food valuation have been reported (Holsen et al., 2006). After intake of a meal, patients with PWS display enhanced activation of limbic regions of the brain when compared to normal weight control subjects (Holsen et al., 2006). Imaging of the frontal cortex, a structure involved in assigning salience to rewards (Fig. 1), by fMRI has shown that patients with PWS show greater activity in the prefrontal cortex than lean controls when viewing food pictures, suggesting that food possesses a higher reward value in those with PWS (Miller et al., 2007). A second fMRI imaging study reported similar results, with enhanced hypothalamic and orbital frontal cortical activation in PWS patients relative to controls when presented with high-calorie versus low-calorie foods (Dimitropoulos and Schultz, 2008).
In 2002, a potential mechanism for the increased motivation for food was discovered when it was reported that patients with PWS display significant elevations in the appetite-stimulating hormone ghrelin (Cummings et al., 2002; DelParigi et al., 2002). Ghrelin is an octanoylated peptide mainly produced by the stomach that serves as the endogenous ligand for the ghrelin receptor [or GHSR (growth hormone secretagogue receptor)]. The developmental progression of PWS has been suggested to be linked to this elevation in ghrelin, as early elevations accompany a failure to thrive followed by the development of hyperphagia as early as the second year of life (Gunay-Aygun et al., 2001). Several studies have demonstrated a large elevation of ghrelin in PWS children relative to lean and obese controls (Feigerlová et al., 2008; Bizzarri et al., 2010). After meal ingestion, ghrelin levels decrease slightly yet remain high and may be responsible for the decreased satiety evident in PWS patients (Feigerlová et al., 2008; Bizzarri et al., 2010). These studies also suggest that perturbations in ghrelin signaling may be causative in the development of obesity, as the elevation precedes weight gain (Feigerlová et al., 2008; Bizzarri et al., 2010). Other investigations, however, do not show early elevations in ghrelin (Haqq et al., 2008) in all PWS subjects, which might be explained by the inherent difficulty in ghrelin measurement coupled with small sample size. Alternately, PWS may exhibit variability in the expression of hyperghrelinemia. During adolescence and adulthood, ghrelin levels decrease similar to that observed in the control population (Feigerlová et al., 2008; Bizzarri et al., 2010). Finally, the use of somatostatin to suppress ghrelin levels in individuals with PWS did not reduce appetite, suggesting that inhibition of ghrelin signaling alone may not be enough to normalize food intake and body weight (Tan et al., 2004; De Waele et al., 2008).
With respect to the regulation of food reward, in rodents, the ghrelin receptor is expressed widely throughout the brain, including neurons in both the hypothalamus and dopaminergic neurons in the ventral tegmental area (VTA) (Zigman et al., 2006). The mesolimbic dopamine circuit is composed of dopaminergic neurons originating in the VTA that project to neurons in the nucleus accumbens. This circuit regulates many aspects of reward-related behaviors, including psychomotor activation, behavioral arousal, and conditioned learning (remembering food-associated cues) (Berridge, 2009; Lutter and Nestler, 2009). Ghrelin acts directly on VTA neurons to stimulate dopamine release and increase food intake (Abizaid et al., 2006). Recently, we have shown that ghrelin also increases food reward-related behaviors by enhancing both food CPP and operant responding for high-fat diet under a progressive ratio schedule (Perello et al., 2010). Also, ghrelin action is required to observe the enhancement in food CPP normally seen after calorie restriction in animals (Perello et al., 2010). Functional MRI signaling in humans confirms that ghrelin-induced enhancement in appetite correlates with activation of brain reward regions, including the orbitofrontal cortex, anterior insula, amygdala, and striatum (Fig. 1) (Malik et al., 2008).
