

Abstract
Rationale
Critically ill patients are highly susceptible to hospital-acquired infection. Neutrophil function in critical illness remains poorly understood.
Objectives
To characterize and define mechanisms of peripheral blood neutrophil (PBN) dysfunction in critically ill patients. To determine whether the inflamed lung contributes additional phagocytic impairment.
Methods
Prospective collection of blood and bronchoalveolar lavage fluid from patients with suspected ventilator-associated pneumonia and from age- and sex-matched volunteers; laboratory analysis of neutrophil functions.
Measurements and Main Results
Seventy-two patients and 21 volunteers were included. Phagocytic capacity of PBNs was 36% lower in patients than in volunteers (P < 0.0001). From several biologically plausible candidates only activated complement was significantly associated with impaired PBN phagocytosis (P < 0.0001). Phagocytosis was negatively correlated with serum C3a and positively correlated with expression of C5a receptor type 1 (CD88) on PBNs. C5a recapitulated impaired PBN phagocytosis and significantly down-regulated CD88 expression in vitro. C5a-mediated phagocytic impairment was prevented by blocking either CD88 or phosphoinositide 3-kinase, and completely reversed by granulocyte-macrophage colony-stimulating factor. C5a also impaired killing of Pseudomonas aeruginosa by, and migration of, PBNs, indicating that effects were not restricted to phagocytosis. Bronchoalveolar lavage fluid leukocytes from patients also demonstrated significantly impaired function, and lavage supernatant reduced phagocytosis in healthy neutrophils by 43% (P = 0.0001). However, lavage fluid did not affect CD88 expression and lavage-mediated impairment of phagocytosis was not blocked by anti-CD88 antibody.
Conclusions
Critically ill patients have significant dysfunction of PBNs, which is mediated predominantly by activated complement. Further, profound complement-independent neutrophil dysfunction occurs in the inflamed lung.
Keywords: complement, natural immunity, intensive care, phagocytosis
Patients with critical illness are highly susceptible to developing hospital-acquired infections (HAIs) (1, 2). HAIs are thought to affect 2 million American patients, resulting in 77,000 additional deaths, per annum (3, 4). Various factors may contribute to the risk of infection including the nature of the underlying illness (5), medical interventions (e.g., intubation [6], insertion of vascular lines [7], drugs [8]), and the acquisition and transmission of relatively virulent pathogens in the intensive care unit (ICU) (9, 10). Running parallel to these themes is the observation that innate immunity is impaired in critically ill patients. Neutrophils are critical for the clearance of most ICU-related pathogens, and defective phagocytosis by peripheral blood neutrophils (PBNs) has been described in mechanically ventilated patients (11, 12). However, surprisingly little is known about the mechanisms underlying defective neutrophil function in the critically ill. Identification of such mechanisms would be expected to suggest novel targets for the prevention and treatment of HAI.
AT A GLANCE COMMENTARY.
Scientific Knowledge on the Subject
Neutrophil dysfunction occurs in critical illness but remains poorly characterized. Animal models of sepsis implicate C5a in this process.
What This Study Adds to the Field
This study provides the first human demonstration of C5a-mediated dysfunction of peripheral blood neutrophils in critical illness. The findings demonstrate that C5a-mediated dysfunction of peripheral blood neutrophils is preventable and reversible ex vivo, suggesting potential therapeutic avenues. Provides evidence that neutrophil dysfunction persists, but is independent of C5a, in the inflamed lung.
The aims of this study were therefore to characterize neutrophil dysfunction and to define the underlying mechanism(s) in patients with a high clinical suspicion for ventilator-associated pneumonia (VAP), the most commonly fatal HAI (13). We hypothesized that profound systemic inflammation characteristic of critical illness drives impairment of neutrophil phagocytic capacity. We also sought to determine whether the local, inflamed environment of the lung imparts additional dysfunction to neutrophils migrating from the blood. Our work proposes a significant role for activated complement (in particular high concentrations of C5a) in neutrophil dysfunction in critically ill patients. Elevated concentrations of C5a have been previously described in sepsis and have been shown to impair neutrophil function in rodents (14–17). Some of the results of these studies have been previously reported in the form of an abstract (18).
METHODS
Reagents
Recombinant human C5a (an anaphylotoxin generated by cleavage of complement factor 5), formyl methionyl leucyl phenylalanine (fMLP), and platelet-activating factor (PAF) were from Sigma (Gillingham, UK), and granulocyte-macrophage colony-stimulating factor (GM-CSF) was from Affinity Bioreagents (Golden, CO). S5/1, a specific antibody against the C5a receptor CD88, was from Abcam (Cambridge, UK). Fluorophore-labeled antibodies against CD11b and the Fcγ receptors CD32 and CD64 were obtained from Invitrogen (Paisley, UK). Isoproterenol (a β1- and β2-adrenoceptor agonist), the adenylate cyclase inhibitor SQ22536, and the phosphoinositide 3-kinase (PI3K) inhibitors wortmannin and LY294002 were from Tocris Bioscience (Bristol, UK).
Patients and Volunteers
Patients were recruited from two teaching hospital ICUs if they had clinically suspected VAP, that is, mechanical ventilation for at least 48 hours, new and persisting infiltrates on a chest radiograph, and at least two of the following: purulent tracheal secretions, temperature greater than 38°C, or white cell count greater than 11 × 109/L. Patients provided blood and had fiberoptic bronchoscopy and bronchoalveolar lavage (BAL) performed by a standardized technique (see the online supplement for details). This study considered all patients with clinically suspected VAP together as a single group; however, subgroups with microbiologically confirmed VAP and adult respiratory distress syndrome are analyzed separately in the online supplement.
Age- and sex-matched volunteers (MVs) were recruited from a local primary care practice and also underwent blood sampling, bronchos-copy, and BAL. Exclusion criteria and additional details pertaining to patients and volunteers are in the online supplement. Separate university staff and students provided blood to generate healthy PBNs for in vitro experiments. Informed, witnessed assent was obtained from a relative or main carer for all patients. Informed, written consent was obtained from all volunteers. The study was approved by the relevant research ethics committees.
Processing of Bronchoalveolar Lavage Fluid and Blood
Details of processing are in the online supplement. Bronchoalveolar lavage fluid (BALF) was separated into a cellular fraction and supernatant. Serum and neutrophils were derived fresh from whole blood (19).
Inflammatory Cytokines and Complement Factors
Concentrations of C3a des-Arg and C5a des-Arg, the breakdown products of complement factors C3a and C5a, were estimated with a cytometric bead array kit (BD Biosciences, Oxford, UK). GM-CSF, IL-6, IL-8, and tumor necrosis factor (TNF)-α levels were assessed by the same method.
Lavage concentrations of these molecules were corrected for dilution by measuring urea in BALF and plasma (20).
Phagocytosis Assays
PBNs were adhered in Iscove’s modified Dulbecco’s medium (IMDM) containing 10% autologous serum. Cells were exposed for 1 hour to zymosan particles (Sigma) that had been preincubated with 50% autologous serum. PBNs were air dried, fixed with methanol, and stained with Reastain Quik-Diff (Reagena, Toivala, Finland). Light microscopy was used to distinguish PBNs containing two or more zymosan particles. Duplicate counts were performed on four randomly selected fields (minimum of 100 neutrophils per field). Variations of this core protocol were performed to explore mechanisms underlying defective phagocytosis, and to assess killing of Pseudomonas aeruginosa strain PA01 (details are in the online supplement).
