

Abstract
T cells can inhibit tumor growth, but their function in the tumor microenvironment is often suppressed. Many solid tumors exhibit abundant macrophage infiltration and low oxygen tension, yet how hypoxic conditions may affect innate immune cells and their impact on tumor progression is poorly understood. Targeted deletion of the hypoxia responsive transcription factor HIF-1α in macrophages in a progressive murine model of breast cancer resulted in reduced tumor growth, although VEGF-A and vascularization was unchanged. Tumor associated macrophages can suppress tumor infiltrating T cells by several mechanisms, and we found that hypoxia powerfully augmented macrophage-mediated T cell suppression in vitro in a manner dependent on macrophage expression of HIF-1α. Our findings link the innate immune hypoxic response to tumor progression through induction of T cell suppression in the tumor microenvironment.
Introduction
T cells can have potent effects on tumor progression (1, 2); however, it is evident that many solid tumors are resistant to immune responses and immune cell attack. Although much effort has been devoted to increasing immune responses against tumors, this is hampered by localized immunosuppression of the adaptive immune system (3) (4).
The tumor microenvironment differs from that of normal tissues in a number of respects. Tumors are frequently marked by regions of hypoxia, as rapidly dividing malignant cells outpace the capacity of the established vasculature to deliver oxygen and nutrients (5, 6). HIF-1α is a constitutively expressed bHLH transcription factor expressed in nearly all mammalian cell types, including macrophages and neutrophils (7). Tissue-specific genetic deletion of HIF-1α largely ablates the cellular transcriptional response to decreasing oxygen tension in macrophages (8, 9). Initial characterization of the role of HIF-1α in myeloid cells showed that it was essential for the capacity to mount a full immune response, suggesting a mechanism to amplify innate immune responses under low oxygen tensions – conditions typically found in wounds or infected tissues (8, 10). A number of studies have demonstrated the immunosuppressive nature of macrophages and myeloid-derived suppressor cells (MDSC) in tumor bearing hosts (11–15). Hypoxia is a hallmark of neoplastic growth; however, it is unclear how cellular hypoxic response, mediated at the transcriptional level by the Hypoxia-Inducible Factor-1α (HIF-1α), acts on the suppressive capacity of tumor-infiltrating myeloid cells.
Two L-arginine consuming enzymes have been implicated in myeloid T cell suppression: the inducible nitric oxide synthase (iNOS/NOS2, NM010927) and arginase I (ArgI, NM007482). Activation of myeloid iNOS acts to suppress T cells by production of nitric oxide, which then inhibits signal transduction (16, 17). Other groups have also documented the role of ArgI mediated L-arginine depletion in T cell suppression (13, 18). Myeloid cells are capable of a striking increase in iNOS and ArgI enzyme levels following specific signaling events, and this increase is further potentiated by low oxygen tensions found in tumors, suggesting a role for HIF-1α dependent hypoxic regulation of iNOS and ArgI in myeloid cell-mediated T cell suppression(9).
We show here that loss of HIF-1α in myeloid cells directly relieves a hypoxia-induced suppression of T cell activation. We also show that loss of HIF-1α in myeloid cells slows tumor progression, and that T cells isolated from tumors in myeloid HIF-1α null mice are more responsive to stimulation, indicating a release from immunosuppression. Our data demonstrate that there is a hypoxia-induced and HIF-1α-dependent suppression of the adaptive immune system by the innate immune system in solid tumors.
Methods
Cell culture, cell lines, and hypoxic incubation
Resident peritoneal macrophages were obtained through peritoneal lavage with 10 mls of cold PBS without Ca2+/Mg2+. Resulting cells were pelleted, red blood cells lysed with ACK buffer, and resuspended in RPMI 1640/10% FBS/1% PenStrep and plated on 15 cm Petri dishes overnight. Media was then aspirated and plates were washed with DPBS two times before addition of cold PBS +15 mM EDTA. After incubation for 10–15 minutes, adherent cells were removed by pipetteing, which removed the majority of the cells followed by light scraping to maximize yield. Bone Marrow Derived Macrophages (BMDM) were obtained by incubating the lavage of femur and tibia from rodents of the indicated genotype with RPMI 1640/20% FBS/30% L929 cell supernatant/1% PenStrep/1% Amphotericin B in two 15 cm Petri dishes. After 6 days in culture, media was aspirated and the dish washed 1× in PBS before harvesting in the same manner as resident peritoneal macrophages detailed above. For gene expression or western analysis, cells were then plated in RPMI media overnight before experimental manipulation. Hypoxic incubation was performed in a water jacketed humidified multi-gas tissue culture incubator equipped with nitrogen and carbon dioxide which flushes out oxygen until reaching a final concentration of 1% oxygen/5% carbon dioxide. Parallel normoxic incubation was carried out in standard humidified 5% carbon dioxide TC incubators. Co-culture experiments were performed in 0.4µm pore Transwell Plates (Corning Life Sciences, Lowell, MA) in either the 6-well or 24 well formats following the manufacturers instructions. PyMT MEC (Mammary Epithelial Cells) were isolated from endpoint animals by growing out adherent epithelial cells after collagenase digestion of PyMT tumors, and thus are polyclonal with a homogeneous, adherent, epithelioid appearance.