Bulimia nervosa
Similar to PWS, functional imaging techniques have been used to identify neural correlates of disordered eating in patients with bulimia nervosa. Several studies have consistently identified deficits in corticostriatal neural circuits in patients with bulimia nervosa (Fig. 1) (Uher et al., 2004; Marsh et al., 2009; Wagner et al., 2010). In contrast to PWS, no increase in ghrelin levels has been observed for bulimia nervosa. However, several studies have reported reduced levels of the adipocyte-derived hormone leptin (Jimerson et al., 2000; Monteleone et al., 2000). Leptin is a potent anorexigenic signal essential in the regulation of the energy balance. Leptin receptors are highly expressed throughout the brain, including several nuclei in the hypothalamus, the brainstem, and VTA (Elias et al., 2000; Hommel et al., 2006). Leptin acts directly on VTA dopaminergic neurons to reduce firing rate and suppress food intake (Hommel et al., 2006). Importantly, reduction in leptin receptor levels in the VTA by RNAi enhances motivated behaviors such as locomotor activity and preference for highly palatable foods such as sucrose and a high-fat diet (Hommel et al., 2006). A recent study has identified a novel population of leptin-responsive neurons in the lateral hypothalamus that project to the VTA. Activation of these neurons by local administration of leptin inhibits feeding, increases expression of tyrosine hydroxylase in the VTA (the rate limiting step of dopamine synthesis), and restores dopamine content within the nucleus accumbens (Leinninger et al., 2009). By using two patients with congenital deficiency in leptin, researchers have demonstrated that leptin modulates the perception of food reward by acting on brain reward circuits (Farooqi et al., 2007). These findings suggest that leptin action on VTA function likely involves a complex interplay of multiple neural circuits to differentially regulate behavioral outcomes.
Binge eating disorder
After exposure to food images, patients with binge eating disorder demonstrated increased reward sensitivity and activation of the orbital frontal cortex, anterior cingulated cortex, and insula (Fig. 1) (Schienle et al., 2009). These regions have previously been associated with processing of hedonic value of food (Berridge, 2009). A recent study suggests a role for two G-protein-coupled receptors, the dopamine 2 receptor (D2R) and μ-opioid receptor (mOR), in the regulation of hedonic aspects of overeating in patients with binge eating disorder. Compared to weight-matched control subjects with obesity, genetic evaluation of individuals with binge eating disorder found higher frequencies of genetic polymorphisms associated with increased activity of D2R and mOR, both of which are associated with increased reward sensitivity, creating a “hedonic-enhanced” state in response to highly palatable foods (Davis et al., 2009). Although this genetic association is still preliminary and needs to be replicated, the results are tantalizing given that extensive work performed in mice has highlighted the importance of mOR signaling in food reward (Berridge, 2009). The opioid peptide β-endorphin [derived from pro-opiomelanocortin] is the endogenous ligand to μ- and δ-opioid receptors (Glass et al., 1999). Although opioid signaling increases food intake, the effect appears to occur by promoting a preference for the palatability of food rather than its energy content (Erlanson-Albertsson, 2005). Opioid agonists act on the nucleus accumbens to affect many different aspects of food reward, such as increasing food-seeking behaviors and motivation for palatable foods (Erlanson-Albertsson, 2005).
Human neuroimaging techniques have been used to investigate hypotheses first generated from human genetic and mouse behavioral studies. Researchers examined individuals lacking the A1 allele of the D2R using fMRI (Felsted et al., 2010). This genotype is associated with increased levels of D2R within the striatum and displays an increased frequency in individuals with binge eating disorder compared to obese controls (Davis et al., 2009). After ingesting a highly palatable food (a milkshake), individuals without the A1 allele displayed enhanced reward sensitivity corresponding with increased activation of several brain regions, including the midbrain, thalamus, and orbital frontal cortex (Davis et al., 2009). This finding supports the idea that individuals with binge eating disorder experience a “hedonic-enhanced” state that promotes overeating.
Obesity
Human genetics has been a powerful tool in identifying genetic contributions to specific behaviors such as hyperphagia (Hinney et al., 2010). Although several sporadic mutations have been reported, the two most common genes implicated in obesity because of hyperphagia are MC4R and FTO. Frameshift mutations in MC4R associated with an autosomal codominant form of inherited obesity were originally described in 1998 (Yeo et al., 1998; Farooqi et al., 2003b). Since that time, several other mutations affecting MC4R have been described that promote the development of obesity (Farooqi et al., 2003b). Although great controversy exists regarding whether MC4R mutations are associated with an increased risk for developing eating disorders such as bulimia nervosa or binge eating disorder (Branson et al., 2003; Farooqi et al., 2003a; Gotoda, 2003; Herpertz et al., 2003; Hebebrand et al., 2004), clearly, individuals with these mutations consume significantly more calories in test meals than unaffected relatives even after correction for lean body mass, especially in children (Farooqi et al., 2003b).