Superoxide Production and Transmigration Assays
Neutrophil superoxide production was assayed by cytochrome c reduction assay (21). Transmigration was quantified by histological assessment of neutrophils traversing a Transwell filter (pore size, 3 μm; Corning, Lowell, MA). Further details are described in the online supplement.
Flow Cytometry
PBNs were incubated with primary, fluorophore-tagged antibody or isotype control (details of labeling and of the antibodies used are in the online supplement). Cells were fixed with 10% formalin and analyzed by FACSCalibur (BD Biosciences). Expression was quantified as geometric mean fluorescence.
Epithelial Cell Inflammation Assay
A549 cells (American Type Culture Collection, Manassas, VA) (derived from human type II alveolar epithelial cells [22]) were grown to confluence. Freshly isolated neutrophils (500,000) were applied. LPS O127:B8 (Sigma), derived from Escherichia coli, was added (final concentration, 100 ng/ml) for 24 hours at 37°C. To distinguish soluble from membrane-mediated effects experiments were repeated with permeable membranes (Transwell; pore size, 0.4 μm), preventing direct contact between neutrophils and A549 cells.
Statistical Analysis
Analysis was conducted with Prism (Graphpad Software, La Jolla, CA). Nonparametric data were analyzed by the Mann-Whitney U test for two variables and by the Kruskal-Wallis test for greater than two variables, using Dunn’s post-hoc analysis test. Normally distributed data were analyzed by Student’s t test or analysis of variance with post-hoc analysis by Bonferroni’s test. Correlations were assessed by Spearman’s rank test. Categorical data were assessed by chi-square test. P < 0.05 was considered statistically significant.
RESULTS
Of the subjects enrolled, 72 patients and 21 volunteers had recoverable BALF, and so entered the analysis. Clinical and demographic details for patients and volunteers are described in Table 1. Details relating to subgroups of patients are shown in the online supplement.
TABLE 1.
DEMOGRAPHIC AND CLINICAL DATA FOR PATIENTS AND MATCHED CONTROL SUBJECTS
| All Patients (n = 72) |
Matched Volunteers (n = 21) |
P Value | |
|---|---|---|---|
| Mean age, years (range) | 58 (26–87) | 60 (24–84) | 0.89* |
| Percentage of patients male | 64 | 79 | 0.14† |
| Mean (95% CI) APACHE II score | 22 (20–23) | NA | |
| Median (IQR) days of ventilation before enrollment | 8 (6–10) | NA | |
| ICU mortality, % | 35 | NA | |
| Percentage with surgical diagnosis on admission | 52 | NA | |
| Percentage with ≥1 comorbidity | 54 | NA | |
| Percentage receiving immunosuppressant drugs (including corticosteroids) |
11 | 0 | |
| Median (IQR) serum C3a des-Arg, ng/ml | 606 (388–847) | 346 (216–400) | 0.0006‡ |
| Median (IQR) serum C5a des-Arg, ng/ml | 90 (59–141) | 36 (25–43) | <0.0001‡ |
| Median (IQR) serum GM-CSF, pg/ml | 3 (2–4) | 2 (0–5) | 0.34‡ |
| Median (IQR) BALF C3a, ng/ml | 15 (4–130) | 1 (0–1) | <0.0001‡ |
| Median (IQR) BALF C5a, ng/ml | 6 (0.5–30) | 1 (0–2) | 0.005‡ |
| Median (IQR) BALF GM-CSF, pg/ml | 2 (1–9) | 2 (0–4) | 0.23‡ |
| Median (IQR) BALF neutrophils, × 105/ml | 2.8 (0.4–16) | 0.03 (0.01–0.09) | <0.0001‡ |
| Median (IQR) BALF macrophages, × 105/ml | 1.8 (0.3–3.9) | 2.6 (1.9–6.24) | 0.04‡ |
Definition of abbreviations: APACHE II = Acute Physiology and Chronic Health Evaluation II; BALF = bronchoalveolar lavage fluid; CI = confidence interval; C3a/C5a des-Arg = breakdown products of complement factors C3a/C5a; GM-CSF = granulocyte-macrophage colony-stimulating factor; ICU = intensive care unit; IQR = interquartile range; NA = not applicable.
By t test.
By chi-square test.
By Mann-Whitney U test.
The capacity of PBNs to phagocytose zymosan was significantly lower in patients than in volunteers (Figure 1). Various strategies were employed to investigate potential mechanisms underlying the phagocytic defect in patients. To test the hypothesis that deficient opsonization explained the observed impairment, zymosan was incubated in serum from patients (known to have defective PBN phagocytosis) or volunteers (known to have efficient PBN phagocytosis), and then added to healthy PBNs from four separate donors. The rates of phagocytosis were 61% (95% confidence interval [CI], 55–68%) and 65% (95% CI, 55–74%) in patient- and volunteer-opsonized zymosan, respectively (P = 0.5). Further experiments relating to opsonization are reported in the online supplement.
Figure 1.
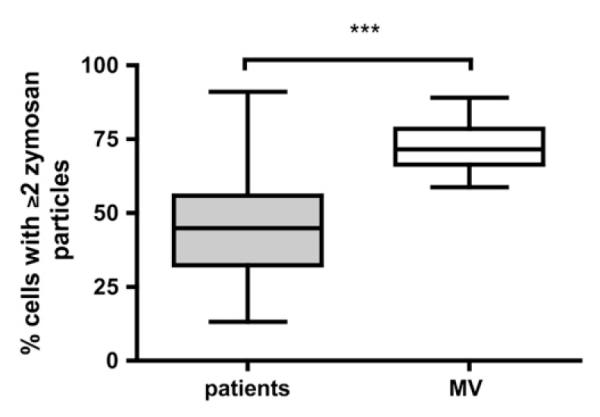
Neutrophil phagocytosis is impaired in patients with suspected ventilator-associated pneumonia (VAP) compared with matched volunteers (MVs). Phagocytosis by peripheral blood neutrophils (PBNs) from all patients was compared with that of matched volunteers. Data are presented as medians, interquartile ranges (box), and range (whiskers); ***P < 0.0001 by Mann-Whitney U test. Data are derived from 89 subjects (68 patients and 21 MVs); for the remaining 4 patients phagocytosis data were not available because of insufficient PBN adherence to tissue culture plates.
To test whether a serum factor (preventing ligation of microbes to phagocytic receptors) may explain the observed phagocytic impairment, healthy PBNs from four separate donors were preincubated in serum from patients (known to have poor phagocytosis) and volunteers (known to have efficient phagocytosis), and then exposed to zymosan. No significant differences in phagocytosis were observed (P = 0.94).
PBNs from the patients and volunteers in Figure 1 were used to study correlations between phagocytosis and expression of neutrophil phagocytic receptors (23). No significant correlation was found for CD11b (r = −0.3, P = 0.12), activated CD11b (r = −0.2, P = 0.45), CD32 (r = 0.3, P = 0.2), or CD64 (r = −0.34, P = 0.1). In addition, no evidence was found for defective expression or activation of the predominant complement-mediated phagocytic receptor CD11b (see the online supplement).