T cell macrophage co-culture proliferation assay
Resident peritoneal macrophages or bone marrow derived macrophages were isolated from HIF-1α WT or HIF DF/LysM-cre+ littermates as described above. Splenocytes were isolated from a separate WT C57Bl/6J mouse by removal of the spleen, gentle squeezing between frosted glass slides, and brief ACK hypotonic red blood cell lysis. T cells were purified using the CD4+ T cell isolation kit from Miltenyi Biotec, which uses negative selection. The resulting purified T cell suspension lacking platelets and red blood cells was labeled with CFSE (Invitrogen, Carlsbad, CA) to later quantify T cell proliferation. 1×105 CFSE labeled T cells were plated in 24 well CD3+ antibody coated (5µg/ml) plates with CD28 soluble antibody (1µg/ml) added to the media to induce T cell proliferation either with or without indicated ratios of macrophages, which were added 3 hours after T cell activation (protocol modified from (19). Cells were then incubated in normoxia or hypoxia (1% oxygen) for 60h with or without the iNOS specific inhibitor 1400W at 100µM. Analysis of cells was performed on a FACScalibur flow cytometer after labeling with fluorescent antibodies and propidium iodide to exclude dead cells.
PyMT Model of carcinogenesis in backcrossed C57BL/6J/HIF-1α+f/+f murine hosts
Backcrossed mice carrying the polyoma middle T oncogene (PyMT) under the control of the Murine Mammary Tumor Virus long terminal repeat (MMTV) (20), here referred to simply as PyMT, were obtained from the lab of Dr. Leslie Ellies (UCSD Medical Center, La Jolla, CA). Individual transgenic alleles for LysM-cre (21) and HIF-1α flox (22) were backcrossed to C57BL/6J to > 99% using Jackson labs speed backcrossing SNP genotyping service (Bar Harbor, ME). After one further cross to C57BL/6J, mice were crossed to obtain female HIF-1α+f/+f /PyMT+/−/LysM-cre+/− (experimental) and female littermate controls HIF-1α+f/+f /PyMT+/−/LysM-cre−/−. The LysozymeM-cre, efficiently excises floxed sequences in neutrophils and macrophages, but not CD11c+ dendritic cells (8, 21). We observed abundant macrophage infiltration but little to no neutrophil infiltration of tumors (data not shown). We use the terms "myeloid" or "macrophage deletion" in this work to refer to the pattern of deletion generated by the LysM-cre.
Enzyme assays
Tumors were rapidly dissected out of endpoint hosts and flash frozen in liquid N2. After Polytron homogenization of the sample on ice and pelleting of insoluble material, arginase activity from the same samples was determined by quantifying the abundance of urea after lysate incubation with an excess of L-arginine, as shown by other groups (23). Arginase activity was then normalized to total protein in the lysate as measured by the BCA assay (Thermo Fisher Scientific/Pierce, Rockford, IL).
Flow cytometry, histology, immunohistochemistry, antibodies, recombinant cytokines, and real-time PCR reagents
Single cell suspensions of tumors were generated by chopping with a razor blade followed by incubation at 300 RPM at 37 degrees for 60 minutes in 5 mls 0.22µm filtered RPMI 1640 media (no serum)/0.05–0.1% Collagenase A (Roche Diagnostics, Indianapolis, IN). RPMI 1640/10% FBS/25mM HEPES/1% Pen/Strep was used for all experiments except where T cells were cultured, where 55µM Beta-mercaptoethanol (Invitrogen) was added as is standard in murine T cell culture.