Once it was established that signaling through the MC4R affected food intake and body weight in humans, mouse models could then be used to further elucidate the neural processes that mediate these effects. Genetic and pharmacologic techniques have been used in mice to demonstrate a role for MC4R signaling in several feeding behaviors, including meal size, macronutrient selection, and instrumental responding for food (Adan et al., 2006; Vaughan et al., 2006). Advanced gene expression techniques in mice have been used to identify the neural circuits that underlie specific functions of MC4R. For instance, a distinct population of neurons within the paraventricular nucleus of the hypothalamus and amygdala control food intake without affecting energy expenditure (Balthasar et al., 2005).
Recently, genome-wide association studies have been used for the identification of candidate genetic variants that can be associated with obesity. Common variants of FTO have been associated with increased body mass in the general population (Dina et al., 2007; Frayling et al., 2007). It is now becoming clear that alteration of eating behaviors may be the reason why variants of FTO are consistently associated with obesity risk (Cecil et al., 2008; Speakman et al., 2008; Timpson et al., 2008; Haupt et al., 2009). Although the exact mechanism of this effect is not clear, one potential way in which variants of FTO might affect food intake is by decreasing self-reported feelings of satiety (Wardle et al., 2008).
Experiments in mice are now being conducted to identify specific neuronal functions of FTO. The FTO gene codes for a demethylase enzyme that is highly expressed in the brain (Gerken et al., 2007). The neuroanatomical distribution of FTO is consistent with a potential role in body weight homeostasis with high expression in hypothalamic regions related to the energy balance, including the paraventricular, arcuate, and dorsomedial nuclei of the hypothalamus (Olszewski et al., 2009). Interestingly, FTO is expressed at very low levels in regions typically associated with food reward such as the VTA and nucleus accumbens (Olszewski et al., 2009). Additional studies will need to elucidate the role of FTO in neuronal function and to determine whether reward modulation is affected in individuals harboring pro-obesity variants of the gene.
Conclusions and future directions
Food intake in humans is a complex interplay of hormonal, neural, and psychological processes. Although disordered eating syndromes often present with similar behavioral disturbances, a combination of approaches can be used to identify pathophysiological processes. Future studies will focus on better understanding the role of specific genes on neural circuits that control aspects of food intake and food reward. Additionally, a great need exists to determine how engaging in specific eating behaviors, such as repeated episodes of binging, affects neuronal function. A good example of this concept is the recent paper that demonstrates the effect of repeated exposure to highly palatable food on dopaminergic signaling in the ventral striatum (Johnson and Kenny, 2010). Hopefully, new insights into these processes will allow for the development of novel treatments for individuals suffering from these disorders.
Footnotes
Editor's Note: Disease Focus articles provide brief overviews of a neural disease or syndrome, emphasizing potential links to basic neural mechanisms. They are presented in the hope of helping researchers identify clinical implications of their research. For more information, see http://www.jneurosci.org/misc/ifa_minireviews.dtl.
This work was supported by National Institutes of Health Grants K08 MH084058, 1RL1DK081185-01, and 1R01DA024680 and the Klarman Family Foundation, National Alliance for Research on Schizophrenia and Depression Young Investigator Award, The Disease Oriented Clinical Scholars Program, Florencio Fiorini Foundation, and 2010 Returning Home Fellowship of the International Brain Research Organization.