No correlation was found between phagocytosis and serum concentrations of inflammatory cytokines including IL-6, IL-8, and TNF-α (Figures 2A–2C). In contrast, serum concentration of C3a des-Arg demonstrated a significant inverse correlation with the phagocytic capacity of PBNs (Figure 2D). As neutrophils have considerably more binding sites for C5a than for C3a, leading to rapid C5a clearance from the serum, C3a is a more reliable serum marker of complement activation and anaphylotoxin exposure (24). Serum levels of C3a des-Arg and C5a des-Arg were significantly elevated in patients compared with matched volunteers (see Table 1).
Figure 2.

Phagocytosis in peripheral blood neutrophils correlates with markers of activated complement but not with serum IL-6, IL-8, or tumor necrosis factor (TNF)-α. (A) Correlation between serum TNF-α and neutrophil phagocytosis (n = 79 study participants§). (B) Correlation between serum IL-8 and neutrophil phagocytosis (n = 79 study participants). (C) Correlation between serum IL-6 and neutrophil phagocytosis (n = 79 study participants). (D) Correlation between serum C3a (a stable marker of complement activation) and neutrophil phagocytosis (n = 79 study participants). All r and P values by Spearman’s correlation. §From the n = 93 study participants in Table 1; phagocytosis data were not available for 4, and cytokine data were not available for 10.
We therefore studied the relationship between phagocytosis and neutrophil expression of CD88 (synonymous with C5a receptor type 1 [25]). CD88 expression is known to be down-regulated by C5a (24). In patients and volunteers phagocytosis by PBNs correlated significantly with expression of CD88 (Figure 3A).
Figure 3.

Effects of recombinant C5a applied ex vivo to neutrophils. (A) Correlation between neutrophil surface C5a receptor (CD88) expression (as determined by flow cytometry) and phagocytic capacity. Data are pooled for patients with suspected ventilator-associated pneumonia (VAP), and matched volunteers (n = 22 consecutive study participants, i.e., an interim analysis suggested an effect of C5a on phagocytosis, and CD88 was measured in all consecutive subjects thereafter). r and P values by Spearman’s correlation. (B) Correlation between phagocytosis and CD88 expression in healthy volunteer neutrophils exposed to escalating concentrations of C5a (n = 3 peripheral blood neutrophil [PBN] donors). The neutrophils of three separate donors were exposed to four separate doses of C5a, that is, each symbol × is from a different donor’s neutrophils exposed to 1000 nM C5a, and so on. (C) Effect of recombinant C5a on the phagocytic capacity of healthy human neutrophils in vitro. Neutrophils from healthy volunteers were incubated with either S5/1 (a murine monoclonal antibody blocking CD88) or with control antibody. Neutrophils were then exposed to C5a and the capacity for phagocytosis of zymosan was estimated. Data are derived from duplicates (n = 5 PBN donors) and presented as medians and interquartile range. P < 0.0001 by Kruskal-Wallis, *P < 0.05, **P < 0.01 by Dunn’s post hoc test. NS = not significant.
On the basis of these observations the effect of C5a on neutrophil phagocytic function was investigated further. Recombinant human C5a applied to healthy PBNs induced simultaneous down-regulation of CD88 and impairment of phagocytosis in a dose-dependent manner (Figure 3B), producing a similar correlation to that seen in patients. The C5a-mediated impairment of phagocytosis could be prevented by prior treatment of PBNs with S5/1, an antibody specifically blocking CD88 (26) (Figure 3C).
To establish whether C5a-mediated effects on PBNs were restricted to impairment of phagocytosis, additional neutrophil functions were studied. Production of superoxide by PBNs followed a strikingly similar pattern to phagocytosis, in that patients produced significantly less superoxide than control subjects (Figure 4A), whereas superoxide production showed a significant negative correlation with serum C3a des-Arg (Figure 4B) and a positive correlation with PBN surface CD88 expression (r = 0.47, P = 0.03). Addition of C5a to healthy PBNs resulted in significant impairment of transmigration (Figure 4C). Similarly, addition of C5a to healthy PBNs significantly impaired the capacity to kill P. aeruginosa, a common nosocomial pathogen (Figure 4D).
Figure 4.

C5a-mediated neutrophil defects are not restricted to phagocytosis. (A) Production of superoxide radical by neutrophils primed with 100 nM platelet-activating factor (PAF) and stimulated with 100 nM formyl methionyl leucyl phenylalanine (fMLP). P = 0.0076 by Mann-Whitney test. Data are derived from 16 patients and 12 matched volunteers, reflecting consecutive subjects recruited after interim analysis suggesting an effect of C5a on phagocytosis. (B) Correlation between PAF/fMLP-stimulated superoxide production by neutrophils and serum C3a concentrations in patients and matched volunteers. r and P values by Spearman’s correlation. Data are derived from 20 consecutive study participants (i.e., recruited after interim analysis suggesting an effect of C5a on phagocytosis). (C) Transmigration of neutrophils across a polystyrene membrane (pore size, 3 μm) to a target of 100 nM fMLP. Neutrophils from healthy volunteers were preincubated in control medium, or 100 nM C5a, **P = 0.008 by Mann-Whiney test (duplicates from n = 5 healthy peripheral blood neutrophil [PBN] donors). (D) Effect of C5a on the bactericidal capacity of neutrophils. P. aeruginosa strain PA01 was added for 30 minutes to wells containing no cells (left column), or to healthy neutrophils preincubated with either 100 nM C5a (middle column) or control medium (right column). Residual bacteria were quantified and expressed as colony-forming units (CFU) above those at t0.**P = 0.003 by Mann-Whitney (duplicates of n = 5 healthy PBN donors).
To ascertain whether the dysfunction outlined previously simply represented global inactivity of PBNs in critically ill patients, we investigated the effects of PBNs from patients and age/sex-matched volunteers in a model of epithelial inflammation using an epithelial cell line (A549 cells) (22). A549 cells cocultured with PBNs released negligible amounts of IL-6 and IL-8, irrespective of whether the PBNs came from patients or matched volunteers (data not shown). However, when the cocultures were stimulated with LPS the patient PBNs produced significantly greater IL-6 and IL-8 release than those from matched volunteers (Figures 5A and 5B). The effect of LPS-stimulated PBNs on IL-6 and IL-8 release required direct PBN–epithelial interaction, because when contact was prevented by separating the cells with membranes (pore size, 0.4 μm) there was significantly reduced IL-6 and IL-8 release (see insets in Figures 5A and 5B).
Figure 5.

LPS-stimulated neutrophils from critically ill patients drive increased release of inflammatory mediators from A549 cells. (A) IL-6 release induced by LPS-stimulated neutrophils from patients and matched volunteers (MVs). Data are expressed as a ratio of IL-6 release by stimulated A549 cells to IL-6 release from unstimulated A549 cells. The inset shows IL-6 release when the neutrophils were separated from A549 cells by a Transwell (TW) membrane (pore size, 0.4 μm). Data are shown as median and interquartile range, *P = 0.017 by Mann-Whitney U test. Data are derived from 71 study participants (57 patients and 14 MVs, i.e., all subjects from whom sufficient neutrophils and A549 cells were available to complete the assay). The Transwell data are derived from 12 study participants. (B) IL-8 release in the same experiments shown in (A). **P = 0.0098 by Mann-Whitney U test.