All fluorescent antibodies, recombinant mouse IFNγ, and Interleukin-4 were from eBioscience (San Diego, CA) with the exception of F4/80 (AbD Serotec, Raleigh, NC). ABC Elite HRP or AP kits from Vector Labs (Burlingame, CA) were used for IHC following the manufactures instructions. Biotin anti-CD31 and Biotin anti-PCNA were from BD Pharmingen (La Jolla, CA). Rabbit anti-arginase I was from Santa Cruz (Santa Cruz, CA). Rabbit anti-iNOS was from Calbiochem/EMD Biosciences (San Diego, CA). Colorimetric TUNEL staining was performed using the Promega DEADend kit (Madison, WI).
Gene expression was carried out on an ABI 7700 normalized to levels of 18S rRNA. Target and reference real time reactions were run in duplicate and the average used to calculate ΔΔCT. Error bars indicate the SEM of multiple samples.
Measurement of IFNγ Production by Tumor Infiltrating Lymphocytes
Single cell suspensions of PyMT tumors from myeloid HIF WT or KO tumors were added to anti-CD3 antibody coated plates. BD Biosciences Golgistop and CD28 antibody were added to the media and 8 hours later, cells were stained for CD4/CD8 on the surface and then intracellularly with anti-IFNγ PE using the Cytofix/Cytoperm kit from BD Biosciences (La Jolla, CA) before flow cytometry as described above.
Results
Creation of myeloid HIF-1α null mammary tumor transgenics
To gain insight into the role of the myeloid hypoxic response in the tumor microenvironment, we employed a well-characterized model that allows the study of tumor progression - the murine mammary tumor virus long terminal repeat promoter driving the polyoma middle T oncogene specifically in mammary tissue - the MMTV-PyMT model of mammary carcinogenesis (20, 24). As part of our initial characterization of HIF-1α+f/+f/PyMT+/−/LysM-cre+/− mice, we determined that mammary gland development was normal in mice lacking HIF-1α. There were no differences in epithelial ductal tree formation detected between the genotypes; both displayed branching structures that extended through the entire fat pad - evidence of normal mammary gland development (data not shown). After tumor growth, initial immunohistochemical staining of myeloid WT mice revealed a prominent tumor macrophage F4/80+ infiltrate, consistent with reports by others (25) (data not shown).
We found that there is efficient deletion of HIF-1α in myeloid cells found in tumor bearing mice, including Gr1+CD11b+ myeloid derived suppressor cells (MDSC) (Supplemental Figure 1) (8, 21). We also detected tumor infiltration with CD8+ T cells (data not shown). The PyMT model exhibits several important characteristics consistent with clinically relevant tumor scenarios, including stage-wise histological progression, marked macrophage infiltration, and a spontaneous endogenous T cell response which is eventually suppressed during tumor growth (24, 26). To investigate the role of myeloid HIF-1α in this relevant setting, we designed a breeding strategy that would yield backcrossed, myeloid cell-specific knockouts of HIF-1α in the PyMT model of mammary carcinogenesis.
Myeloid deletion of HIF-1α results in reduced tumor mass, and retarded tumor progression
One important control for early tumor promoting events as well as for the efficacy of inbred strain backcrossing, is latency, or time to the first detectable tumor growth; we found no difference in latency between genotypes (Figure 1A). In this model, 2–3 months pass from detection of first tumor growth by palpation, to experimental endpoint; during this time tumors undergo growth and stage-wise histological progression. Myeloid deletion of HIF-1α resulted in a marked decrease in endpoint tumor mass (Figure 1B), despite similar levels of tumor infiltration with F4/80+ macrophages (Figure 1C). Remarkably, histological analysis revealed striking differences between the tumors, with HIF-1α myeloid null tumors having a more nodular and cell dense appearance. Analysis by immunohistochemistry for PCNA, a marker for cell proliferation, showed extensive dividing cell populations in the advanced histological stage/dense cell masses, which were more abundant in the wild type control animals (data not shown). The PyMT model has been characterized in regard to human equivalents of tumor stages/progression (24). We used this stage classification technique to grade tumors into three levels of histological progression by quantifying the area occupied by each stage from midline cross sections of the transformed glands. The progression follows from what we refer to as a "precancerous stage" - hyperplasia and adenoma/MIN premalignant lesions, which still retain some normal ductal and acinar mammary gland morphology, to a more epithelial cell dense early carcinoma with some stromal invasion, and finally an invasive, high mitotic index, late stage carcinoma. As reported by careful work from Lin and Pollard et.al., similar human equivalents of these stages were are florid ductal hyperplasia, ductal carcinoma in situ with early stromal invasion, and poorly differentiated invasive ductal carcinoma, respectively. In our study, this histological stage analysis revealed that tumors from wild type mice were more advanced, with increased amounts of late carcinoma tumor mass. Mice lacking myeloid cell HIF-1α had significant increases in precancerous stages relative to wild type animals (Figure 1D, E) indicating that there is a delay in histological progression of tumors in the mutant mice.