References
- Abizaid A, Liu ZW, Andrews ZB, Shanabrough M, Borok E, Elsworth JD, Roth RH, Sleeman MW, Picciotto MR, Tschöp MH, Gao XB, Horvath TL. Ghrelin modulates the activity and synaptic input organization of midbrain dopamine neurons while promoting appetite. J Clin Invest. 2006;116:3229–3239. doi: 10.1172/JCI29867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adan RA, Tiesjema B, Hillebrand JJ, la Fleur SE, Kas MJ, de Krom M. The MC4 receptor and control of appetite. Br J Pharmacol. 2006;149:815–827. doi: 10.1038/sj.bjp.0706929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- American Psychiatric Association. Diagnostic and statistical manual of mental disorders. Ed 4. Washington, DC: American Psychiatric Association; 1994. [Google Scholar]
- Balthasar N, Dalgaard LT, Lee CE, Yu J, Funahashi H, Williams T, Ferreira M, Tang V, McGovern RA, Kenny CD, Christiansen LM, Edelstein E, Choi B, Boss O, Aschkenasi C, Zhang CY, Mountjoy K, Kishi T, Elmquist JK, Lowell BB. Divergence of melanocortin pathways in the control of food intake and energy expenditure. Cell. 2005;123:493–505. doi: 10.1016/j.cell.2005.08.035. [DOI] [PubMed] [Google Scholar]
- Batterham RL, ffytche DH, Rosenthal JM, Zelaya FO, Barker GJ, Withers DJ, Williams SC. PYY modulation of cortical and hypothalamic brain areas predicts feeding behaviour in humans. Nature. 2007;450:106–109. doi: 10.1038/nature06212. [DOI] [PubMed] [Google Scholar]
- Berridge KC. ‘Liking’ and ‘wanting’ food rewards: brain substrates and roles in eating disorders. Physiol Behav. 2009;97:537–550. doi: 10.1016/j.physbeh.2009.02.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bizzarri C, Rigamonti AE, Luce A, Cappa M, Cella SG, Berini J, Sartorio A, Müller EE, Salvatoni A. Children with Prader-Willi syndrome exhibit more evident meal-induced responses in plasma ghrelin and peptide YY levels than obese and lean children. Eur J Endocrinol. 2010;162:499–505. doi: 10.1530/EJE-09-1033. [DOI] [PubMed] [Google Scholar]
- Bragulat V, Dzemidzic M, Bruno C, Cox CA, Talavage T, Considine RV, Kareken DA. Food-related odor probes of brain reward circuits during hunger: a pilot fMRI study. Obesity (Silver Spring) 2010;18:1566–1571. doi: 10.1038/oby.2010.57. [DOI] [PubMed] [Google Scholar]
- Branson R, Potoczna N, Kral JG, Lentes KU, Hoehe MR, Horber FF. Binge eating as a major phenotype of melanocortin 4 receptor gene mutations. N Engl J Med. 2003;348:1096–1103. doi: 10.1056/NEJMoa021971. [DOI] [PubMed] [Google Scholar]
- Cecil JE, Tavendale R, Watt P, Hetherington MM, Palmer CN. An obesity-associated FTO gene variant and increased energy intake in children. N Engl J Med. 2008;359:2558–2566. doi: 10.1056/NEJMoa0803839. [DOI] [PubMed] [Google Scholar]
- Cummings DE, Clement K, Purnell JQ, Vaisse C, Foster KE, Frayo RS, Schwartz MW, Basdevant A, Weigle DS. Elevated plasma ghrelin levels in Prader Willi syndrome. Nat Med. 2002;8:643–644. doi: 10.1038/nm0702-643. [DOI] [PubMed] [Google Scholar]
- Davis CA, Levitan RD, Reid C, Carter JC, Kaplan AS, Patte KA, King N, Curtis C, Kennedy JL. Dopamine for “wanting” and opioids for “liking”: a comparison of obese adults with and without binge eating. Obesity (Silver Spring) 2009;17:1220–1225. doi: 10.1038/oby.2009.52. [DOI] [PubMed] [Google Scholar]
- de Castro JM. Eating behavior: lessons from the real world of humans. Nutrition. 2000;16:800–813. doi: 10.1016/s0899-9007(00)00414-7. [DOI] [PubMed] [Google Scholar]
- DelParigi A, Tschöp M, Heiman ML, Salbe AD, Vozarova B, Sell SM, Bunt JC, Tataranni PA. High circulating ghrelin: a potential cause for hyperphagia and obesity in Prader-Willi syndrome. J Clin Endocrinol Metab. 2002;87:5461–5464. doi: 10.1210/jc.2002-020871. [DOI] [PubMed] [Google Scholar]