To further elucidate the mechanism by which C5a mediates the observed defect in phagocytosis we examined the actions of two other agents. fMLP is a bacterial peptide that acts via a receptor coupled to the same Gα subunit (Gαi2) as CD88 (27, 28). Incubation of fMLP with neutrophils produced a defect in phagocytosis similar to that seen with C5a (Figure 6A). C5a and fMLP activate several intracellular signaling pathways, including pathways dependent on cyclic adenosine monophosphate/protein kinase A (cAMP/PKA) (29) and PI3K (30). Both pathways have been shown to influence phagocytosis and cell migration (31, 32). Blockade of cAMP failed to prevent the effects of C5a and fMLP on PBN phagocytosis (see the online supplement). However, preincubation of neutrophils with the PI3K inhibitor LY294002 blocked the effects of both C5a and fMLP on phagocytosis (Figures 6B and 6C). A similar effect was achieved with another PI3K inhibitor, wortmannin (data not shown).
Figure 6.

C5a and formyl methionyl leucyl phenylalanine (fMLP) induce similar effects on phagocytosis, which appear to be dependent on phosphoinositide 3-kinase (PI3K) signaling. (A) The effects of C5a and fMLP on phagocytosis by healthy neutrophils. Data are presented as median and interquartile ranges, P < 0.0001 by Kruskal-Wallis, **P < 0.01 by Dunn’s post-hoc test (n = 6 individual peripheral blood neutrophil [PBN] donors, all experiments performed in duplicate). (B) The effect of preincubation of healthy neutrophils with 6.6 μM LY294002 (IC50 for PI3Kγ) before exposure to 100 nM C5a. Data are illustrated as median and interquartile range, P = 0.0002 by Kruskal-Wallis, **P < 0.01 by Dunn’s post-hoc test (n = 6 individual PBN donors, all experiments performed in duplicate). (C) The effect of preincubation of healthy neutrophils with 6.6 μM LY294002 (IC50 for PI3Kγ) before exposure to 100 nM fMLP. Data are illustrated as median and interquartile range, P = 0.0007 by Kruskal-Wallis, **P < 0.01, by Dunn’s post-hoc test (n = 6 individual PBN donors, all experiments performed in duplicate). NS = not significant.
In considering the clinical implications of our findings it could be argued that an agent capable of reversing deficient phagocytosis would be useful. On the basis that GM-CSF has been associated with preservation of phagocytosis in patients with sepsis (33), we postulated that GM-CSF may reverse the phagocytic impairment described. GM-CSF conferred no additional phagocytic capacity to healthy PBNs, but fully restored C5a-mediated impairment of phagocytosis (Figure 7). This principle could be extended to neutrophils from patients with suspected VAP, GM-CSF producing a 33% increase in phagocytosis (P = 0.018 by Mann-Whitney, n = 6 separate patient neutrophil populations). Of note, the levels of GM-CSF in patient serum and lavage were low and did not differ between patients and control subjects (see Table 1).
Figure 7.
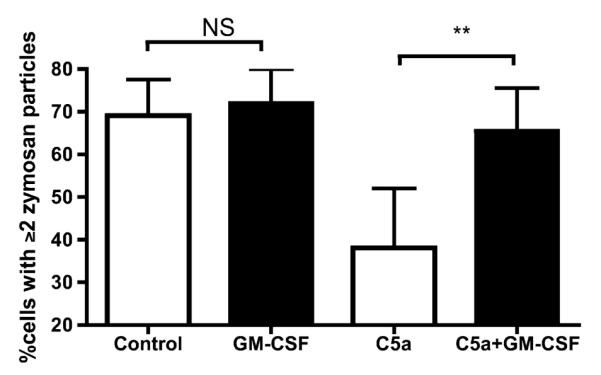
Granulocyte-macrophage colony-stimulating factor (GM-CSF) rescues phagocytic function in neutrophils. Peripheral blood neutrophils (PBNs) from healthy volunteers were incubated with C5a or control, and exposed to GM-CSF. The capacity for phagocytosis of zymosan was estimated. Data are derived from duplicates (n = 5 PBN donors) and presented as means and standard deviation. P = 0.0001 by Kruskal-Wallis, **P < 0.01 by Dunn’s post hoc test. NS = not significant.
The studies in PBNs and serum were extended to cells and supernatant derived from BALF, with the aim of determining whether neutrophils accessing the alveolar compartment function in a manner reminiscent of blood neutrophils. Impaired phagocytic function was observed in cells derived from BALF (Figure 8A), resembling the pattern in PBNs (Figure 1). However, whereas the PBNs in Figure 1 comprised a pure population of neutrophils, the BALF cells from patients were a mixture of alveolar macrophages (AMs) and neutrophils (see Table 1). As neutrophils are inherently more phagocytic than AMs (compare the MV column with the inset in Figure 8A), patient BALF cells were expected to have a phagocytic “advantage” over volunteer BALF cells (which are almost exclusively AMs). That the opposite trend was observed suggests a profound impairment of phagocytosis in BALF retrieved from patients.
Figure 8.

Lavage from patients with suspected ventilator-associated pneumonia (VAP) induces a range of neutrophil defects in a C5a-independent manner. (A) Phagocytosis by cells retrieved from bronchoalveolar lavage fluid (BALF) in patients and in matched volunteers (MVs). The predominant cell in patients was the neutrophil, whereas in MVs it was the alveolar macrophage (see Table 1). The inset shows the level of phagocytosis by the equivalent number of neutrophils from the same MVs (making the point that phagocytosis is more avidly performed by peripheral blood neutrophils than by alveolar macrophages). Data are presented as medians, interquartile ranges (box), and range (whiskers), P = 0.03 by Mann-Whitney U test. Data are derived from 44 study participants (33 patients and 11 MVs, i.e., all subjects for whom sufficient numbers of phagocytes were available and adhered to tissue culture plates). (B) Effect on phagocytosis of incubating healthy peripheral blood neutrophils (PBNs) with BALF from MVs or patients with clinically suspected VAP. Data represent duplicates from five separate healthy PBN donors and are expressed as medians, interquartile ranges (box), and range (whiskers), ***P = 0.0001 by Mann-Whitney U test. (C) Comparison of the effects of patients’ lavage or C5a on CD88 expression by healthy neutrophils. Data represent duplicates from three separate healthy PBN donors and are expressed as mean and standard deviation, P < 0.0001 by analysis of variance, **P < 0.01 by Bonferroni’s post hoc test. (D) The effect of preincubating neutrophils with the anti-CD88 antibody S5/1 on the ability of patient lavage to induce a defect in phagocytosis in healthy donor neutrophils. Data are shown as median and interquartile ranges of duplicates from n = 3 separate PBN donors, P < 0.001 by Kruskal-Wallis, *P < 0.05 by Dunn’s post hoc test. (E) The effect of preincubating healthy PBNs with lavage from MVs or patients on transmigration toward formyl methionyl leucyl phenylalanine (fMLP). Data are shown as median and interquartile ranges of duplicates from n = 3 separate PBN donors (exposed to lavage from one MV and six patients), P = 0.0046 by Kruskal-Wallis, **P < 0.01 by Dunn’s post-hoc test. (F) The effect of preincubating healthy PBNs with lavage from MVs or patients on the killing of P. aeruginosa. Data are shown as median and interquartile ranges of duplicates from n = 3 separate PBN donors (exposed to lavage from one MV and six patients), **P = 0.013 by Mann-Whitney U test.