Figure 1. Deletion of HIF-1α in macrophages results in lower endpoint tumor mass despite equivalent latency and macrophage infiltration.

(A) latency, or time to first tumor (B) mass of all tumors at 20-week endpoint, n=8 (C) % of F4/80+ macrophages in single cell suspensions generated from tumors, n=4 (D) macroscopic hematoxylyn and eosin staining of typical tumors (E) histological stage analysis of tumors. All error bars are S.E.M.
Loss of HIF-1α does not affect VEGF-A levels or vascularization of mammary tumors
HIF-1α regulates the angiogenic factor VEGF-A (22, 26, 27). Although VEGF-A is expressed by tumor-associated macrophages (28), whole tumor lysate VEGF-A levels were nearly identical in wild type and myeloid HIF-1α null tumors (Figure 2A). CD31+ staining of tumor sections to determine stage-dependent vascular density revealed no significant changes in a stage-specific analysis (Figure 2B); thus, loss of HIF-1α does not result in an altered level of tumor vascularization. We have previously shown that mice lacking myeloid cell VEGF-A expression have increased tumor mass and progression(28); this is a further argument against VEGF-A expression as a determinant of the phenotype described here.
Figure 2. Myeloid HIF-1α null tumors have similar VEGF-A mRNA levels and similar stage-specific microvascular densities, but different levels of cell death.

(A) VEGF-A mRNA from whole tumor lysates relative to 18S rRNA, n=8 (B) stage specific quantitation of CD31 staining of random fields, n=8 (C) stage specific quantitation of TUNEL positivity of random fields, n=8. All error bars are S.E.M.
Increased tumor apoptosis following loss of myeloid HIF-1α
Staining for cell death and apoptosis by TUNEL revealed that there was a highly significant increase in cell death in the myeloid HIF-1α null at early and middle stages of tumor progression (Figure 2C). This indicated that loss of HIF-1α in the myeloid lineage was causing an increase in tumor cell death; this would act to slow the progression of the tumors, and reduce their mass. Because angiogenesis did not appear to be affecting tumor growth, and T cell cytotoxicity can result in TUNEL positivity, we set out to test how T cell function was affected by macrophages and hypoxia.
Macrophages inhibit T cell proliferation under hypoxia
T cell activation and proliferation are essential steps in the adaptive immune response, and increase the clonal frequency of antigen specific T cells, as well as inducing differentiation into effector and memory cells. Subsequent activation of effector or memory cells results in cytokine release and antigen-specific cytotoxicity. Given the infiltration of the experimental mammary tumors with macrophages and T cells, we tested the capacity of T cells co-cultured with macrophages to proliferate under normoxia and hypoxia. In this assay, T cell division can be detected by flow cytometry. Notably, only the highest ratios of macrophages inhibited T cell proliferation under normoxia (Figure 3A–C). Careful titration of macrophage:T cell ratios, however, revealed reduced oxygen levels augmented T cell inhibition of macrophages. T cells are able to proliferate under hypoxia (1% oxygen) after CD3/28 stimulation (Figure 3B, 1:80 condition). However, when macrophages were 5% of the total cell number, cell cycle progression was markedly blocked (Figure 3A–C, 1:20 conditions), whereas normoxic proliferation at this ratio was largely unaffected. Figure 3C quantitates this hypoxic potentiation of suppressive capacity over a range of macrophage:T cell ratios, and demonstrates that lowering oxygen tension increases macrophage inhibition of T cell proliferation and viability.
Figure 3. Macrophage suppression of T cell proliferation is augmented under hypoxia.

