

Abstract
Innate immunodeficiency has recently been reported resulting from the Q293X IRAK-4 mutation, with consequent defective TLR/IL-1R signalling. Here we report a method for the rapid allele-specific detection of this mutation and demonstrate both cell-type specificity and ligand specificity in defective IRAK-4-deficient cellular responses, indicating differential roles for this protein in human peripheral blood mononuclear cells and primary dermal fibroblasts, and in LPS, IL-1β and TNF-α signalling. We demonstrate transcriptional and post-transcriptional defects, despite NF-κB signalling and intact MyD88-independent signalling, and propose that dysfunctional Complex 1 (IRAK1/TRAF6/TAK1) signalling, as a consequence of IRAK-4-deficiency, generates specific defects in mitogen-activated protein kinase activation that could underpin this patient’s innate immunodeficiency. These studies demonstrate the importance of studying primary human cells bearing a clinically relevant mutation; they underscore the complexity of innate immune signalling and illuminate novel roles for IRAK-4 and the fundamental importance of accessory pro-inflammatory signalling to normal human innate immune responses and immunodeficiencies.
Keywords: Human, Immunodeficiency diseases, Inflammation
Introduction
Toll-like receptors (TLR) are innate pattern recognition receptors that initiate distinct inflammatory responses to specific microbial components (1). Upon stimulation, TLR and interleukin-1 receptor (IL-1R) family members induce inflammatory signalling cascades, via cytoplasmic Toll/IL-1R (TIR) domains, utilizing adaptor proteins such as MyD88, TIRAP, TRIF, and TRAM (2). Classically, MyD88 recruits IL-1R associated kinase-1 (IRAK-1) and IRAK-4 through death-domain (DD) interactions, with subsequent IRAK-4-dependent phosphorylation of IRAK-1. Phosphorylated IRAK-1 then associates with TNF receptor associated factor-6 (TRAF6), initiating Transforming Growth Factor-β-activated Kinase-1 (TAK-1)-dependent signalling that ultimately activates the I-κB-kinase (IKK) complex, enables nuclear translocation of NF-κB, and the induction of specific inflammatory gene transcription.
Activation of TAK-1 also initiates less well characterised accessory pro-inflammatory signalling through the phosphorylation and activation of mitogen-activated protein kinases (MAPK) (3,4). These MAPK can modulate multiple transcriptional and post-transcriptional mechanisms (5-8). MAPK therefore represent a potentially critical level of control of inflammatory responses and have the potential to provide stimulus-specificity to the responses. However, the upstream controlling elements for these pathways and their full significance in the orchestration of an appropriately targeted immune response are not fully understood. While IRAK-4 is proposed to be critical for canonical MyD88-dependent cytokine production (9), its role in regulating MAPK-dependent responses remains unclear.
IRAK-4 deficiency has recently been described as a rare form of innate immunodeficiency; patients present with pyogenic bacterial infections and bacteraemia, particularly with Streptococcus pneumoniae (10-14). We have previously described a patient presenting with recurrent S. pneumoniae bacteraemia, and a clinical phenotype strikingly similar to that reported in IRAK-4 deficiency (15). We now report a homozygous Q293X mutation in IRAK-4 in this patient, and provide the first definitive report of the lethality of this mutation, occurring in his sibling. Furthermore, studying IRAK-4-deficient primary dermal fibroblasts and peripheral blood mononuclear cells (PBMC) we demonstrate cell-type-specific and ligand-specific defective cytokine responses, dysfunctional at transcriptional and/or post-transcriptional levels despite NF-κB signalling and intact MyD88-independent signalling.
Materials and Methods
History and consent
The clinical and immunological descriptions of the patient studied, and his deceased brother have been previously reported (15). Blood was collected from healthy adult volunteers and the patient’s immediate family, with informed consent (UBC Ethics protocol C02-009). Blood samples from the patient and five healthy 2-year-olds undergoing elective surgery were collected with parental consent according to Ethics protocol C02-0138.
Media and reagents
Tissue culture reagents were all from Invitrogen (ON, Canada) unless stated. Human recombinant IL-1β and GM-CSF were from Research Diagnostics Inc. (NJ, USA), recombinant human M-CSF and TNF-α were from R&D systems (MN, USA), Poly(I:C) was from Sigma (ON, Canada), and CpG2006 from Coley Pharmaceutical Group (MA, USA). All reagents were tested for endotoxin by Limulus amebocyte lysate assay. Escherichia coli lipopolysaccharide (LPS) from Sigma was repurified by phenol-chloroform-extraction (16) and titrated against Polymyxin B, or purchased as TLR4-specific Ultra-Pure LPS (InvivoGen, CA, USA). IRAK-4 antibodies were a kind gift from Tularik, USA (17), or purchased from Upstate Cell Signalling Solutions (MA, USA) and Cell Signalling Technology (MA, USA). Additional antibodies were polyclonal rabbit anti-IRAK-1 and anti-NF-κB p65 (Santa Cruz Biotechnology, CA, USA), mouse monoclonal anti-FLAG (Sigma), mouse anti-human CD80, CD86, and CD40 (BD Biosciences ON, Canada), or polyclonal rabbit antibodies from Cell Signalling Technology.
Peripheral blood leukocytes
Peripheral blood mononuclear cells (PBMC) were isolated by Ficoll-Paque™ Plus (Amersham Pharmacia Biotech, Uppsala, Sweden) density gradient centrifugation. Monocyte-derived dendritic cells (MDDC) were cultured as described (18). Neutrophils were isolated as described (19).
Culture of primary and immortalised fibroblasts
Primary dermal fibroblast lines were prepared from skin biopsies of the patient and a fully anonymised age-matched child (undergoing an unrelated diagnostic test). Adult human dermal fibroblasts were from Cascade Biologics (OR, USA). No difference was observed in inflammatory responses between adult and control child cells, so adult cells were routinely used thenceforth. Immortalised cell lines were also generated from these primary cell lines, by transfection with PSV3-Neo (American Type Tissue Collection, VA, USA), using FuGENE 6 transfection reagent (Roche Diagnostics, QC, Canada).
Fibroblast transfection
Immortalised fibroblasts were cultured in DMEM with 10% FCS until ~60% confluency and transfected with pFLAGRK7-IRAK-4 expression vector (a kind gift from Tularik (17)) or control empty vector, using FuGENE 6 transfection reagent (Roche Diagnostics), 48 hr before exposure to inflammatory stimuli.
Real time IRAK-4 genotyping
Primers and probes were designed using Primer Express® Software Version 2.0. Probes (6FAM-ACCCTGAGCAATC- MGBNFQ, and VIC-CACCCTAAGCAATC- MGBNFQ) were purchased from Applied Biosystems (ON, Canada), and primers (5′-ACC ACT TTC TTG GCA CAT GAG A-3′, 5′-CAT GTA GAA AAT TGA TGC CAT TAG CT-3′) were synthesised by UBC Nucleic Acid Protein Service Unit (NAPS) (BC, Canada). Resulting PCR products were sequenced to confirm validity. Triplicate reactions and allelic discrimination analysis were performed using an ABI PRISM 7000 Sequence Detection System (Applied Biosystems) on genomic DNA templates from the patient, his immediate family, 60 fully anonymised mixed archived adult and child samples, and template-free controls.
DNA Sequencing
Genomic DNA from PBMC, cell lines, or from a formalin-fixed, paraffin-embedded, archived diagnostic tissue specimen was prepared using DNeasy Tissue Kits (Qiagen, ON, Canada). Each coding exon of IRAK-4 was amplified by PCR using exon-spanning intronic primers (UBC NAPS; sequences upon request), the products purified using Qiaquick PCR purification kits (Qiagen) and sequenced on an ABI 3100 using Big Dye terminators (Applied Biosystems). Sequences from repeated PCR reactions, aligned into contigs, were analysed using Phred, Phrap and Consed (version 11.0), and compared against the IRAK-4 gene sequence accession number AF445802.