BALF supernatant from patients significantly reduced the capacity of healthy PBNs to phagocytose zymosan when compared with BALF supernatant from volunteers (Figure 8B). However, lavage fluid did not influence the expression of CD88 on healthy PBNs (Figure 8C), and application of anti-CD88 antibody S5/1 could not prevent the impairment of phagocytosis induced by patient lavage fluid (Figure 8D). Although the levels of C3a and C5a in lavage were elevated in patients compared with matched volunteers, the levels were 10 times lower than in serum (Table 1), and no correlation was found between lavage C3a and lavage cell phagocytosis (r = 0.21, P = 0.2).
The impairment of neutrophil function induced by patient lavage was not limited to phagocytosis. Patient lavage abolished transmigration of healthy PBNs (Figure 8E) and significantly abrogated their potential to kill P. aeruginosa (Figure 8F) when compared with lavage from matched volunteers.
DISCUSSION
Although abnormal function of neutrophils has been observed previously in critical illness (11, 12), our data extend these findings by proposing a central role for activated complement in mediating dysfunction of neutrophils in the circulation. We have also identified a similar profound impairment of neutrophil function in BALF that does not appear to be mediated by C5a, but by an as yet unidentified factor(s).
C5a in relatively low concentrations is traditionally regarded as an inflammatory anaphylotoxin promoting chemotaxis and activation of neutrophils (17). However, it is increasingly recognized that C5a is overexpressed in sepsis (14), and that in this setting C5a exerts alternative influences on neutrophil function. Thus important animal models and in vitro experiments suggest C5a is an important mediator of neutrophil dysfunction (15–17), acting via the major C5a receptor CD88 (25). CD88 is not directly phagocytic but is a marker of exposure to C5a, which inhibits phagocytosis through previously undefined mechanisms. This study provides a first clinical extension of these observations, showing that phagocytosis correlated with CD88 expression on patient neutrophils, that C5a applied to healthy PBNs could recapitulate the phenotype of patient PBNs, that blockade of CD88 prevented impairment of phagocytosis, that the PI3K pathway is involved in regulating phagocytic impairment mediated by C5a, and that GM-CSF could restore phagocytosis to levels observed before C5a exposure.
Our data cannot exclude an important influence of mediators other than C5a in driving neutrophil dysfunction. However, we were unable to demonstrate an association between phagocytic impairment and alterations in biological factors classically linked to phagocytosis (e.g., expression of CD11b, CD32, and CD64). CD11b is up-regulated on “activated” neutrophils (34) and the failure of CD11b expression to correlate with phagocytosis suggests that the correlation observed with CD88 is not simply the result of generalized “neutrophil activation.” CD88 down-regulation can occur after exposure to other inflammatory mediators than C5a (35),such as IL-8 and TNF-α. However, the finding that C3a des-Arg correlated significantly and negatively with both phagocytosis and ROS production, when TNF-α, IL-8, and IL-6 showed no such relationship, again suggests that impaired phagocytosis is related to anaphylotoxin generation rather than a nonspecific result of systemic inflammation.
Although the main focus of this work was on PBN phagocytosis we demonstrated that C5a, in concentrations relevant to critically ill patients, impaired a range of other neutrophil functions including reactive oxygen species (ROS) generation, chemotactic migration, and the capacity to kill the nosocomial pathogen P. aeruginosa. Thus our data are not restricted to an effect on zymosan or to an effect on phagocytosis. Importantly, whereas neutrophils from patients exhibit diminished function with respect to two vital antibacterial activities (i.e., phagocytosis and ROS production), they demonstrate enhanced proinflammatory effects toward host cells. This suggests that patient neutrophils are not in a nonspecific and generalized “anergic state,” but in a complex dysfunctional state. The evolution of a compensatory antiinflammatory syndrome (CARS) is well described in critical illness (36). This concept arose largely in relation to monocyte deactivation in septic patients (37), but other key elements of immune function are now known to be influenced in a similar way (11, 12). The emerging consensus is that CARS and severe inflammatory response syndrome (SIRS) are not temporally distinct phases, and may coexist and overlap within the same patient. The finding that neutrophils may be driven into a hypofunctional state by a classically proinflammatory mediator (C5a) supports dynamic, and potentially maladaptive, interactions between proinflammatory molecules and neutrophils. A practical consequence of our findings is that CD88 may potentially serve as a marker of neutrophil dysfunction much in the way HLA-DR expression is used in monocytes (38).
In pursuing the mechanisms by which C5a may impair neutrophil phagocytosis we were unable to demonstrate a role for cAMP. In contrast, inhibition of PI3K abolished the effects of C5a. This finding was perhaps surprising, given the work by Wrann and colleagues suggesting that PI3K blockade does not rescue phagocytosis and is detrimental in a murine model of sepsis (39). However, their work did not specifically demonstrate C5a-mediated reduction in phagocytosis, in contrast to our findings and previous findings from the same group (15–17, 40). It also remains plausible that responsiveness to PI3K inhibitors differs between mice and humans. Although PI3K is generally thought to facilitate proinflammatory functions (41), chronic PI3K activation has demonstrated “antiinflammatory” functions (42). We hypothesize that when neutrophils are exposed to pathological levels of C5a, as occurs in systemic inflammation and sepsis, this “overloads” the PI3K pathways and paralyzes cellular functions such as phagocytosis. Further work is being undertaken to elucidate the temporal interplay between C5a generation, PI3K activation, and neutrophil function.
Given that significant levels of activated complement are generated in most critically ill patients (14, 43), it is tempting to suggest that C5a-induced neutrophil dysfunction is common to most ICU patients. Two important implications arise from this scenario. First, generation of activated complement could explain the almost ubiquitous immune impairment seen in the ICU, despite the wide variation in clinical presentations. Second, restoration of phagocytosis may be a potential target in the design of strategies for preventing infection in critically ill patients. In this regard anti-C5 and anti-CD88 therapies do exist (44, 45). However, these have not yet been used in clinical trials related to critical illness/sepsis, and of concern is the finding that CD88 knockout mice fail to clear bacteria (46). On the basis of data presented here, GM-CSF can potentially restore C5a-mediated neutrophil dysfunction. As endogenous levels in serum and BALF were low in our hands, GM-CSF may be an alternative therapy worthy of further investigation. Clinical trials of GM-CSF in nonneutropenic critical illness have been limited, and have not stratified patients by immune phenotype (33, 47). GM-CSF could potentially have a stronger effect in preventing nosocomial infections in patients with confirmed neutrophil dysfunction and, encouragingly, GM-CSF–mediated preservation of neutrophil function in critically ill patients has been demonstrated (33).