(A and B) representative quantitation of T cell proliferation as measured by dilution of CFSE. Purified T cells were loaded with CFSE and activated in vitro by CD3/28 and co-cultured with the indicated ratio of bone-marrow derived macrophages for 60h under (A) normoxia or (B) 1% oxygen (hypoxia) (C) an independent replicate experiment graphically depicting hypoxia potentiated macrophage suppression of T cell proliferation (n=2). The increased hypoxia/macrophage dependent suppression was observed in 8 independent experiments with various macrophage populations including resident peritoneal (3 times), thioglycollate-elicted (2 times), and bone-marrow derived macrophages (3 times). All error bars are S.E.M.
Macrophage T suppression in hypoxia is HIF-1α/iNOS dependent
To test the role of HIF-1α in macrophage-mediated hypoxic T cell suppression, we carried out co-cultures with macrophages derived from HIF-1α+f/+f/LysM-cre+/− (macrophage HIF KO) bone marrow (Figure 4). In Figure 4A, we show that under normoxia, no change in suppressive capacity between the genotypes exists over a wide range of macrophage:T cell ratios (green and black lines). In hypoxia, however, HIF-1α null macrophages were poor inhibitors of T cell proliferation at 1:20 and higher ratios, ratios where wild type macrophages induced a potent cell cycle arrest (Figure 4A, red and blue lines, CFSE traces Figure 4B). Using propidium iodide exclusion to count viable cells collected during a fixed time and flow rate, we show in Figure 4C that T cells incubated with wild type macrophages at 1:20 have only 30% of the viability of those incubated with an equal number of HIF-1α null macrophages. These results show that macrophage-mediated suppression of T cell proliferation in hypoxia is HIF-1α dependent.
Figure 4. HIF-1α null macrophages fail to augment T cell suppression under hypoxia, and WT suppressive effect is dependent on HIF-1α upregulation of macrophage iNOS and can be blocked by neutralizing classical proinflammatory cytokines or specific iNOS inhibition.

(A) CFSE dilution after T cell activation 60h in normoxia or 1% oxygen with the indicated number of macrophages added to the cultures. (B) representative overlays of CFSE dilution in T cells after activation and co-culture at 1:20 macrophage:T cell as in A; samples are from an independent experiment yet can be identified by the same color scheme as in A. Control unstimulated CFSE loaded T cells appear in grey (C) viable T cells recovered in hypoxic conditions as a % of the amount recovered in normoxic conditions at 1:20 macrophage:T cells; n=2, all results representative of 3 independent experiments. (D) representative CFSE dilution of purified T cells after activation and co-culture for 60h with WT bone-marrow derived macrophages with the cytokine neutralizing antibodies (α to indicate antibody) or the iNOS inhibitor 1400W added as indicated. Similar results were observed in 3 independent experiments.
Activated T cells rapidly produce Th1 and Th2 cytokines, e.g., IFNγ and IL-4, following stimulation. Macrophages have a specific and inverse transcriptional response to these cytokines, increasing iNOS and downregulating ArgI expression after IFNγ stimulation, for example, while increasing ArgI and downregulating iNOS expression after IL-4 stimulation (29); both of these occur in a HIF-dependent fashion when macrophages are hypoxic (9). Although both iNOS and ArgI have been extensively reported to suppress T cell function (30), iNOS can act acutely to block T cell proliferation by nitric oxide and subsequent peroxynitrite formation, whereas ArgI creates immunosuppressive microenvironments by depleting local L-arginine first - an indirect effect unlikely to affect T cell proliferation in an acute in vitro assay (31). Because activated T cells make a mixture of Th1 and Th2 cytokines, neutralizing antibodies to IFNγ and IL-12 can generate a Th2-type cytokine profile (by leaving IL-4 as the dominant T cell derived cytokine), whereas neutralizing antibodies to IL-4 generate a Th1 profile.