Cell stimulation
All cells were cultured at 37°C in 5% CO2. Fibroblasts were seeded at 2 × 104/ml in α-MEM with 10% FCS 48 hr before RNA extraction and analysis of supernatant cytokines. Fibroblasts for protein extraction were cultured to confluence before use. PBMC were cultured in R10 media (RPMI-1640 medium, 2 mM L-glutamine, 1mM sodium pyruvate, 10% FCS) for 1 hr prior to stimulation, at 2 × 105/ml for analysis of supernatant cytokines and 1-5 × 106/ml for extraction of RNA or protein. Cells were exposed in triplicate to 100 ng/ml of either E. coli LPS, TNF-α, IL-1β, or left unstimulated, and incubated for 24 hr for supernatant cytokine analysis, or the time specified for other analyses. MDDC, cultured at 1×105 cells/ml R10 in Teflon™ 24-well inserts (Savillex Corporation, MN, USA), were exposed for 24 hr, in triplicate, to either 100 ng/ml E. coli LPS, 10 μg/ml CpG 2006, 10 μg/ml Poly(I:C), or incubated with confluent monolayers of either untransfected or CD40L-transfected 3T3 fibroblasts (kindly provided by Dr Gregor Reid, University of British Columbia ).
Culture supernatants were spun down, aliquoted and stored at −80°C. IL-6 and IL-8 concentrations were determined in triplicate from each biological replicate using ELISA Ready-SET-go (eBioscience, CA, USA) and OptEIA ELISA kits (Pharmingen, ON, Canada) respectively. MDDC were analysed by FACS as previously described (18).
Transcriptional analysis
The transcription of IL-6 and IL-8 mRNA was assessed by semi-quantitative RT-PCR. In addition, transcription was directly monitored by analysis of the levels of partially spliced heteronuclear RNA (hnRNA) transcripts by PCR, a method reported to have greater sensitivity than nuclear run-off transcription assays (19). Total RNA was isolated using RNeasy Midi kits (Qiagen), treated to remove DNA (DNA-free™; Ambion, TX, USA), and eluted in RNase-free water (Ambion). Concentration-corrected RNA samples were divided into two portions for reverse-transcription of cDNA representing either mRNA (using Oligo dT as primer), or total RNA, including hnRNA (using random hexamers as primers), using Superscript™II (Invitrogen). PCR was performed using intron-spanning primer pairs for mRNA (exonic PCR), or primer pairs specific for an exon and an intron of the unspliced hnRNA (intronic PCR). PCR with a primer pair specific to genomic promoter regions were also performed to exclude genomic DNA contamination. All primer sequences are available on request. Each RT-PCR reaction was performed in at least duplicate, from n ≥ 3 independent experiments, analyzed by densitometry in the linear phase of amplification, using analysis of amplification reactions with incrementally increasing cycle number, and normalized to the control gene glyceraldehyde-3-phosphate dehydrogenase (GAPDH).
Western Immunoblotting
Protein preparation and Western blotting was performed as previously described (21), or with use of Complete EDTA-free Protease Inhibitor Cocktail Tablets (Roche Diagnostics), lysate concentrations were quantified using a Micro BCA Protein assay kit (Pierce Biotechnology, IL, USA) with lysate volumes used corrected accordingly to ensure equal loading, and Immuno-blot PVDF membranes were used (Bio-Rad, CA, USA). Nuclear extracts were generated using the NE-PER™ Nuclear and Cytoplasmic Extraction Reagents (Pierce Biotechnology).
Statistical analysis
Figures show mean values ± SEM. Student’s t test was used to compare patient and healthy control responses. A p value of less than 0.05 was considered to indicate statistical significance.
Results
IRAK-4 mutation and diagnosis
Initial characterisation in this patient implicated a defect downstream of TLR/IL1-R activation (15). Allelic discrimination real-time PCR clearly demonstrated homozygous substitution of thymidine for cytidine at position 877 in exon 8 of IRAK-4 in the patient, hemizygous C877T substitution in two brothers (Siblings 2 and 3) and both parents, and no C877T substitution in 60 normal controls (Fig. 1). In addition, using genomic DNA extracted from a diagnostic pathology specimen, a homozygous C877T substitution was demonstrated in the patient’s oldest brother (Sibling 1), whose medical history was also suggestive of IRAK-4 mutation, and died at age 5 years with rapidly progressive pneumococcal meningitis. This C877T substitution results in a Q293X mutation with replacement of a glutamine by a designated stop codon, as described (10). All mutations were confirmed by sequencing of Exon 8 PCR products and further sequencing of the complete IRAK-4 gene revealed no additional exonic mutations in this patient.
Figure 1. Detection of IRAK-4 Q293X mutation.
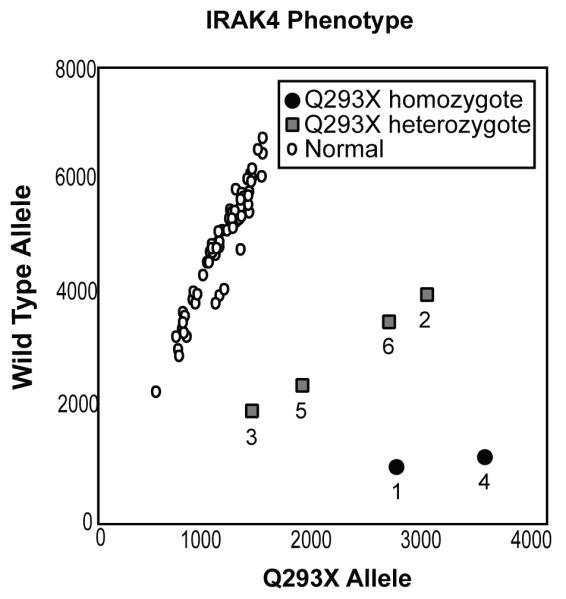
A real-time PCR allelic discrimination assay demonstrated homozygous C877T substitution in the patient (4) and deceased sibling (1), hemizygous C877T substitution in siblings (2 and 3) and both parents (5 and 6), but no C877T substitution in 60 normal controls.
The Q293X mutation in IRAK-4 results in defective expression of full-length IRAK-4 mRNA, as previously demonstrated by Northern blot in patients homozygous for this mutation (10). Prior to sequencing the IRAK-4 gene, we initially excluded the diagnosis of IRAK-4-deficiency in our patient based upon RT-PCR amplification of apparently normal levels of IRAK-4 from MDM mRNA (15). However, mRNA extracted from IRAK-4-deficient patient cells has been shown to contain sufficient partial IRAK-4 mRNA transcripts to enable cDNA sequencing of the IRAK-4 gene (11). This resulted in apparently normal levels of IRAK-4 mRNA expression when assessed by quantitative and semi-quantitative RT-PCR amplification (data not shown). Thus, although rapid degradation of transcript by nonsense mediated RNA decay (22) can be observed by RT-PCR in some diseases caused by premature stop codons (23), these studies demonstrate that such approaches are inappropriate to diagnose IRAK-4-deficiency. Interestingly, RT-PCR of IRAK-4 exons 4 – 9 (spanning the mutation) amplified two products; one of the expected size, and an additional truncated product. Sequence analysis of the smaller product revealed an 86bp deletion at the end of exon-5 (bases 566 to 651 inclusive) (Fig. 2). This truncated product may represent a previously unknown alternatively spliced form, since there is a potential cryptic splice donor site (GTG|GTAATA) at the 5′ end of the deletion. If translated, this deletion would result in a frame-shift after amino acid 188, resulting in the addition of two new amino acids followed by a premature stop codon. Translation would generate a truncated protein (predicted ~21 kDa) of unknown stability or biological significance. The C877T substitution in exon 8 in the patient was observed in both splice forms.
Figure 2. RT-PCR of IRAK-4 revealed a splice variant.
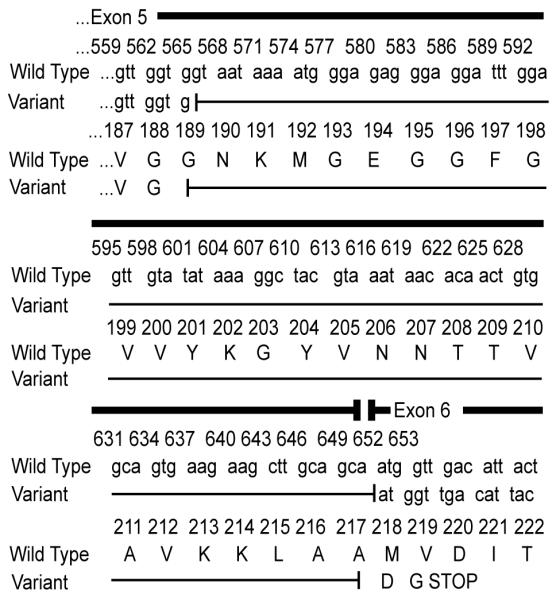
RT-PCR of partial IRAK-4 transcripts using PBMC mRNA amplified the expected 660 bp product and a 574 bp product in both patient and control samples. Both products from the patient and a control were sequenced, and the 574 bp product revealed an 86 bp deletion at the end of exon-5, resulting in a premature stop codon.