- De Waele K, Ishkanian SL, Bogarin R, Miranda CA, Ghatei MA, Bloom SR, Pacaud D, Chanoine JP. Long-acting octreotide treatment causes a sustained decrease in ghrelin concentrations but does not affect weight, behaviour and appetite in subjects with Prader-Willi syndrome. Eur J Endocrinol. 2008;159:381–388. doi: 10.1530/EJE-08-0462. [DOI] [PubMed] [Google Scholar]
- Dimitropoulos A, Schultz RT. Food-related neural circuitry in Prader-Willi syndrome: response to high- versus low-calorie foods. J Autism Dev Disord. 2008;38:1642–1653. doi: 10.1007/s10803-008-0546-x. [DOI] [PubMed] [Google Scholar]
- Dina C, Meyre D, Gallina S, Durand E, Körner A, Jacobson P, Carlsson LM, Kiess W, Vatin V, Lecoeur C, Delplanque J, Vaillant E, Pattou F, Ruiz J, Weill J, Levy-Marchal C, Horber F, Potoczna N, Hercberg S, Le Stunff C, et al. Variation in FTO contributes to childhood obesity and severe adult obesity. Nat Genet. 2007;39:724–726. doi: 10.1038/ng2048. [DOI] [PubMed] [Google Scholar]
- Elias CF, Kelly JF, Lee CE, Ahima RS, Drucker DJ, Saper CB, Elmquist JK. Chemical characterization of leptin-activated neurons in the rat brain. J Comp Neurol. 2000;423:261–281. [PubMed] [Google Scholar]
- Erlanson-Albertsson C. How palatable food disrupts appetite regulation. Basic Clin Pharmacol Toxicol. 2005;97:61–73. doi: 10.1111/j.1742-7843.2005.pto_179.x. [DOI] [PubMed] [Google Scholar]
- Farooqi IS, Yeo GS, O'Rahilly S. Binge eating as a phenotype of melanocortin 4 receptor gene mutations. N Engl J Med. 2003a;349:606–609. author reply 606–609. [PubMed] [Google Scholar]
- Farooqi IS, Keogh JM, Yeo GS, Lank EJ, Cheetham T, O'Rahilly S. Clinical spectrum of obesity and mutations in the melanocortin 4 receptor gene. N Engl J Med. 2003b;348:1085–1095. doi: 10.1056/NEJMoa022050. [DOI] [PubMed] [Google Scholar]
- Farooqi IS, Bullmore E, Keogh J, Gillard J, O'Rahilly S, Fletcher PC. Leptin regulates striatal regions and human eating behavior. Science. 2007;317:1355. doi: 10.1126/science.1144599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feigerlová E, Diene G, Conte-Auriol F, Molinas C, Gennero I, Salles JP, Arnaud C, Tauber M. Hyperghrelinemia precedes obesity in Prader-Willi syndrome. J Clin Endocrinol Metab. 2008;93:2800–2805. doi: 10.1210/jc.2007-2138. [DOI] [PubMed] [Google Scholar]
- Felsted JA, Ren X, Chouinard-Decorte F, Small DM. Genetically determined differences in brain response to a primary food reward. J Neurosci. 2010;30:2428–2432. doi: 10.1523/JNEUROSCI.5483-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finlayson G, King N, Blundell JE. Is it possible to dissociate ‘liking’ and ‘wanting’ for foods in humans? A novel experimental procedure. Physiol Behav. 2007;90:36–42. doi: 10.1016/j.physbeh.2006.08.020. [DOI] [PubMed] [Google Scholar]
- Frayling TM, Timpson NJ, Weedon MN, Zeggini E, Freathy RM, Lindgren CM, Perry JR, Elliott KS, Lango H, Rayner NW, Shields B, Harries LW, Barrett JC, Ellard S, Groves CJ, Knight B, Patch AM, Ness AR, Ebrahim S, Lawlor DA, et al. A common variant in the FTO gene is associated with body mass index and predisposes to childhood and adult obesity. Science. 2007;316:889–894. doi: 10.1126/science.1141634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerken T, Girard CA, Tung YC, Webby CJ, Saudek V, Hewitson KS, Yeo GS, McDonough MA, Cunliffe S, McNeill LA, Galvanovskis J, Rorsman P, Robins P, Prieur X, Coll AP, Ma M, Jovanovic Z, Farooqi IS, Sedgwick B, Barroso I, et al. The obesity-associated FTO gene encodes a 2-oxoglutarate-dependent nucleic acid demethylase. Science. 2007;318:1469–1472. doi: 10.1126/science.1151710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glass MJ, Billington CJ, Levine AS. Opioids and food intake: distributed functional neural pathways? Neuropeptides. 1999;33:360–368. doi: 10.1054/npep.1999.0050. [DOI] [PubMed] [Google Scholar]