To our knowledge the phagocytic capacity of cells from the alveolar compartment has not been systematically assessed in patients with suspected VAP. The data presented suggest that, in contrast to neutrophils from the blood, C5a is not the mediating factor in lavage. Levels of C3a and C5a were far lower in BALF than in serum, antibody blockade of CD88 failed to prevent the reduction in phagocytosis, and the application of lavage did not alter CD88 expression. The implication is that phagocytic impairment is compartmentalized and multifactorial, with contributing influences in the local inflammatory milieu and in the circulation. It is intriguing to note that the ratio of C3a to C5a in the inflamed lung is on the order of 2:1, in contrast to the serum, where it is 6:1, in keeping with previous literature on this topic (48, 49). This suggests that complement activation may be differently regulated in these two compartments, as indeed are other processes such as fibrinolysis (50).
The strength of the neutrophil impairment induced by BALF was perhaps surprising, given that BALF is generally considered to be 100 times more dilute than epithelial lining fluid (20). The effect of patient BALF on phagocytosis by, and transmigration of, healthy neutrophils was particularly striking (Figures 8B and 8E, respectively). The presence of a potent pulmonary inhibitor(s) of neutrophil function may partly explain the high proportion, and clinical importance, of pneumonias among HAIs in the critically ill (1).
Identification of distinct influences on neutrophil function in the blood and in the lung is pertinent to the design of therapeutic strategies discussed earlier. Thus, restoration of phagocytosis in PBNs may not be effective in preventing pneumonia if a local alveolar environment capable of deactivating recruited PBNs has already evolved. It remains unknown whether neutrophils at other sites susceptible to HAI (e.g., the urinary tract, skin wounds, sinuses) can be deactivated by local factors or remain dependent on influences in the blood. These considerations suggest both that a greater understanding of the pathogenesis of HAI is required before “neutrophil restoration” therapy is realized, and that the timing of administration of any such therapy is likely to crucial.
Extending these themes, certain cautions should be exercised in extrapolating data from our study. The patients studied here had been mechanically ventilated in ICU for a median of 8 days and by definition already had chest X-ray infiltrates. Our study does not have the capacity to dissect whether neutrophil dysfunction contributed to the propensity for severe and ongoing illness, or whether it was a consequence of the specific setting (suspected VAP) studied here. Indeed, the timing of acquisition of neutrophil dysfunction in critically ill patients has been the focus of relatively few studies. In addition, this study was not designed to determine the subsequent rate of HAI in our cohort of patients. Despite these caveats, however, our patients were well characterized, satisfied strict predefined criteria for clinically suspected VAP, and are representative of an important group of patients seen in all ICUs. We are currently undertaking a study to investigate these issues further and to determine whether C5a is a mediator of neutrophil dysfunction in other groups of critically ill patients. Furthermore, although our volunteers were closely matched to the patients in age and sex, they were not mechanically ventilated. We cannot exclude the possibility that ventilation or underlying comorbidities may explain some of the neutrophil dysfunction demonstrated in patients. However, this does not detract from the purpose of the study, which was to characterize neutrophil impairment in critically ill patients.
In conclusion, critically ill patients with suspected VAP have C5a-mediated dysfunction of peripheral blood neutrophils. A further, complement-independent impairment of neutrophil function resides in the inflamed alveolar compartment. These findings have significant implications for the understanding of the pathogenesis of HAI, and for the design of novel strategies for the prevention of HAI.
Supplementary Material
Acknowledgment
The authors are grateful to the following people: Dr. David Swann, Ms. Pam Ramsay and Mr. Gordon Mills (ICU, Royal Infirmary of Edinburgh). and Professor Peter Andrews (ICU, Western General Hospital, Edinburgh) for general help and advice; Dr. Ian Laurenson (Department of Medical Microbiology, Royal Infirmary of Edinburgh) for general help and advice and Professor John Govan (Centre for Infectious Diseases, University of Edinburgh) for providing P. aeruginosa strain PA01; and Professor Ian Dransfield (MRC Centre for Inflammation Research, University of Edinburgh) for general help and advice on specific neutrophil assays.
Supported by a grant from the Sir Jules Thorn Charitable Trust. Donald J. Davidson is a Wellcome Trust Career Development Fellow (Fellowship #078265).
Footnotes
This article has an online supplement, which is accessible from this issue’s table of contents at www.atsjournals.org
Conflict of Interest Statement: A.C.M. does not have a financial relationship with a commercial entity that has an interest in the subject of this manuscript. K.K. does not have a financial relationship with a commercial entity that has an interest in the subject of this manuscript. T.S.W. does not have a financial relationship with a commercial entity that has an interest in the subject of this manuscript. K.D. does not have a financial relationship with a commercial entity that has an interest in the subject of this manuscript. L.F. does not have a financial relationship with a commercial entity that has an interest in the subject of this manuscript. T.W. declares that he is a recipient of an unrestricted educational grant form Wyeth pharmaceuticals for work concerning epidemiology of ICU-acquired infection. S.J.M. does not have a financial relationship with a commercial entity that has an interest in the subject of this manuscript. H.R. does not have a financial relationship with a commercial entity that has an interest in the subject of this manuscript. D.J.D. does not have a financial relationship with a commercial entity that has an interest in the subject of this manuscript. C.H. does not have a financial relationship with a commercial entity that has an interest in the subject of this manuscript. A.G.R. does not have a financial relationship with a commercial entity that has an interest in the subject of this manuscript. J.-M.S. does not have a financial relationship with a commercial entity that has an interest in the subject of this manuscript. A.J.S. declares that he has received expenses from AztraZeneca and GlaxoSmithKline (for travel and accommodation) to attend international educational conferences.