In the absence of a diagnostically applicable RT-PCR assay, detection of IRAK-4 protein by Western immunoblot was evaluated from a diagnostic perspective, using three different antibodies specific to IRAK-4. IRAK-4 protein (~ 52 kDa) could be clearly detected after transfection of immortalised control fibroblasts with pFLAGRK7-IRAK-4 (Fig 3a), demonstrating the effectiveness of these antibodies in cells over-expressing IRAK-4 protein. However, this was not observed in the absence of transfection (Fig. 3a), indicating that under normal conditions, these antibodies could not detect the native IRAK-4 expressed in these cells. Although multiple bands within a similar size range were detected with these antibodies, no significant differences were observed between the patient’s cells and controls, using whole cell lysates from untransfected immortalised fibroblasts (Fig. 3a), PBMC (Fig. 3b) or primary fibroblasts (data not shown). These data indicate the non-specificity of these bands, and the unsuitability of Western immunoblot with these antibodies as a diagnostic approach for IRAK-4-deficiency. Detection of IRAK-4 has previously been demonstrated utilising whole cell lysates of immortalised lymphoblastoid B-cells from healthy donors, and shown to be defective in those derived from IRAK-4-deficient patients (10), suggesting differences in IRAK-4 expression levels, or the quality of protein extracted from different cell types. However, our data are compatible with another report in which IRAK-4 protein could not be demonstrated in PBMC from either IRAK-deficient patient or controls, while IRAK-4 was detected when over-expressed in cell lines (11).
Figure 3. Western immunoblot analysis for IRAK-4 protein expression in fibroblasts and PBMC does not discriminate between patient and controls.
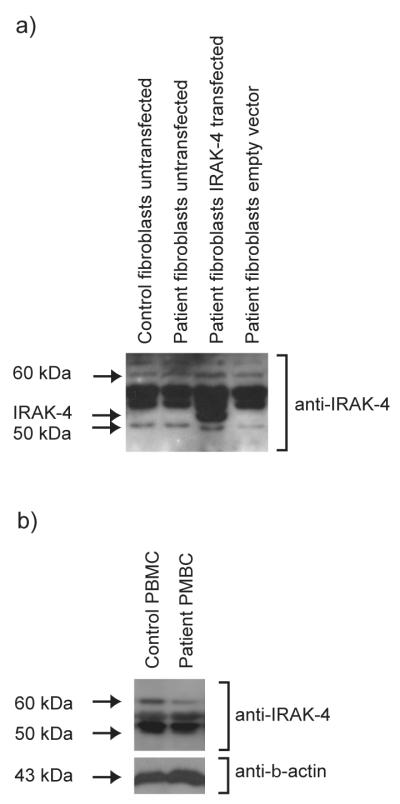
Western immunoblot analysis was performed with antibodies specific for IRAK-4. Representative images are shown using a) Upstate Cell Signalling Solutions anti-IRAK-4 and b) Tularik anti-IRAK-4. a) Lysates from immortalised dermal fibroblast lines from the patient, transfected with pRK-7-IRAK-4 expression vector (but not control empty vector) expressed a ~52 kDa protein that was not detected in untransfected patient or control cells. Representative of n=2 biological repeats for this antibody. b) Lysates from PBMC demonstrated non-specific bands, and Western immunoblots were not discriminatory for patient cells when compared to controls. Representative of n=4 biological repeats for this antibody, with an anti-β-actin antibody used as loading control
These data emphasise the importance of appropriate laboratory approaches in evaluating possible IRAK-4-deficient individuals, indicating the need for functional studies of TLR/IL-1R-induced cytokine production, diagnostic genotyping of specific mutations and/or gene sequencing.
Cytokine responses are differentially affected in Q293X mutant primary fibroblasts and PBMC
We next characterised the IL-6 and IL-8 responses of our patient’s primary dermal fibroblasts and PBMC following LPS, IL-1β, or TNF-α stimulation. LPS induced significant production of IL-6 and IL-8 by PBMC from healthy controls, whereas the patient’s PBMC produced no IL-6 in response to LPS and approximately 10-fold less IL-8, significantly differing from the controls (p<0.05, p<0.01 respectively; Fig. 4a & b). Notably, we observed expression of both CD14 and TLR4 by RT-PCR in primary dermal fibroblasts from healthy controls (Fig. 5c) and these cells also expressed significant levels (p<0.05) of both IL-6 and IL-8 in response to highly purified LPS (Fig. 5a & b). The patient’s dermal fibroblasts expressed comparable levels of CD14 and TLR4 (Fig. 5c) but did not produce any IL-6 or IL-8 after LPS stimulation (Fig. 5a & b).
Figure 4. Defective cytokine responses in IRAK-4-deficient PBMC.

Primary PBMC from the patient and controls were exposed to 100 ng/ml each of LPS, IL-1β, or TNF-α, or unstimulated and incubated at 37°C in 5% CO2 for 24 hr. Supernatants were examined by ELISA, for IL-6 (a) and IL-8 (b). Mean (± SEM) from n≥3 repeats for each stimulus, with n=3 replicates per study. ** p < 0.01, * p<0.05.
Figure 5. Defective cytokine responses in IRAK-4-deficient fibroblasts.

a) & b) Primary fibroblasts from the patient and controls were exposed to 100 ng/ml each of LPS, IL-1β, or TNF-α, or unstimulated and incubated at 37°C in 5% CO2 for 24 hr. Supernatants were examined by ELISA, for IL-6 (a) and IL-8 (b). Mean (± SEM) from n≥3 repeats for each stimulus, with n=3 replicates per study. ** p < 0.01, * p<0.05. c) Semi-quantitative RT-PCR was performed to determine the expression of CD14 and TLR4 in unstimulated primary dermal fibroblasts from the patient and control. d) Immortalised dermal fibroblasts from the patient were transfected with pFLAGRK7-IRAK-4 or control empty vector, and exposed to 100 ng/ml IL-1β or TNF-α, or unstimulated and incubated at 37°C in 5% CO2 for 24 hr. Supernatants were examined by ELISA for IL-6. Mean responses (± SEM) from n=3 biological replicates in a study representative of n=2 experimental repeats.
Healthy control dermal fibroblasts (Fig. 5a & b) and PBMC (Fig. 4a & b) showed substantial IL-6 and IL-8 protein responses to IL-1β and TNF-α. In contrast, neither the patient’s fibroblasts (Fig. 5a & b) nor his PBMC (Fig. 4a & b) showed any IL-6 or IL-8 protein expression in response to IL-1β. While the patient’s fibroblasts showed a normal IL-6 and IL-8 response to TNF-α (Fig. 5a & b), his PBMC did not express any significant quantities of IL-6 and approximately 3-fold less IL-8 than PBMC from controls in response to this stimulus, significantly differing from the controls (p<0.05, p<0.01 respectively; Fig. 4a & b). These data demonstrate both cell-type-specific and stimulus-specific defects as a consequence of IRAK-4 mutation.
To confirm the causative nature of the Q293X mutation in this patient’s cellular phenotype, immortalised dermal fibroblasts from our patient were transfected with pFLAGRK7-IRAK-4. FLAG-tagged wild-type IRAK-4 restored IL-1β-induced expression of IL-6 (Fig. 5d) and IL-8 (data not shown) in keeping with previous observations (10). This provided functional confirmation of IRAK-4-deficiency and correction with wild-type IRAK-4 in our patient’s cells.
Transcriptional and post-transcriptional defects in IRAK-4 mutant fibroblasts and PBMC
In light of the defects observed in IL-6 and IL-8 production by our patient’s cells, and the differences between our patient’s fibroblasts and PBMC, we next examined the immediate expression of these cytokines at the mRNA level using semi-quantitative RT-PCR. LPS, IL-1β and TNF-α all induced the rapid expression (within 1h) of IL-6 and IL-8 mRNA by healthy control fibroblasts and PBMC (Fig. 6 and 7a). In contrast, no IL-6 or IL-8 mRNA expression was detected in the patient’s fibroblasts in response to IL-1β. However, IL-6 and IL-8 mRNA expression was detected in the patient’s fibroblasts in response to TNF-α, and also after exposure to LPS (Fig. 6). The latter observation contrasted with the absence of LPS-induced cytokine production by patient fibroblasts (Fig. 5). PBMC from the patient once again showed a different response to the fibroblasts, with all three stimuli inducing IL-8 transcription (Fig. 7a). Although the levels of IL-8 transcriptional responses to LPS and IL-1β in the patient PBMC were diminished, the responses were not significantly different in comparison to controls. The induction of IL-6 mRNA expression in patient PBMC was only observed in response to TNF-α exposure, and at a greatly reduced level (Fig. 7a).