- Goldstone AP. Prader-Willi syndrome: advances in genetics, pathophysiology and treatment. Trends Endocrinol Metab. 2004;15:12–20. doi: 10.1016/j.tem.2003.11.003. [DOI] [PubMed] [Google Scholar]
- Gotoda T. Binge eating as a phenotype of melanocortin 4 receptor gene mutations. N Engl J Med. 2003;349:606–609. doi: 10.1056/NEJM200308073490615. author reply 606–609. [DOI] [PubMed] [Google Scholar]
- Gunay-Aygun M, Schwartz S, Heeger S, O'Riordan MA, Cassidy SB. The changing purpose of Prader-Willi syndrome clinical diagnostic criteria and proposed revised criteria. Pediatrics. 2001;108:E92. doi: 10.1542/peds.108.5.e92. [DOI] [PubMed] [Google Scholar]
- Haqq AM, Grambow SC, Muehlbauer M, Newgard CB, Svetkey LP, Carrel AL, Yanovski JA, Purnell JQ, Freemark M. Ghrelin concentrations in Prader-Willi syndrome (PWS) infants and children: changes during development. Clin Endocrinol (Oxf) 2008;69:911–920. doi: 10.1111/j.1365-2265.2008.03385.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haupt A, Thamer C, Staiger H, Tschritter O, Kirchhoff K, Machicao F, Häring HU, Stefan N, Fritsche A. Variation in the FTO gene influences food intake but not energy expenditure. Exp Clin Endocrinol Diabetes. 2009;117:194–197. doi: 10.1055/s-0028-1087176. [DOI] [PubMed] [Google Scholar]
- Hebebrand J, Geller F, Dempfle A, Heinzel-Gutenbrunner M, Raab M, Gerber G, Wermter AK, Horro FF, Blundell J, Schäfer H, Remschmidt H, Herpertz S, Hinney A. Binge-eating episodes are not characteristic of carriers of melanocortin-4 receptor gene mutations. Mol Psychiatry. 2004;9:796–800. doi: 10.1038/sj.mp.4001491. [DOI] [PubMed] [Google Scholar]
- Herpertz S, Siffert W, Hebebrand J. Binge eating as a phenotype of melanocortin 4 receptor gene mutations. N Engl J Med. 2003;349:606–609. author reply 606–609. [PubMed] [Google Scholar]
- Herzig V, Capuani EM, Kovar KA, Schmidt WJ. Effects of MPEP on expression of food-, MDMA- or amphetamine-conditioned place preference in rats. Addict Biol. 2005;10:243–249. doi: 10.1080/13556210500223272. [DOI] [PubMed] [Google Scholar]
- Hinney A, Vogel CI, Hebebrand J. From monogenic to polygenic obesity: recent advances. Eur Child Adolesc Psychiatry. 2010;19:297–310. doi: 10.1007/s00787-010-0096-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holsen LM, Zarcone JR, Brooks WM, Butler MG, Thompson TI, Ahluwalia JS, Nollen NL, Savage CR. Neural mechanisms underlying hyperphagia in Prader-Willi syndrome. Obesity (Silver Spring) 2006;14:1028–1037. doi: 10.1038/oby.2006.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hommel JD, Trinko R, Sears RM, Georgescu D, Liu ZW, Gao XB, Thurmon JJ, Marinelli M, DiLeone RJ. Leptin receptor signaling in midbrain dopamine neurons regulates feeding. Neuron. 2006;51:801–810. doi: 10.1016/j.neuron.2006.08.023. [DOI] [PubMed] [Google Scholar]
- Jarosz PA, Kessler JT, Sekhon P, Coscina DV. Conditioned place preferences (CPPs) to high-caloric “snack foods” in rat strains genetically prone vs. resistant to diet-induced obesity: resistance to naltrexone blockade. Pharmacol Biochem Behav. 2007;86:699–704. doi: 10.1016/j.pbb.2007.02.017. [DOI] [PubMed] [Google Scholar]
- Jimerson DC, Mantzoros C, Wolfe BE, Metzger ED. Decreased serum leptin in bulimia nervosa. J Clin Endocrinol Metab. 2000;85:4511–4514. doi: 10.1210/jcem.85.12.7051. [DOI] [PubMed] [Google Scholar]