References
- 1.Vincent J-L, Bihari DJ, Suter PM, Bruining HA, White J, Nicolas-Chanoin MH, Wolff M, Spencer RC, Hemmer M, EPIC International Advisory Committee The prevalence of nosocomial infection in intensive care units in Europe: results of the European Prevalence of Infection in Intensive Care (EPIC) Study. JAMA. 1995;274:639–644. [PubMed] [Google Scholar]
- 2.Richards M, Edwards J, Culver D, Gaynes R. Nosocomial infections in medical intensive care units in the United States. Crit Care Med. 1999;27:887–892. doi: 10.1097/00003246-199905000-00020. [DOI] [PubMed] [Google Scholar]
- 3.Haley RW, Culver DH, White JW, Morgan WM, Emori TG. The nationwide nosocomial infection rate: a new need for vital statistics. Am J Epidemiol. 1985;121:159–163. doi: 10.1093/oxfordjournals.aje.a113988. [DOI] [PubMed] [Google Scholar]
- 4.Martone WJ, Jarvis WR, Culver DH, Haley RW. Incidence and nature of endemic and epidemic nosocomial infections. In: Bennett JV, Brachman PS, editors. Hospital infections. 3rd ed Little Brown; Boston: 1992. pp. 577–596. [Google Scholar]
- 5.Cook DJ, Walter SD, Cook RJ, Griffith LE, Guyatt GH, Leasa D, Jaeschke RZ, Brun-Buisson C. Incidence of and risk factors for ventilator-associated pneumonia in critically ill patients. Ann Intern Med. 1998;129:433–440. doi: 10.7326/0003-4819-129-6-199809150-00002. [DOI] [PubMed] [Google Scholar]
- 6.Nourdine K, Combes P, Carton M-J, Beuret P, Cannamela A, Ducreux J-C. Does noninvasive ventilation reduce the ICU nosocomial infection risk? A prospective clinical survey. Intensive Care Med. 1999;25:567–573. doi: 10.1007/s001340050904. [DOI] [PubMed] [Google Scholar]
- 7.McLaws M-L, Berry G. Nonuniform risk of bloodstream infection with increasing central venous catheter-days. Infect Control Hosp Epidemiol. 2005;26:715–719. doi: 10.1086/502608. [DOI] [PubMed] [Google Scholar]
- 8.Rello J, Diaz E, Roque M, Valles J. Risk factors for developing pneumonia within 48 hours of intubation. Am J Respir Crit Care Med. 1999;159:1742–1746. doi: 10.1164/ajrccm.159.6.9808030. [DOI] [PubMed] [Google Scholar]
- 9.Lynch JP. Hospital-acquired pneumonia: risk factors, microbiology, and treatment. Chest. 2001;119:373S–384S. doi: 10.1378/chest.119.2_suppl.373s. [DOI] [PubMed] [Google Scholar]
- 10.Gastmaier P, Sohr D, Geffers C, Behnke M, Rüden H. Risk factors for death due to nosocomial infection in intensive care unit patients: findings from the Krankenhaus Infektions Surveillance System. Infect Control Hosp Epidemiol. 2007;28:466–472. doi: 10.1086/510810. [DOI] [PubMed] [Google Scholar]
- 11.Kaufmann I, Hoelzl A, Schliephake F, Hummel T, Chouker A, Peter K, Thiel M. Polymorphonuclear leukocyte dysfunction syndrome in patients with increasing sepsis severity. Shock. 2006;26:254–261. doi: 10.1097/01.shk.0000223131.64512.7a. [DOI] [PubMed] [Google Scholar]
- 12.Simms HH, D’Amico R. Polymorphonuclear leukocyte dysregulation during the systemic inflammatory response syndrome. Blood. 1994;83:1398–1407. [PubMed] [Google Scholar]
- 13.Chastre J, Fagon J-Y. Ventilator-associated pneumonia. Am J Respir Crit Care Med. 2002;65:867–903. doi: 10.1164/ajrccm.165.7.2105078. [DOI] [PubMed] [Google Scholar]
- 14.Bengtson A, Heideman M. Anaphylatoxin formation in sepsis. Arch Surg. 1988;123:645–649. doi: 10.1001/archsurg.1988.01400290131023. [DOI] [PubMed] [Google Scholar]
- 15.Huber-Lang M, Sarma VJ, Lu KT, McGuire SR, Padgaonkar VA, Guo RF, Younkin EM, Kunkel RG, Ding J, Erickson R, et al. Role of C5a in multiorgan failure during sepsis. J Immunol. 2001;166:1193–1199. doi: 10.4049/jimmunol.166.2.1193. [DOI] [PubMed] [Google Scholar]
- 16.Huber-Lang M, Younkin EM, Sarma VJ, McGuire SR, Lu KT, Guo RF, Padgaonkar VA, Curnutte JT, Erickson R, Ward PA. Complement-induced impairment of innate immunity during sepsis. J Immunol. 2002;169:3223–3231. doi: 10.4049/jimmunol.169.6.3223. [DOI] [PubMed] [Google Scholar]
- 17.Ward PA. The dark side of C5a in sepsis. Nat Rev Immunol. 2004;4:133–142. doi: 10.1038/nri1269. [DOI] [PubMed] [Google Scholar]
- 18.Reid H, Morris A Conway, Kefala K, Wilkinson T, Farrell L, Walsh TS, MacKenzie S, Sallenave JM, Dhaliwal K, Haslett C, et al. Neutrophils from patients with ventilator associated pneumonia: pro-inflammatory and cytotoxic interactions with alveolar epithelium [abstract] Thorax. 2008;63:A143. [Google Scholar]
- 19.Haslett C, Guthrie LA, Kopaniak MM, Johnston RB, Henson PM. Modulation of multiple neutrophil functions by preparative methods or trace concentrations of bacterial lipopolysaccharide. Am J Pathol. 1985;119:101–110. [PMC free article] [PubMed] [Google Scholar]
- 20.Rennard SI, Basset G, Lecossier D, O’Donnell KM, Pinkston P, Martin PG, Crystal RG. Estimation of volume of epithelial lining fuid recovered by lavage using urea as marker of dilution. J Appl Physiol. 1986;60:532–538. doi: 10.1152/jappl.1986.60.2.532. [DOI] [PubMed] [Google Scholar]
- 21.Condliffe AM, Chilvers ER, Haslett C, Dransfield I. Priming differentially regulates neutrophil adhesion molecule expression/function. Immunology. 1996;89:105–111. doi: 10.1046/j.1365-2567.1996.d01-711.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Giard DJ, Aaronson SA, Todaro GJ. In vitro cultivation of human tumors: establishment of cell lines derived from a series of solid tumors. J Natl Cancer Inst. 1973;51:1417–1423. doi: 10.1093/jnci/51.5.1417. [DOI] [PubMed] [Google Scholar]
- 23.Lee WL, Harrison RE, Grinstein S. Phagocytosis by neutrophils. Microbes Infect. 2003;5:1299–1306. doi: 10.1016/j.micinf.2003.09.014. [DOI] [PubMed] [Google Scholar]
- 24.Oppermann M, Götze O. Plasma clearance of the human C5a anaphylatoxin by binding to leucocyte C5a receptors. Immunology. 1994;82:516–521. [PMC free article] [PubMed] [Google Scholar]
- 25.Chenoweth DE, Hugli TE. Demonstration of specific C5a receptor on intact human polymorphonuclear leukocytes. Proc Natl Acad Sci USA. 1978;75:3943–3947. doi: 10.1073/pnas.75.8.3943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Oppermann M, Raedt U, Hebell T, Schmidt B, Zimmermann B, Götze O. Probing the human receptor for C5a anaphylatoxin with site-directed antibodies: identification of a potential ligand binding site on the NH2-terminal domain. J Immunol. 1993;151:3785–3794. [PubMed] [Google Scholar]