Figure 6. Transcriptional defects in IRAK-4-deficient fibroblasts.
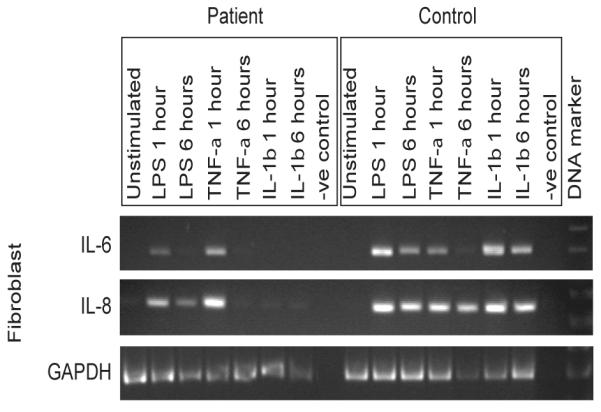
Primary dermal fibroblasts from the patient and controls were exposed to 100 ng/ml each of LPS, IL-1β, or TNF-α, or unstimulated and incubated at 37°C in 5% CO2 for 24 hr, before isolation of total RNA and semi-quantitative RT-PCR analyses, with primers designed to evaluate IL-6 and IL-8 mRNA expression, and amplification of the house-keeping gene GAPDH used as a corrective control for semi-quantitative analysis. Representative images show amplified products run on an agarose gel, with a RT-negative control, from n≥3 experimental repeats and n≥2 replicates per sample, using n=2 different normal controls.
Figure 7. Transcriptional defects in IRAK-4-deficient PBMC.

PBMC from the patient and controls were exposed to 100 ng/ml each of LPS, IL-1β, or TNF-α, or unstimulated and incubated at 37°C in 5% CO2 for 24 hr, before isolation of total RNA. a) cDNA representing mRNA was synthesised using Oligo-dT and used in semi-quantitative RT-PCR analyses, with primers designed to evaluate IL-6 and IL-8 mRNA expression. Quantitation was performed by densitometry, and corrected for GAPDH expression. Mean responses (± SEM) are shown from n≥3 replicates, and difference between patient and controls assessed for significance, ** p < 0.01, * p<0.05, b) cDNA representing total RNA, including heteronuclear RNA (hnRNA), was synthesised using random primers, and intronic PCR amplification performed using primers designed to evaluate IL-6 hnRNA, or primers designed to amplify a region of IL-6 promoter used to exclude DNA contamination (IL-6 genomic). a) and b) Representative images show amplified products run on an agarose gel, with a RT-negative control, from n≥3 experimental repeats and n≥2 replicates per sample, using n=2 different normal controls.
Intronic RT-PCR analysis revealed unspliced IL-6 hnRNA in healthy control PBMC exposed to stimuli, in marked contrast to patient PBMC (Fig 7b). This suggested that a primary failure of transcription of IL-6, rather than reduced mRNA stability, underpinned the defective IL-6 response to LPS and IL-1β, and diminished response to TNF-α. This was also the case for the defective IL-1β-induced IL-6 expression observed in patient fibroblasts (data not shown).
These data suggest that IRAK-4-deficiency has differential effects of upon cytokine expression at transcriptional and post-transcriptional levels, and indicate at least partially intact signalling responses to LPS, capable of inducing gene transcription, but IRAK-4-dependence of cytokine protein production.
NF-κB signalling in IRAK-4 mutant cells
To determine the effect of the IRAK-4-deficiency upon NF-κB signalling, phosphorylation of IκB-α in the patient’s fibroblasts (Fig 8a) and PBMC (Figure 8b) was examined after exposure to stimuli. No response to IL-1β was observed in the patient’s fibroblasts. However, a robust response to LPS was observed despite IRAK-4 mutation, correlating with the LPS-induced transcription of IL-6 and IL-8 in these cells, described above. Not unexpectedly, although slightly diminished, IκB-α responses to TNF-α were demonstrated in the patient’s fibroblasts.
Figure 8. NF-κB and MAPK signalling in IRAK-4-deficient fibroblasts and PBMC.

Primary fibroblasts (a) and PBMC (b) from the patient and control were exposed to 100 ng/ml each of LPS, IL-1β, or TNF-α, or unstimulated for the times indicated. Western immunoblot analyses were performed on whole cell lysates using antibodies specific for phosphorylated IκB-α, IκB-α, phosphorylated p38, total p38, phosphorylated JNK, and β-actin, and nuclear proteins using antibodies specific for p65. Immunoblots are representative of n≥2 experimental repeats, using n=2 different normal controls.
The patient’s PBMC showed a decrease in IκB-α and a parallel increase in phosphorylated IκB-α in response to TNF-α and LPS (albeit diminished in comparison to control cells), with a much lesser, but repeatedly observed response to IL-1β (Fig 8b). Concurrent nuclear localisation of p65 was evident, but diminished when compared to control cells in response to LPS and IL-1β (Fig 8b). These data indicate at least partially intact NF-κB activation pathways in PBMC from this patient.
Activation of MAPK pathways in Q293X mutant PBMC
In contrast to control PBMC, defective phosphorylation (activation) of p38 and JNK MAPK was observed in the patient’s cells (Fig 8b). No MAPK activation was observed in response to IL-1β, while LPS induced a delayed and diminished activation of p38. Activation of p38 in response to TNF-α was also diminished but not delayed compared to control. JNK activation was only observed at a low level in response to prolonged (60 minutes) TNF-α exposure. These data demonstrate defects in activation of key accessory pro-inflammatory signalling pathways involved in the transcriptional and translational control of IL-6 and IL-8 expression, under stimulatory conditions in which NF-κB activation was observed in patient cells.
Alternative LPS signalling pathways in Q293X mutant cells
LPS has been shown to activate MyD88-independent TLR signalling using alternative adaptor proteins. To determine whether these pathways were intact in Q293X mutant cells, LPS-induced transcription of the STAT-1 dependent gene IP-10 was examined in PBMC. No significant difference was observed between patient and control cells (Fig 9a). In addition, LPS-induced RANTES expression in our patient’s peripheral blood monocytes was also found to be comparable to that of control cells (fold change versus control = 1.2, p=0.2), using microarray gene expression-based studies (K. L. Brown et al., manuscript in preparation). Additional studies demonstrated that upregulation of co-stimulatory molecules occurred normally on the patient’s monocyte-derived DC upon activation with TLR agonists capable of utilising MyD88-independent pathways (LPS, and Poly(I:C)), with a similar magnitude of change when compared with the control cells, but with no response to CpG (Fig 9b). Interestingly, the patient’s immature DC had constitutively higher levels of expression of co-stimulatory molecules CD86 and CD80, but normal CD40, when compared with controls. Despite normal co-stimulatory molecule upregulation, the patient’s MDDC remained defective in IL-6 production, except when activated by exposure to CD40L (Fig 9c). These data indicate intact MyD88-independent pathways in patient cells, but demonstrate a necessity for functional IRAK-4 for TLR-induced MDDC IL-6 protein production.
Figure 9. Intact MyD88-independent signalling in IRAK-4-deficient cells.

a) Patient and control PBMC were exposed to 100ng/ml LPS for the times indicated. Semi-quantitative RT-PCR was performed for IP-10 expression, quantified by densitometry and corrected for expression of the housekeeping gene GAPDH. b) Immature MDDC were stimulated as above. The percentage of positive staining cells by FACS is shown (after correction for staining with a parallel isotype-specific control antibody in each sample), representing the percentage of double positivity for CD80 and CD86, or single positivity for CD40. Representative of n=3 experimental repeats, with n=3 different controls. c) Immature MDDC from the patient or control were exposed for 24 hr to either 100 ng/ml LPS, 10 μg/ml CpG2006, 10 μg/ml Poly(I:C), by co-culture with CD40L-transfected murine 3T3 fibroblasts or untreated. Supernatants were examined for IL-6 by ELISA. Mean ± SEM from n = 3 experimental repeats, with n=3 biological replicates per condition.