- Johnson PM, Kenny PJ. Dopamine D2 receptors in addiction-like reward dysfunction and compulsive eating in obese rats. Nat Neurosci. 2010;13:635–641. doi: 10.1038/nn.2519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leinninger GM, Jo YH, Leshan RL, Louis GW, Yang H, Barrera JG, Wilson H, Opland DM, Faouzi MA, Gong Y, Jones JC, Rhodes CJ, Chua S, Jr, Diano S, Horvath TL, Seeley RJ, Becker JB, Münzberg H, Myers MG., Jr Leptin acts via leptin receptor-expressing lateral hypothalamic neurons to modulate the mesolimbic dopamine system and suppress feeding. Cell Metab. 2009;10:89–98. doi: 10.1016/j.cmet.2009.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe MR, Butryn ML, Didie ER, Annunziato RA, Thomas JG, Crerand CE, Ochner CN, Coletta MC, Bellace D, Wallaert M, Halford J. The power of food scale. A new measure of the psychological influence of the food environment. Appetite. 2009;53:114–118. doi: 10.1016/j.appet.2009.05.016. [DOI] [PubMed] [Google Scholar]
- Lutter M, Nestler EJ. Homeostatic and hedonic signals interact in the regulation of food intake. J Nutr. 2009;139:629–632. doi: 10.3945/jn.108.097618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malik S, McGlone F, Bedrossian D, Dagher A. Ghrelin modulates brain activity in areas that control appetitive behavior. Cell Metab. 2008;7:400–409. doi: 10.1016/j.cmet.2008.03.007. [DOI] [PubMed] [Google Scholar]
- Marsh R, Steinglass JE, Gerber AJ, Graziano O'Leary K, Wang Z, Murphy D, Walsh BT, Peterson BS. Deficient activity in the neural systems that mediate self-regulatory control in bulimia nervosa. Arch Gen Psychiatry. 2009;66:51–63. doi: 10.1001/archgenpsychiatry.2008.504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller JL, James GA, Goldstone AP, Couch JA, He G, Driscoll DJ, Liu Y. Enhanced activation of reward mediating prefrontal regions in response to food stimuli in Prader-Willi syndrome. J Neurol Neurosurg Psychiatry. 2007;78:615–619. doi: 10.1136/jnnp.2006.099044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monteleone P, Di Lieto A, Tortorella A, Longobardi N, Maj M. Circulating leptin in patients with anorexia nervosa, bulimia nervosa or binge-eating disorder: relationship to body weight, eating patterns, psychopathology and endocrine changes. Psychiatry Res. 2000;94:121–129. doi: 10.1016/s0165-1781(00)00144-x. [DOI] [PubMed] [Google Scholar]
- Naleid AM, Grace MK, Chimukangara M, Billington CJ, Levine AS. Paraventricular opioids alter intake of high-fat but not high-sucrose diet depending on diet preference in a binge model of feeding. Am J Physiol Regul Integr Comp Physiol. 2007;293:R99–R105. doi: 10.1152/ajpregu.00675.2006. [DOI] [PubMed] [Google Scholar]
- Nasser JA, Evans SM, Geliebter A, Pi-Sunyer FX, Foltin RW. Use of an operant task to estimate food reinforcement in adult humans with and without BED. Obesity (Silver Spring) 2008;16:1816–1820. doi: 10.1038/oby.2008.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogawa S, Lee TM, Nayak AS, Glynn P. Oxygenation-sensitive contrast in magnetic resonance image of rodent brain at high magnetic fields. Magn Reson Med. 1990;14:68–78. doi: 10.1002/mrm.1910140108. [DOI] [PubMed] [Google Scholar]
- Olausson P, Jentsch JD, Tronson N, Neve RL, Nestler EJ, Taylor JR. DeltaFosB in the nucleus accumbens regulates food-reinforced instrumental behavior and motivation. J Neurosci. 2006;26:9196–9204. doi: 10.1523/JNEUROSCI.1124-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olszewski PK, Fredriksson R, Olszewska AM, Stephansson O, Alsiö J, Radomska KJ, Levine AS, Schiöth HB. Hypothalamic FTO is associated with the regulation of energy intake not feeding reward. BMC Neurosci. 2009;10:129. doi: 10.1186/1471-2202-10-129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelchat ML, Johnson A, Chan R, Valdez J, Ragland JD. Images of desire: food-craving activation during fMRI. Neuroimage. 2004;23:1486–1493. doi: 10.1016/j.neuroimage.2004.08.023. [DOI] [PubMed] [Google Scholar]