- 27.Skokowa J, Ali Syed R, Felda O, Kumar V, Konrad S, Shushakova N, Schmidt RE, Piekorz RP, Nürnberg B, Spicher K, et al. Macrophages induce the inflammatory response in the pulmonary Arthus reaction through Gαi2 activation that controls C5aR and Fc receptor co-operation. J Immunol. 2005;174:3041–3050. doi: 10.4049/jimmunol.174.5.3041. [DOI] [PubMed] [Google Scholar]
- 28.Bommakanti RK, Dratz EA, Siemsen DW, Jesaitis AJ. Extensive contact between Gi2 and N-formyl peptide receptor of human neutrophils: mapping of binding sites using receptor-mimetic peptides. Biochemistry. 1995;34:6720–6728. doi: 10.1021/bi00020a017. [DOI] [PubMed] [Google Scholar]
- 29.Tsu RC, Allen RA, Wong YH. Stimulation of type II adenylyl cyclase by chemoattractant formyl peptide and C5a receptors. Mol Pharmacol. 1995;47:835–841. [PubMed] [Google Scholar]
- 30.Riedemann NC, Guo RF, Gao H, Sun L, Hoesel M, Hollmann TJ, Wetsel RA, Zetoune FS, Ward PA. Regulatory role of C5a on macrophage migration inhibitory factor release from neutrophils. J Immunol. 2004;173:1355–1359. doi: 10.4049/jimmunol.173.2.1355. [DOI] [PubMed] [Google Scholar]
- 31.Busse WW, Sosman JM. Isoproterenol inhibition of isolated human neutrophil function. J Allergy Clin Immunol. 1984;73:404–410. doi: 10.1016/0091-6749(84)90416-0. [DOI] [PubMed] [Google Scholar]
- 32.Araki N, Johnson MT, Swanson JA. A role for phosphoinositide 3-kinase in the completion of macropinocytosis and phagocytosis by macrophages. J Cell Biol. 1996;135:1249–1260. doi: 10.1083/jcb.135.5.1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Presneill JJ, Harris T, Stewart AG, Cade JF, Wilson JW. A randomized phase II trial of granulocyte-macrophage colony-stimulating factor therapy in severe sepsis with respiratory dysfunction. Am J Respir Crit Care Med. 2002;166:138–143. doi: 10.1164/rccm.2009005. [DOI] [PubMed] [Google Scholar]
- 34.Berger M, O’Shea J, Cross AS, Folks TM, Chused TM, Brown EJ, Frank MM. Human neutrophils increase expression of C3bi as well as C3b receptors upon activation. J Clin Invest. 1984;74:1566–1571. doi: 10.1172/JCI111572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Furebring M, Håkansson L, Venge P, Sjölin J. C5a, interleukin-8 and tumour necrosis factor-α-induced changes in granulocyte and monocyte expression of complement receptors in whole blood and on isolated leukocytes. Scand J Immunol. 2006;63:208–216. doi: 10.1111/j.1365-3083.2006.01724.x. [DOI] [PubMed] [Google Scholar]
- 36.Bone RC. Sir Isaac Newton, sepsis, SIRS, and CARS. Crit Care Med. 1996;24:1125–1128. doi: 10.1097/00003246-199607000-00010. [DOI] [PubMed] [Google Scholar]
- 37.Döcke WD, Randow F, Syrbe U, Krausch D, Asadullah K, Reinke P, Volk HD, Kox W. Monocyte deactivation in septic patients: restoration by IFN-γ treatment. Nat Med. 1997;3:678–681. doi: 10.1038/nm0697-678. [DOI] [PubMed] [Google Scholar]
- 38.Hershman MJ, Cheadle WG, Wellhausen SR, Davidson PF, Polk HC. Monocyte HLA-DR antigen expression characterizes clinical out-come in the trauma patient. Br J Surg. 1990;77:204–207. doi: 10.1002/bjs.1800770225. [DOI] [PubMed] [Google Scholar]
- 39.Wrann C, Tabriz NA, Barkhausen T, Klos A, van Griensven M, Pape HC, Kendoff DO, Renfeng G, Ward PA, Krettek C, et al. The phosphatidylinositol 3-kinase signaling pathway exerts protective effects during sepsis by controlling C5a-mediated activation of innate immune functions. J Immunol. 2007;178:5940–5948. doi: 10.4049/jimmunol.178.9.5940. [DOI] [PubMed] [Google Scholar]
- 40.Flierl MA, Perl M, Rittirsch D, Bartl C, Schreiber H, Fleig V, Schlaf G, Liener U, Brueckner UB, Gebhard F, et al. The role of C5a in the innate immune response after experimental blunt chest trauma. Shock. 2008;29:25–31. doi: 10.1097/shk.0b013e3180556a0b. [DOI] [PubMed] [Google Scholar]
- 41.Rückle T, Schwarz MK, Rommel C. PI3Kγ inhibition: towards an “aspirin of the 21st century”? Nat Rev Drug Discov. 2006;5:903–918. doi: 10.1038/nrd2145. [DOI] [PubMed] [Google Scholar]
- 42.Blanchet MR, Israel-Assayag E, Daleau P, Beaulieu MJ, Cormier Y. Dimethyphenylpiperazinium, a nicotinic receptor agonist, downregulates inflammation in monocytes/macrophages through PI3K and PLC chronic activation. Am J Physiol Lung Cell Mol Physiol. 2006;291:757–767. doi: 10.1152/ajplung.00409.2005. [DOI] [PubMed] [Google Scholar]
- 43.Fosse E, Pillgram-Larsen J, Svennevig JL, Nordby C, Skulberg A, Mollnes TE, Abdelnoor M. Complement activation in injured patients occurs immediately and is dependent on the severity of the trauma. Injury. 1998;29:509–514. doi: 10.1016/s0020-1383(98)00113-2. [DOI] [PubMed] [Google Scholar]
- 44.Rother RP, Rollins SA, Mojcik CF, Brodsky RA, Bell L. Discovery and development of the complement inhibitor eculizumab for the treatment of paroxysmal nocturnal hemoglobinuria. Nat Biotechnol. 2007;25:1256–1264. doi: 10.1038/nbt1344. [DOI] [PubMed] [Google Scholar]
- 45.Vergunst CE, Gerlag DM, Dinant H, Schulz L, Vinkenoog M, Smeets TJM, Sanders ME, Reedquist KA, Tak PP. Blocking the receptor for C5a in patients with rheumatoid arthritis does not reduce synovial inflammation. Rheumatology. 2007;46:1773–1778. doi: 10.1093/rheumatology/kem222. [DOI] [PubMed] [Google Scholar]
- 46.Höpken UE, Lu B, Gerard NP, Gerard C. The C5a chemoattractant receptor mediates mucosal defence to infection. Nature. 1996;383:86–89. doi: 10.1038/383086a0. [DOI] [PubMed] [Google Scholar]
- 47.Rosenbloom AJ, Linden PK, Dorrance A, Penkosky N, Cohen-Melamed MH, Pinsky MR. Effect of granulocyte-monocyte colony-stimulating factor therapy on leukocyte function and clearance of serious infection in non-neutropenic patients. Chest. 2005;127:2139–2150. doi: 10.1378/chest.127.6.2139. [DOI] [PubMed] [Google Scholar]
- 48.Krug N, Tschernig T, Erpenbeck VJ, Hohlfeld JM, Kohl J. Complement factors C3a and C5a are increased in bronchoalveolar lavage ñuid after segmental allergen provocation in subjects with asthma. Am J Respir Crit Care Med. 2001;164:1841–1843. doi: 10.1164/ajrccm.164.10.2010096. [DOI] [PubMed] [Google Scholar]
- 49.Marc MM, Korosec P, Kosnik M, Kern I, Flezar M, Suskovic S, Sorli J. Complement factors C3a, C4a, and C5a in chronic obstructive pulmonary disease and asthma. Am J Respir Cell Mol Biol. 2004;31:216–219. doi: 10.1165/rcmb.2003-0394OC. [DOI] [PubMed] [Google Scholar]
- 50.Schultz MJ, Millo J, Levi M, Hack CE, Weverling GJ, Garrard CS, van der Poll T. Local activation of coagulation and inhibition of fibrinolysis in the lung during ventilator-associated pneumonia. Thorax. 2004;59:130–135. doi: 10.1136/thorax.2003.013888. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.