To first test the role of the HIF target and Th1-induced gene iNOS in suppression, we blocked its activity with the chemical compound 1400W, an iNOS specific inhibitor (32). These experimental conditions are reported in Figure 4D; as can be seen, wild type macrophages at a 1:10 ratio with T cells arrest T cell proliferation under hypoxia (blue trace), leaving them at proliferation rates comparable to unstimulated cells (purple trace). The addition of neutralizing antibodies to IL-4 fails to arrest T cell proliferation. However, addition of neutralizing antibodies to IFNγ and IL-12 releases the T cells from the macrophage/hypoxia potentiated cell cycle progression arrest. Considering the data discussed above, a suppressive effect that depends on HIF-1α, hypoxia, and IFNγ, yet can be rescued with IL-4, would suggest an iNOS-dependent suppression. Indeed, treatment of a 1:10 macrophage:T cell culture with 1400W fully restores the capacity for robust T cell proliferation under hypoxia (Figure 4D, yellow trace, +1400W). Macrophages derived from iNOS−/− mice were similarly unable to inhibit T cell proliferation in this acute assay (data not shown). It should be noted that T cells and other immune cells engaged in cytotoxic action against tumors typically produce IFNγ.
PyMT tumor infiltrating macrophages have a mixed M1/M2 polarization signature independent of HIF-1α
We next wished to determine if HIF-1α had any affect upon polarization of macrophages, and to characterize the polarization state of tumor-infiltrating macrophages in the MMTV-driven mammary tumors, i.e., determine whether they were classically or alternatively activated (33, 34). To accomplish this, bone marrow-derived macrophages were treated with classical M1 (IFNγ) or alternatively activating M2 (IL-4) cytokines and cell surface expression of markers reported to be upregulated by a M1/classical polarization state were measured, MHC II and CD86. No difference was found in the capacity of HIF-1α wild type or nullizygous macrophages to polarize under IFNγ, IL-4, and IL-10 stimulated conditions under hypoxia, as determined by multiple cell surface markers correlated with activation state. However, macrophages infiltrating tumors displayed a mixed activation phenotype - displaying properties of both classically activated and alternatively activated macrophages (Supplemental Figure 2).
Macrophage HIF-1α dependent induction of ArgI after co-culture with PyMT MECs under hypoxia
To determine whether cancer cells alone combined with hypoxia might induce the iNOS or ArgI genes in macrophages in close proximity, we carried out co-culture studies using Transwell tissue culture plates. Bone marrow derived macrophages were added to the bottom of a Transwell dish and a polyoma middle T mammary epithelial cell (MEC) line derived from a wild type tumor (data not shown) was plated in the upper well. In this culture design, both cells share the same media via a porous membrane (Figure 5A, left). The pores allow diffusion of soluble factors, but not of cells. We tested HIF-1α null macrophages in normoxia or hypoxia in the presence or absence MEC for 24 hours. Subsequent Western blotting revealed that ArgI is markedly induced in macrophages by co-culture with MEC (Figure 5A, right). However, ArgI was abundant only under hypoxia, and was dependent on HIF-1α (Figure 5A, right). In contrast, iNOS showed a modest induction only in hypoxia without MEC at the limit of detection. This demonstrates that under hypoxia, tumor cells are capable of releasing soluble factor(s) that potently activate the ArgI-dependent arm of the immunosuppressive repertoire of macrophages.
Figure 5. HIF-1α dependent ArgI induction by tumor cells and hypoxia is associated with tumor T cell suppression.

(A) in vitro co-culture of macrophages with or without a PyMT mammary epithelial cell line in normoxia or hypoxia for 24h, left, experimental design; right, protein analysis/detection by western blot (B) arginase enzyme activity quantitation of myeloid HIF-1α WT PyMT tumor lysates, n=9 (C) Single cell suspensions of spleens or PyMT tumors WT or KO for myeloid HIF-1α were stimulated in vitro with CD3/28 and the % of T cells making IFNγ after 8h is reported, n=12. All error bars are S.E.M
Macrophage HIF-1α acts to suppress cytotoxic T cell responsiveness within tumors
HIF-1α-regulated enzymes ArgI and iNOS can induce immunosuppression in T cells (18, 35–37). We performed enzyme activity assays on whole PyMT tumor lysates from myeloid HIF-1α wild type or nullizygous mice. ArgI activity was significantly higher in lysates from wild type mice, as expected (Figure 5B). iNOS activity in wild type tumors was higher and with a larger variance, but the difference was not statistically significant (data not shown). These data appear to mirror the PyMT/macrophage co-culture (Figure 5A), which was more effective at inducing ArgI than iNOS. In our analysis of single cell suspensions from tumors, we found no significant difference in the number of infiltrating CD8+ T cells (data not shown). Given the high amount of immunosuppressive ArgI activity in wild type tumors, it was important to test the responsiveness of T cells taken directly from wild type and myeloid HIF-1α null host tumors.