- Perello M, Sakata I, Birnbaum S, Chuang JC, Osborne-Lawrence S, Rovinsky SA, Woloszyn J, Yanagisawa M, Lutter M, Zigman JM. Ghrelin increases the rewarding value of high-fat diet in an orexin-dependent manner. Biol Psychiatry. 2010;67:880–886. doi: 10.1016/j.biopsych.2009.10.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders AC, Hussain AJ, Hen R, Zhuang X. Chronic blockade or constitutive deletion of the serotonin transporter reduces operant responding for food reward. Neuropsychopharmacology. 2007;32:2321–2329. doi: 10.1038/sj.npp.1301368. [DOI] [PubMed] [Google Scholar]
- Schienle A, Schäfer A, Hermann A, Vaitl D. Binge-eating disorder: reward sensitivity and brain activation to images of food. Biol Psychiatry. 2009;65:654–661. doi: 10.1016/j.biopsych.2008.09.028. [DOI] [PubMed] [Google Scholar]
- Speakman JR, Rance KA, Johnstone AM. Polymorphisms of the FTO gene are associated with variation in energy intake, but not energy expenditure. Obesity (Silver Spring) 2008;16:1961–1965. doi: 10.1038/oby.2008.318. [DOI] [PubMed] [Google Scholar]
- Tan TM, Vanderpump M, Khoo B, Patterson M, Ghatei MA, Goldstone AP. Somatostatin infusion lowers plasma ghrelin without reducing appetite in adults with Prader-Willi syndrome. J Clin Endocrinol Metab. 2004;89:4162–4165. doi: 10.1210/jc.2004-0835. [DOI] [PubMed] [Google Scholar]
- Timpson NJ, Emmett PM, Frayling TM, Rogers I, Hattersley AT, McCarthy MI, Davey Smith G. The fat mass- and obesity-associated locus and dietary intake in children. Am J Clin Nutr. 2008;88:971–978. doi: 10.1093/ajcn/88.4.971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treasure J, Claudino AM, Zucker N. Eating disorders. Lancet. 2010;375:583–593. doi: 10.1016/S0140-6736(09)61748-7. [DOI] [PubMed] [Google Scholar]
- Uher R, Murphy T, Brammer MJ, Dalgleish T, Phillips ML, Ng VW, Andrew CM, Williams SC, Campbell IC, Treasure J. Medial prefrontal cortex activity associated with symptom provocation in eating disorders. Am J Psychiatry. 2004;161:1238–1246. doi: 10.1176/appi.ajp.161.7.1238. [DOI] [PubMed] [Google Scholar]
- Vaughan C, Moore M, Haskell-Luevano C, Rowland NE. Food motivated behavior of melanocortin-4 receptor knockout mice under a progressive ratio schedule. Peptides. 2006;27:2829–2835. doi: 10.1016/j.peptides.2006.07.008. [DOI] [PubMed] [Google Scholar]
- Wagner A, Aizenstein H, Venkatraman VK, Bischoff-Grethe A, Fudge J, May JC, Frank GK, Bailer UF, Fischer L, Putnam K, Kaye WH. Altered striatal response to reward in bulimia nervosa after recovery. Int J Eat Disord. 2010;43:289–294. doi: 10.1002/eat.20699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wardle J, Guthrie CA, Sanderson S, Rapoport L. Development of the Children's Eating Behaviour Questionnaire. J Child Psychol Psychiatry. 2001;42:963–970. doi: 10.1111/1469-7610.00792. [DOI] [PubMed] [Google Scholar]
- Wardle J, Carnell S, Haworth CM, Farooqi IS, O'Rahilly S, Plomin R. Obesity associated genetic variation in FTO is associated with diminished satiety. J Clin Endocrinol Metab. 2008;93:3640–3643. doi: 10.1210/jc.2008-0472. [DOI] [PubMed] [Google Scholar]
- Yeo GS, Farooqi IS, Aminian S, Halsall DJ, Stanhope RG, O'Rahilly S. A frameshift mutation in MC4R associated with dominantly inherited human obesity. Nat Genet. 1998;20:111–112. doi: 10.1038/2404. [DOI] [PubMed] [Google Scholar]
- Yonghui L, Xigeng Z, Yunjing B, Xiaoyan Y, Nan S. Opposite effects of MK-801 on the expression of food and morphine-induced conditioned place preference in rats. J Psychopharmacol. 2006;20:40–46. doi: 10.1177/0269881105057250. [DOI] [PubMed] [Google Scholar]
- Zigman JM, Jones JE, Lee CE, Saper CB, Elmquist JK. Expression of ghrelin receptor mRNA in the rat and the mouse brain. J Comp Neurol. 2006;494:528–548. doi: 10.1002/cne.20823. [DOI] [PMC free article] [PubMed] [Google Scholar]