Discussion
Naturally-occurring genetic mutations in humans, causing rare extreme immunodeficiency phenotypes, present powerful opportunities to determine the relationship between specific defects and human disease processes in vivo and to study the underlying mechanisms involved in primary human cells.
Here we present novel findings, using primary cells (PBMC, dermal fibroblasts, MDDC) from a child with autosomal recessive homozygous Q293X IRAK-4 mutation, to demonstrate that IRAK-4-dependent mechanisms control innate immune responses at both transcriptional and post-transcriptional levels, with IRAK-4-deficiency differentially affecting NF-κB activation and cytokine production. We demonstrate that Q293X IRAK-4 mutant primary cells displayed NF-κB activation with functional MyD88-independent pathways, but defective MAPK activation. We report cell type-specific and stimulus-specific signalling dysfunction in IRAK-4-deficiency, including demonstration of IRAK-4-dependent LPS responses in primary dermal fibroblasts. We also describe the first definitive documentation of fatality from pneumococcal disease attributable to autosomal recessive homozygous Q293X IRAK-4 mutation in a sibling of our patient. Our data suggest that this mutation is likely to be an unrecognized primary immunodeficiency in apparently healthy children succumbing to lethal pyogenic bacterial infections. We offer important considerations for diagnostic approaches, and provide key insights into the mechanisms underlying innate immune dysfunction in IRAK-4-deficiency and the normal role of this protein.
Primary dermal fibroblasts from our patient displayed defective IκB-α phosphorylation and cytokine production in response to IL-1β, but intact TNF-α responses, consistent with observations from immortalised fibroblast lines from other homozygous Q293X patients (10). These data indicate that IRAK-4 is not required for TNF-α responses in fibroblasts, and suggest a simple signalling block upstream of TRAF6 inhibiting IL-1β responses. However, assessment of LPS-induced responses introduces further complexity. Conflicting published literature concerning the potential for LPS-responsiveness in fibroblasts (24) may reflect heterogeneous fibroblast subsets (25), differences between primary and immortalised cells (26), and potential LPS contaminants. Primary human dermal fibroblasts have not been extensively characterised, although reports indicate potential for LPS-responsiveness (25). We demonstrated expression of CD14 and TLR4, with IκB-α phosphorylation and the production of IL-6 and IL-8 in response to ultra-pure LPS in our adult and child control primary dermal fibroblasts. In contrast, although our patient’s primary dermal fibroblasts clearly demonstrated intact I-κBα phosphorylation (Fig 8a) and transcription of IL-6 and IL-8 (Fig. 6), these IRAK-4-deficient cells demonstrated a profound failure to produce IL-6 and IL-8 in response to LPS. These data demonstrate a functional LPS-activation pathway in both wild-type and IRAK-4-deficient dermal fibroblasts and likely confirm TLR4 signalling by well characterised MyD88-independent pathways (2), which we demonstrated are intact in our patient’s PBMC and MDDC, and which would not require functional IRAK-4 for LPS-stimulated NF-κB activation. However, critical to these studies, our patient’s primary dermal fibroblasts and MDDC demonstrated a failure of cytokine production in response to LPS, despite NF-κB signalling and cytokine transcription, and activation, respectively. These data indicate that MyD88-independent signalling is insufficient to generate the normal inflammatory response in these cells, and that even in the presence of NF-κB activation, functional IRAK-4 is required for post-transcriptional processing and cytokine production in these cells.
Intriguingly, our data demonstrate cell-type specificity when comparing the defective responses secondary to IRAK-4 Q293X mutation in primary dermal fibroblasts as compared to PBMC. The molecular basis for these observations remains unclear. However, recent studies have demonstrated key signalling differences between myeloid and non-myeloid cells, with differential use of adaptor molecules, and demonstrated MyD88-dependent LPS responses in synovial fibroblasts that may not utilise TLR4 (26,27). These observations suggest that the precise role for IRAK-4 in different cell types may vary, and introduce a complexity that may be critical to understanding the clinical consequences of IRAK-4-deficiency.
In our patient’s PBMC, neither LPS, IL-1β, or TNF-α induced production of IL-6. However, LPS induced NF-κB translocation and IL-8 transcription, but induced 10 fold less IL-8 protein expression than control cells, reiterating the likely significance of IRAK-4 dependent post-transcriptional mechanisms, as observed in the dermal fibroblasts. Nevertheless, the induction of IL-8 protein production (albeit a significantly reduced level) contrasted with the absolute deficiency demonstrated in the patient’s primary dermal fibroblasts. Our recent quantitative RT-PCR studies on purified monocytes from this patient demonstrate a more severe defect in LPS-induced IL-8 transcription in these cells (albeit retaining a low level transcriptional response), when compared to the PBMC (K. L. Brown et al., manuscript in preparation). Future studies will be required to determine the relative significance of responses of each the different cell types present in PBMC preparations, and the possible significance of interaction between these cells.
IL-1β stimulation of our patient’s PBMC was surprisingly able to repeatedly induce low level NF-κB translocation and stimulate a diminished level of IL-8 (but not IL-6) transcription, but was unable to induce IL-8 protein production. IRAK-4 has been demonstrated to be essential for IL-1R-stimulated NF-κB activation (9). Thus, these data might represent IL-1β-induced transcription of IL-8 by an undetermined, primarily NF-κB-independent mechanism in our patient’s PBMC (but not dermal fibroblasts), or partial substitution of IRAK-4 function in NF-κB signalling by alternative molecules or a truncated translation product of IRAK-4. Indeed IRAK-4 is capable of transmitting signals both dependent on and independent of its kinase activity (28), and truncated IRAK-4 protein has been shown to retain DD interactions (29). Chain-termination mutations resulting in mRNAs that contain premature stop-codons rarely produce truncated proteins, as a consequence of nonsense-mediated mRNA decay (22). However, although it could simply reflect an “overwhelming” of the nonsense-mediated mRNA decay system, a truncated IRAK-4 protein has been detected in transfected cells over-expressing IRAK-4 with the C877T mutation, (29), raising the possibility of its expression in patient cells. Additionally, translation of the alternatively spliced IRAK-4 transcript described would produce a protein unaffected by the Q293X mutation, with a disrupted kinase region, but intact DD and undetermined domain (UD). While low level expression in IRAK-4 competent cells might have no effect, this could be significant in the absence of full length IRAK-4. Thus, although truncated IRAK-4 proteins have not been detected in patient cells, we cannot exclude the possibility that low level expression could influence IL-1β signalling in IRAK-4-deficient PBMC.
Our data demonstrate that the innate immune dysfunction in IRAK-4-deficiency is both stimulus-specific, and determined differentially for specific cytokines. This is consistent with near-complete deficiency in LPS-induced TNF-α production (10), and profound transcriptional defect in GM-CSF, but partial defect in Cox-2 transcription (11) in PBMC from other IRAK-4-deficient individuals. In addition, this is consistent with defective LPS-induced IL-6 and IL-12p40 transcription, but post-transcriptional defects in IL-8, TNF-α, and IL-12p35 observed in our patient’s MDM (15). These data also indicate a critical role for IRAK-4 in both transcriptional and post-transcriptional control of cytokine production, even in the presence of intact NF-κB signalling. In this regard, the severe defects in our patient’s MAPK responses may be highly significant.
The MAPK p38 can affect transcription (30) via chromatin restructuring (5) or transactivation of p65 (8). However, the core transcriptional regulator for IL-8 has an NF-κB element required for activation in all cell types studied, with additional transcription factors required for maximal expression being largely dispensable (31,32). Thus, IL-8 transcription was expected where NF-κB activation was observed, regardless of other factors. This was clearly evident in our patient’s cells. However, translocation of NF-κB subunits in addition to p65 remains to be assessed. Critically however, activation of p38 by MKK-3, -4 and -6 also has an essential post-transcriptional function in the stabilisation of mRNAs with 3′ AU rich elements, including IL-8 and IL-6 (7,33), and p38 inhibition can have profound post-transcriptional effects without affecting transcription rates (7). In our patient’s PBMC, p38 activation was undetectable in response to IL-1β, and substantially diminished in response to LPS, correlating with the defect in production of IL-8 protein. This suggests a critical role for p38-dependent post-transcriptional mechanisms in IL-8 production, defective in our patient as a consequence of IRAK-4 mutation.