One key measurement of T cell responsiveness is the ability to produce interferon-gamma (IFNγ) immediately following stimulation. IFNγ production after activation in vitro has been reported to correlate well with T cell anti-tumor cytotoxic potential, and is a key measure of T cell effector capacity (38–40). To test tumor T cell responsiveness from myeloid HIF-1α wild type and null tumors, the fraction of T cells that make IFNγ after a strong, pan-T cell stimulation with anti-CD3/28 antibody of tumor single cell suspensions was carried out. There was a marked and highly significant increase in the fraction of T cells producing IFNγ from myeloid HIF-1α null tumors when compared to T cells from wild type tumors (Figure 5C). This demonstrates that HIF-1α expression in myeloid cells is correlated with suppression of tumor cytotoxic T cells.
Discussion
Immunosuppression of infiltrating T cells is a significant roadblock to tumor immunotherapy. There are numerous mechanisms of immunusuppression, including localized enzymatic activity of iNOS and ArgI (12, 36, 41). Here we show that HIF-1α control of these immunosuppressive enzymes in myeloid cells has profound effects on T cell proliferative capacity and responsiveness to stimulation. There is evidence of a spontaneous CD8+ T cell response in the MMTV-PyMT mammary tumor model which acts to slow tumor growth (26), and this correlates with our finding of reduced tumor progression and mass following deletion of myeloid HIF-1α - however, our data do not exclude mechanisms other than T cell immunosuppression as contributing to the endpoint tumor phenotype.
In resting HIF-1α myeloid null animals, overall T cell development and homeostasis appears normal (Supplemental Figure 3), despite the fact that, in addition to iNOS and ArgI, a remarkable number of other reported immunosuppressive factors are upregulated by HIF-1α (Supplemental Figure 4). This suggests myeloid HIF-1α may regulate T cells via multiple immunosuppressive pathways, and primarily in response to infection, injury, or other pathophysiological states where myeloid cells are recruited and activated.
The observation of similar tumor vascular density in the absence of myeloid HIF-1α in the PyMT model highlights the complexity of HIF function in tumors. In studies where VEGF-A has been deleted from tumor cells, more dramatic effects were seen in tumor vascularity (42). Indeed, we found that loss of VEGF-A in myeloid cells acted to increase tumor size and progression, the opposite of our findings following loss of HIF-1α in myeloid cells (28). As a result of the inverse phenotypes in these two models, loss of HIF-1α mediated VEGF-A induction may have in fact diminished the magnitude of the phenotype described here. However, our data do not reveal the major changes in tumor vessel density observed in the myeloid VEGF-A knockout.
Co-culture of PyMT mammary epithelial cells resulted in a robust induction of ArgI protein in macrophages under hypoxia. In this assay, we did not detect iNOS induction at the protein level. Apart from bacterial and viral components, IFNγ from T cells is one of the most potent inducers of iNOS in murine macrophages. IFNγ is largely produced by T cells when activated or engaged in a cytotoxic response, however there were no T cells present in the MEC co-culture model we describe here. Therefore, the results are consistent with a model where ArgI is induced by soluble factors produced by the tumor cells, whereas iNOS is likely induced transiently by other factors in the tumor, including T cells producing IFNγ. Studies from other groups have shown that iNOS is poorly expressed by mammary tumor macrophages (43, 44) - this data combined with the poor induction of iNOS by PyMT MEC suggests that ArgI may be the dominant immunosuppressive enzyme in vivo, while iNOS may play a role in other scenarios of macrophage-mediated T cell suppression or in more localized macrophage:T cell interactions.
Both iNOS and ArgI enzymes consume the same amino acid, L-arginine; recent reports suggest that reduced activity of both enzymes to may be required to relieve T cell suppression. Mice harboring mutations of the cationic amino acid transporter-2 (CAT2), which is essential for myeloid cell production of significant amounts of nitric oxide via iNOS, or for significant throughput through the ArgI pathway (45, 46), develop spontaneous lung inflammation at 3 weeks of age (47). One group has reported that global iNOS deficiency results in increased T cell activity (48); also of note are reports that wild type mice show an IFNγ-dependent resistance to autoimmunity, whereas iNOS null mice remain susceptible (49, 50). Other authors have drawn attention to the fact that L-arginine metabolism in general results in T cell suppression, again underscoring the idea that both enzymes contribute to T cell suppression (35, 41).