IL-6 transcription is additionally affected by the p38-activated CHOP (30) via inhibition of negative transcriptional regulators (34). Furthermore, inhibition of Jun N-terminal kinase (JNK), a MAPK activated downstream of TAK-1 by MAPK Kinase (MKK)-4, -7, inhibited IL-6 transcription through failure of an undefined interaction between JNK pathways and other signalling pathways such as NF-κB (35). Our patient’s PBMC demonstrated a complete absence of JNK activation in response to LPS and IL-1β, correlating with defective IL-6 transcription, suggesting a critical role for IRAK-4 in IL-6 transcription, through activation of JNK.
In addition to TLR/IL1-R signalling defects, significantly impaired responses to TNF-α were also observed in IRAK-4-deficient PBMC. In contrast, no significant defect was observed in the TNF-α-induced cytokine response in IRAK-4-deficient dermal fibroblasts (despite slightly diminished TNF-α-induced phosphorylation of I-κBα). These data suggest an as yet undetermined role for IRAK-4 in TNF-α signalling, and cell type-specific differences in the TNF-R signalling pathways in PBMC as compared to fibroblasts. In addition, our data revealed more substantial impairment in the production of IL-6 in comparison to IL-8, in TNF-α stimulated IRAK-4-deficient PBMC. Although TNF-α-induced IL-6 responses in PBMC can vary significantly between individuals, our patient was the only complete non-responder we observed, in contrast to ten normal controls with significant IL-6 responses (all p<0.05). Interestingly, defective TNF-α-induced activation of MAPK was demonstrated in our patient’s PBMC, with JNK activation more notably deficient than p38, correlating with more substantial impairment of IL-6 protein production in comparison to IL-8. In addition, low level transcription of IL-6 was observed in response to TNF-α, the only stimulus to which any JNK activation was observed. Although IRAK-4 is upstream of the proposed convergence of TLR/IL-1R and TNF-R signalling pathways at TRAF6 (36), a role for IRAK-1 has been demonstrated in TNF-R signalling (37) and in the absence of MyD88 function MDM (but not fibroblasts) have been shown to have impaired cytokine production in response to TNF-α, despite intact NF-κB activation (26). Our data suggest a critical role for IRAK-4 in the activation of MAPK downstream of TNF-R activation in PBMC.
We propose that dysfunctional accessory pro-inflammatory signals, rather than NF-κB activation alone, underpins the cellular phenotype in this patient. TLR/IL-1R stimulation of NF-κB via activated TAK-1 is relatively well characterised. Hyperphosphorylation of IRAK-1 by activated IRAK-4, results in Pellino-IRAK-4-IRAK-1-TRAF6 complex (Complex 1) formation and receptor release. Complex 1 interacts with membrane-bound TAK-1-TAB-1-TAB-2, with resultant cytosolic translocation of TRAF6-TAK-1-TAB-1-TAB-2, and TAK-1 activation of IKK complex (38). In addition to NF-κB pathways, TAK-1 also activates MAPK via MKK (3,39). However, despite the common requirement for TRAF6 and TAK-1, divergence of these pathways occurs at, or perhaps upstream of IRAK-1 (40), with the relative overlap of proximal signalling components unclear. Different IRAK-1 regions mediate TRAF6 and TAB-2 translocation (41), and despite inability to interact with TRAF6, the UD of IRAK-1 is sufficient (but not required) for activation of JNK, but not NF-κB (42). This suggests that IRAK-1 mediates interaction of other undefined signalling components with TRAF6 to activate JNK signalling. Thus, Complex 1 dysregulation in IRAK-4-deficient individuals could disrupt MAPK pathways independently from intact TAK-1/IKK/NF-κB signalling, as observed in our patient’s cells. Interestingly, we have observed IRAK-1 kinase function perturbation in our patient’s neutrophils, with high baseline activity diminishing after LPS exposure, in contrast to LPS-enhanced kinase activity in controls (data not shown), consistent with findings in an IRAK-4 compound heterozygote (11). In addition, our patient’s PBMC cultured in M-CSF generated multinucleated giant cells, positive for tartrate-resistant acid phosphatase, in addition to MDM (data not shown). This is suggestive of osteoclastic differentiation, a process normally requiring additional RANKL stimulation, acting via TRAF6 signalling, and also suggesting dysregulated Complex 1 function (43,44). Intriguingly, both this patient and his deceased IRAK-4 Q293X homozygote sibling were above 95th percentile for height, in contrast to their heterozygotic siblings (between 50 and 75th percentiles for age). Thus, we propose that dysfunctional Complex 1 activity in response to TLR/IL-1R activation, resulting in defective MAPK activation, underpins the innate immunodeficient phenotype in IRAK-4 deficient primary cells.
Ongoing studies utilising cells from IRAK-4-deficient individuals are expected to further elucidate the precise nature of the selective signalling defect downstream of Complex 1, and the specific nature of the susceptibility to infection in IRAK-4–deficient individuals. It is interesting to note the relative selectivity of infecting organisms in untreated IRAK-4–deficient individuals, particularly prior to diagnosis, with an apparent particular susceptibility to S. pneumoniae and to Gram positive organisms (although not exclusively)(45). Prophylactic antibiotic therapy after diagnosis clearly precludes longitudinal characterisation of the range of infections which IRAK-4–deficient individuals might have suffered from if left untreated. Nevertheless, in the absence of antibiotics our patient and others were highly susceptible to infection, and in the case of our patient’s eldest brother, this condition was fatal. This clearly indicates the importance of TLR signalling to innate immunity, but the phenotype perhaps contrasts with the acute, broad-spectrum immunodeficiency that might have been predicted for a severe TLR signalling deficiency. The specific reasons for this remain uncertain. However, our new data indicates that the major IRAK-4 mutation described (Q293X) does not result in a complete block to TLR signalling, but rather results in cell type-specific, and ligand-specific defects, differentially affecting different cytokines at transcriptional and or post-transcriptional levels, and in which defects in MAPK pathways may be more significant than the largely intact NF-κB pathway. This suggests levels of complexity that may contribute to the relative selectivity in increased susceptibility to infection in these patients. It is also interesting to note that some older IRAK-4-deficient patients have been taken off prophylactic antibiotics and remained healthy (45), suggesting the possibility that the immunodeficiency associated with IRAK-4 deficiency can be compensated for in those who survive childhood and may be relatively less critical in adult life.
In conclusion, we demonstrate that IRAK-4 mutation in primary human cells from a naturally-occurring, clinically-relevant human “model” of disease does not result in an absolute null phenotype for TLR/IL-1R signalling. Rather, it establishes dysfunctional cellular signalling with partially intact NF-κB pathways, but defective MAPK signalling and dysregulated Complex 1 function, affecting transcriptional and post-transcriptional control of TLR/IL-1R responses. Specificity of accessory pro-inflammatory pathways affected, combined with cytokine-specific mechanisms regulating expression could explain the complex cell type-specific, stimulus-dependent, and cytokine-specific defects observed and further illuminate the unusual pattern of disease susceptibility in IRAK-4–defective patients.
Acknowledgements
The authors would like to thank Wendy Robinson, Gregor Reid, Anne Junker and Simon Dobson for advice and assistance, Hilary Vallance, Renee Wang and Mary Kinloch for technical assistance, and the patient and his family for their cooperation.
Footnotes
Grant Support: DJD and RSD are funded by the Wellcome Trust (060168, 060161), DJD, AJC and JB were supported by the Canadian Cystic Fibrosis Foundation, DMEB is supported by a Canadian Institutes for Health Research (CIHR) studentship, KLB holds a fellowship from CIHR-University of British Columbia Strategic Training Program for Translational Research in Infectious Diseases, CMR holds studentships from CIHR and the Michael Smith Foundation for Health Research, REWH holds a Canada Research Chair and was supported by a Pathogenomics of Innate Immunity grant provided by Genome BC, DPS is supported by operating grants from CIHR and the Canadian Bacterial Diseases Network.
Publisher's Disclaimer: This is an author-produced version of a manuscript accepted for publication in The Journal of Immunology (The JI). The American Association of Immunologists, Inc. (AAI), publisher of The JI, holds the copyright to this manuscript. This version of the manuscript has not yet been copyedited or subjected to editorial proofreading by The JI; hence, it may differ from the final version published in The JI (online and in print). AAI (The JI) is not liable for errors or omissions in this author-produced version of the manuscript or in any version derived from it by the U.S. National Institutes of Health or any other third party. The final, citable version of record can be found at www.jimmunol.org.
References
- 1.Janeway CA, Jr., Medzhitov R. Innate immune recognition. Annu Rev Immunol. 2002;20:197–216. doi: 10.1146/annurev.immunol.20.083001.084359. [DOI] [PubMed] [Google Scholar]
- 2.Akira S, Takeda K. Toll-like receptor signalling. Nat Rev Immunol. 2004;4:499–511. doi: 10.1038/nri1391. [DOI] [PubMed] [Google Scholar]
- 3.Ninomiya-Tsuji J, Kishimoto K, Hiyama A, Inoue J, Cao Z, Matsumoto K. The kinase TAK1 can activate the NIK-I kappaB as well as the MAP kinase cascade in the IL-1 signalling pathway. Nature. 1999;398:252–256. doi: 10.1038/18465. [DOI] [PubMed] [Google Scholar]
- 4.Holtmann H, Enninga J, Kalble S, Thiefes A, Dorrie A, Broemer M, Winzen R, Wilhelm A, Ninomiya-Tsuji J, Matsumoto K, Resch K, Kracht M. The MAPK kinase kinase TAK1 plays a central role in coupling the interleukin-1 receptor to both transcriptional and RNA-targeted mechanisms of gene regulation. J Biol Chem. 2001;276:3508–3516. doi: 10.1074/jbc.M004376200. [DOI] [PubMed] [Google Scholar]
- 5.Saccani S, Pantano S, Natoli G. p38-Dependent marking of inflammatory genes for increased NF-kappa B recruitment. Nat Immunol. 2002;3:69–75. doi: 10.1038/ni748. [DOI] [PubMed] [Google Scholar]
- 6.Winzen R, Kracht M, Ritter B, Wilhelm A, Chen CY, Shyu AB, Muller M, Gaestel M, Resch K, Holtmann H. The p38 MAP kinase pathway signals for cytokine-induced mRNA stabilization via MAP kinase-activated protein kinase 2 and an AU-rich region-targeted mechanism. Embo J. 1999;18:4969–4980. doi: 10.1093/emboj/18.18.4969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Miyazawa K, Mori A, Miyata H, Akahane M, Ajisawa Y, Okudaira H. Regulation of interleukin-1beta-induced interleukin-6 gene expression in human fibroblast-like synoviocytes by p38 mitogen-activated protein kinase. J Biol Chem. 1998;273:24832–24838. doi: 10.1074/jbc.273.38.24832. [DOI] [PubMed] [Google Scholar]
- 8.Madrid LV, Mayo MW, Reuther JY, Baldwin AS., Jr. Akt stimulates the transactivation potential of the RelA/p65 Subunit of NF-kappa B through utilization of the Ikappa B kinase and activation of the mitogen-activated protein kinase p38. J Biol Chem. 2001;276:18934–18940. doi: 10.1074/jbc.M101103200. [DOI] [PubMed] [Google Scholar]
- 9.Suzuki N, Suzuki S, Duncan GS, Millar DG, Wada T, Mirtsos C, Takada H, Wakeham A, Itie A, Li S, Penninger JM, Wesche H, Ohashi PS, Mak TW, Yeh WC. Severe impairment of interleukin-1 and Toll-like receptor signalling in mice lacking IRAK-4. Nature. 2002 doi: 10.1038/nature736. [DOI] [PubMed] [Google Scholar]
- 10.Picard C, Puel A, Bonnet M, Ku CL, Bustamante J, Yang K, Soudais C, Dupuis S, Feinberg J, Fieschi C, Elbim C, Hitchcock R, Lammas D, Davies G, Al-Ghonaium A, Al-Rayes H, Al-Jumaah S, Al-Hajjar S, Al-Mohsen IZ, Frayha HH, Rucker R, Hawn TR, Aderem A, Tufenkeji H, Haraguchi S, Day NK, Good RA, Gougerot-Pocidalo MA, Ozinsky A, Casanova JL. Pyogenic bacterial infections in humans with IRAK-4 deficiency. Science. 2003;299:2076–2079. doi: 10.1126/science.1081902. [DOI] [PubMed] [Google Scholar]
- 11.Medvedev AE, Lentschat A, Kuhns DB, Blanco JC, Salkowski C, Zhang S, Arditi M, Gallin JI, Vogel SN. Distinct Mutations in IRAK-4 Confer Hyporesponsiveness to Lipopolysaccharide and Interleukin-1 in a Patient with Recurrent Bacterial Infections. J Exp Med. 2003;198:521–531. doi: 10.1084/jem.20030701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chapel H, Puel A, von Bernuth H, Picard C, Casanova JL. Shigella sonnei meningitis due to interleukin-1 receptor-associated kinase-4 deficiency: first association with a primary immune deficiency. Clin Infect Dis. 2005;40:1227–1231. doi: 10.1086/428733. [DOI] [PubMed] [Google Scholar]
- 13.Enders A, Pannicke U, Berner R, Henneke P, Radlinger K, Schwarz K, Ehl S. Two siblings with lethal pneumococcal meningitis in a family with a mutation in Interleukin-1 receptor-associated kinase 4. J Pediatr. 2004;145:698–700. doi: 10.1016/j.jpeds.2004.06.065. [DOI] [PubMed] [Google Scholar]
- 14.Yang K, Puel A, Zhang S, Eidenschenk C, Ku CL, Casrouge A, Picard C, von Bernuth H, Senechal B, Plancoulaine S, Al-Hajjar S, Al-Ghonaium A, Marodi L, Davidson D, Speert D, Roifman C, Garty BZ, Ozinsky A, Barrat FJ, Coffman RL, Miller RL, Li X, Lebon P, Rodriguez-Gallego C, Chapel H, Geissmann F, Jouanguy E, Casanova JL. Human TLR-7-, -8-, and -9- Mediated Induction of IFN-alpha/beta and -lambda Is IRAK-4 Dependent and Redundant for Protective Immunity to Viruses. Immunity. 2005;23:465–478. doi: 10.1016/j.immuni.2005.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Currie AJ, Davidson DJ, Reid GS, Bharya S, MacDonald KL, Devon RS, Speert DP. Primary immunodeficiency to pneumococcal infection due to a defect in Toll-like receptor signaling. J Pediatr. 2004;144:512–518. doi: 10.1016/j.jpeds.2003.10.034. [DOI] [PubMed] [Google Scholar]
- 16.Hirschfeld M, Ma Y, Weis JH, Vogel SN, Weis JJ. Cutting edge: repurification of lipopolysaccharide eliminates signaling through both human and murine toll-like receptor 2. J Immunol. 2000;165:618–622. doi: 10.4049/jimmunol.165.2.618. [DOI] [PubMed] [Google Scholar]
- 17.Li S, Strelow A, Fontana EJ, Wesche H. IRAK-4: A novel member of the IRAK family with the properties of an IRAK-kinase. Proc Natl Acad Sci U S A. 2002;99:5567–5572. doi: 10.1073/pnas.082100399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Davidson DJ, Currie AJ, Reid GS, Bowdish DM, MacDonald KL, Ma RC, Hancock RE, Speert DP. The cationic antimicrobial peptide LL-37 modulates dendritic cell differentiation and dendritic cell-induced T cell polarization. J Immunol. 2004;172:1146–1156. doi: 10.4049/jimmunol.172.2.1146. [DOI] [PubMed] [Google Scholar]
- 19.Bylund J, Campsall PA, Ma RC, Conway BA, Speert DP. Burkholderia cenocepacia induces neutrophil necrosis in chronic granulomatous disease. J Immunol. 2005;174:3562–3569. doi: 10.4049/jimmunol.174.6.3562. [DOI] [PubMed] [Google Scholar]
- 20.Walz G, Stevens C, Zanker B, Melton LB, Clark SC, Suthanthiran M, Strom TB. The role of interleukin-6 in mitogenic T-cell activation: Detection of interleukin-2 heteronuclear RNA by polymerase chain reaction. Cellular Immunology. 1991;134:511–519. doi: 10.1016/0008-8749(91)90322-3. [DOI] [PubMed] [Google Scholar]
- 21.Bowdish DM, Davidson DJ, Speert DP, Hancock RE. The human cationic peptide LL-37 induces activation of the extracellular signal-regulated kinase and p38 kinase pathways in primary human monocytes. J Immunol. 2004;172:3758–3765. doi: 10.4049/jimmunol.172.6.3758. [DOI] [PubMed] [Google Scholar]
- 22.Culbertson MR. RNA surveillance. Unforeseen consequences for gene expression, inherited genetic disorders and cancer. Trends Genet. 1999;15:74–80. doi: 10.1016/s0168-9525(98)01658-8. [DOI] [PubMed] [Google Scholar]
- 23.Peltola M, Chiatayat D, Peltonen L, Jalanko A. Characterization of a point mutation in aspartylglucosaminidase gene: evidence for a readthrough of a translational stop codon. Hum Mol Genet. 1994;3:2237–2242. doi: 10.1093/hmg/3.12.2237. [DOI] [PubMed] [Google Scholar]
- 24.Wang PL, Ohura K. Porphyromonas gingivalis lipopolysaccharide signaling in gingival fibroblasts-CD14 and Toll-like receptors. Crit Rev Oral Biol Med. 2002;13:132–142. doi: 10.1177/154411130201300204. [DOI] [PubMed] [Google Scholar]
- 25.Tardif F, Ross G, Rouabhia M. Gingival and dermal fibroblasts produce interleukin-1 beta converting enzyme and interleukin-1 beta but not interleukin-18 even after stimulation with lipopolysaccharide. J Cell Physiol. 2004;198:125–132. doi: 10.1002/jcp.10400. [DOI] [PubMed] [Google Scholar]
- 26.Andreakos E, Sacre SM, Smith C, Lundberg A, Kiriakidis S, Stonehouse T, Monaco C, Feldmann M, Foxwell BM. Distinct pathways of LPS-induced NF-kappa B activation and cytokine production in human myeloid and nonmyeloid cells defined by selective utilization of MyD88 and Mal/TIRAP. Blood. 2004;103:2229–2237. doi: 10.1182/blood-2003-04-1356. [DOI] [PubMed] [Google Scholar]
- 27.Sacre SM, Andreakos E, Feldmann M, Foxwell BM. Endotoxin signaling in human macrophages: signaling via an alternate mechanism. J Endotoxin Res. 2004;10:445–452. doi: 10.1179/096805104225005878. [DOI] [PubMed] [Google Scholar]
- 28.Lye E, Mirtsos C, Suzuki N, Suzuki S, Yeh WC. The role of interleukin 1 receptor-associated kinase-4 (IRAK-4) kinase activity in IRAK-4-mediated signaling. J Biol Chem. 2004;279:40653–40658. doi: 10.1074/jbc.M402666200. [DOI] [PubMed] [Google Scholar]
- 29.Medvedev AE, Thomas K, Awomoyi A, Kuhns DB, Gallin JI, Li X, Vogel SN. Cutting Edge: Expression of IL-1 Receptor-Associated Kinase-4 (IRAK-4) Proteins with Mutations Identified in a Patient with Recurrent Bacterial Infections Alters Normal IRAK-4 Interaction with Components of the IL-1 Receptor Complex. J Immunol. 2005;174:6587–6591. doi: 10.4049/jimmunol.174.11.6587. [DOI] [PubMed] [Google Scholar]
- 30.Wang XZ, Ron D. Stress-induced phosphorylation and activation of the transcription factor CHOP (GADD153) by p38 MAP Kinase. Science. 1996;272:1347–1349. doi: 10.1126/science.272.5266.1347. [DOI] [PubMed] [Google Scholar]
- 31.Mukaida N, Mahe Y, Matsushima K. Cooperative interaction of nuclear factor-kappa B- and cis-regulatory enhancer binding protein-like factor binding elements in activating the interleukin-8 gene by pro-inflammatory cytokines. J Biol Chem. 1990;265:21128–21133. [PubMed] [Google Scholar]
- 32.Hoffmann E, Dittrich-Breiholz O, Holtmann H, Kracht M. Multiple control of interleukin-8 gene expression. J Leukoc Biol. 2002;72:847–855. [PubMed] [Google Scholar]
- 33.Holtmann H, Winzen R, Holland P, Eickemeier S, Hoffmann E, Wallach D, Malinin NL, Cooper JA, Resch K, Kracht M. Induction of interleukin-8 synthesis integrates effects on transcription and mRNA degradation from at least three different cytokine- or stress-activated signal transduction pathways. Mol Cell Biol. 1999;19:6742–6753. doi: 10.1128/mcb.19.10.6742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hattori T, Ohoka N, Hayashi H, Onozaki K. C/EBP homologous protein (CHOP) up-regulates IL-6 transcription by trapping negative regulating NF-IL6 isoform. FEBS Lett. 2003;541:33–39. doi: 10.1016/s0014-5793(03)00283-7. [DOI] [PubMed] [Google Scholar]
- 35.de Haij S, Bakker AC, van der Geest RN, Haegeman G, Vanden Berghe W, Aarbiou J, Daha MR, van Kooten C. NF-kappaB mediated IL-6 production by renal epithelial cells is regulated by c-jun NH2-terminal kinase. J Am Soc Nephrol. 2005;16:1603–1611. doi: 10.1681/ASN.2004090781. [DOI] [PubMed] [Google Scholar]
- 36.Wu H, Arron JR. TRAF6, a molecular bridge spanning adaptive immunity, innate immunity and osteoimmunology. Bioessays. 2003;25:1096–1105. doi: 10.1002/bies.10352. [DOI] [PubMed] [Google Scholar]
- 37.Vig E, Green M, Liu Y, Donner DB, Mukaida N, Goebl MG, Harrington MA. Modulation of tumor necrosis factor and interleukin-1-dependent NF-kappaB activity by mPLK/IRAK. J Biol Chem. 1999;274:13077–13084. doi: 10.1074/jbc.274.19.13077. [DOI] [PubMed] [Google Scholar]
- 38.Jiang Z, Johnson HJ, Nie H, Qin J, Bird TA, Li X. Pellino 1 is required for interleukin-1 (IL-1)-mediated signaling through its interaction with the IL-1 receptor-associated kinase 4 (IRAK4)-IRAK-tumor necrosis factor receptor-associated factor 6 (TRAF6) complex. J Biol Chem. 2003;278:10952–10956. doi: 10.1074/jbc.M212112200. [DOI] [PubMed] [Google Scholar]
- 39.Wang C, Deng L, Hong M, Akkaraju GR, Inoue J, Chen ZJ. TAK1 is a ubiquitin-dependent kinase of MKK and IKK. Nature. 2001;412:346–351. doi: 10.1038/35085597. [DOI] [PubMed] [Google Scholar]
- 40.Janssens S, Beyaert R. Functional diversity and regulation of different interleukin-1 receptor-associated kinase (IRAK) family members. Mol Cell. 2003;11:293–302. doi: 10.1016/s1097-2765(03)00053-4. [DOI] [PubMed] [Google Scholar]
- 41.Qian Y, Commane M, Ninomiya-Tsuji J, Matsumoto K, Li X. IRAK-mediated translocation of TRAF6 and TAB2 in the interleukin-1-induced activation of NFkappa B. J Biol Chem. 2001;276:41661–41667. doi: 10.1074/jbc.M102262200. [DOI] [PubMed] [Google Scholar]
- 42.Li X, Commane M, Jiang Z, Stark GR. IL-1-induced NFkappa B and c-Jun N-terminal kinase (JNK) activation diverge at IL-1 receptor-associated kinase (IRAK) Proc Natl Acad Sci U S A. 2001;98:4461–4465. doi: 10.1073/pnas.071054198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mizukami J, Takaesu G, Akatsuka H, Sakurai H, Ninomiya-Tsuji J, Matsumoto K, Sakurai N. Receptor activator of NF-kappaB ligand (RANKL) activates TAK1 mitogen-activated protein kinase kinase kinase through a signaling complex containing RANK, TAB2, and TRAF6. Mol Cell Biol. 2002;22:992–1000. doi: 10.1128/MCB.22.4.992-1000.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li H, Cuartas E, Cui W, Choi Y, Crawford TD, Ke HZ, Kobayashi KS, Flavell RA, Vignery A. IL-1 receptor-associated kinase M is a central regulator of osteoclast differentiation and activation. J Exp Med. 2005;201:1169–1177. doi: 10.1084/jem.20041444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ku CL, Yang K, Bustamante J, Puel A, von Bernuth H, Santos OF, Lawrence T, Chang HH, Al-Mousa H, Picard C, Casanova JL. Inherited disorders of human Toll-like receptor signaling: immunological implications. Immunol Rev. 2005;203:10–20. doi: 10.1111/j.0105-2896.2005.00235.x. [DOI] [PubMed] [Google Scholar]