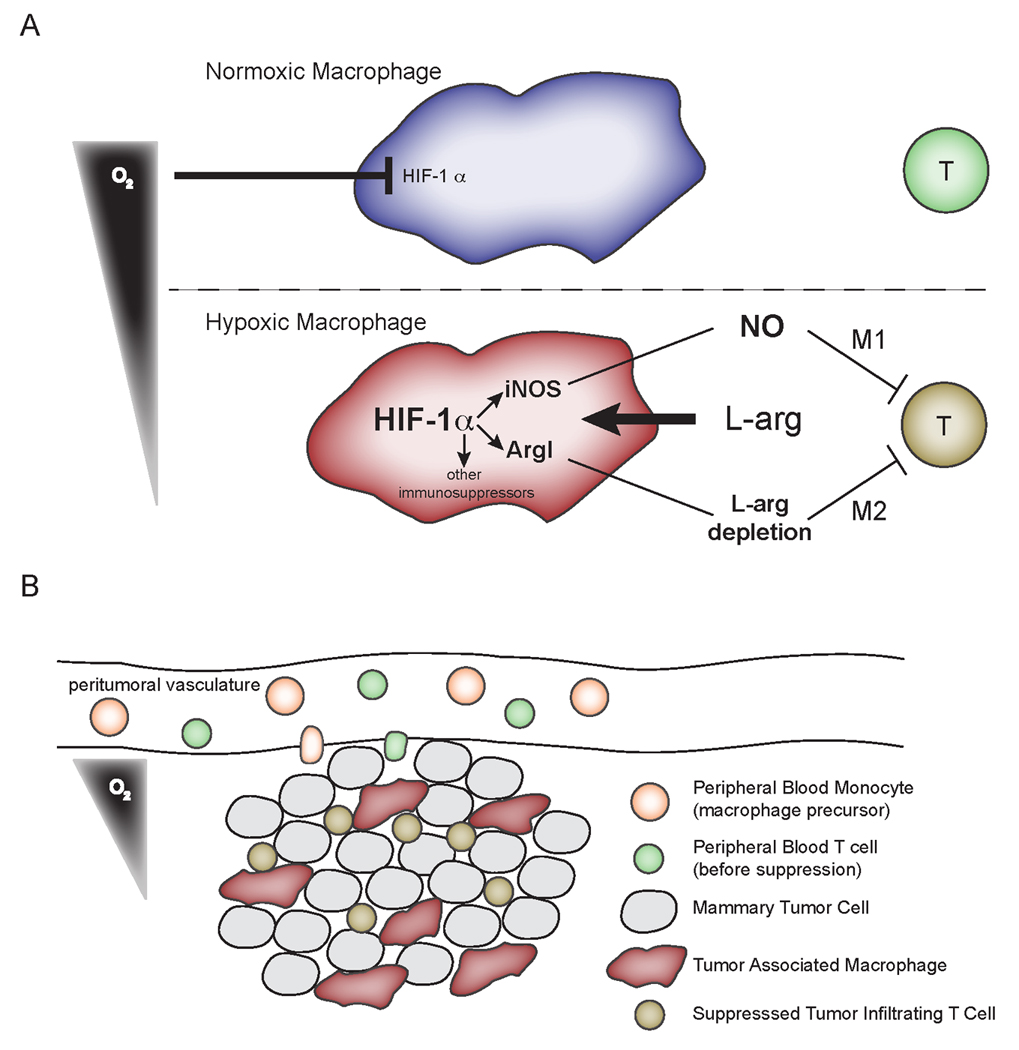
The data presented here demonstrates that myeloid HIF-1α control of ArgI and iNOS acts to induce T cell suppression in the hypoxic microenvironment (modeled in Figure 6). Low oxygen tensions are a hallmark of the tumor microenvironment, as is infiltration with macrophages. The tumor-dependent increase of T cell suppressive pathways under control of HIF-1α and hypoxia demonstrates that tumor microenvironmental conditions and infiltrating cells synergize to subvert adaptive immune responses. Moreover, the localized, HIF-1α-dependent, and hypoxia-potentiated suppression of the adaptive immune system by myeloid cells described here has important implications for the treatment of cancer with HIF inhibitors. Most research indicates cancer cell HIF-1α acts in a protumor fashion, promoting survival and angiogenesis. Our data identifies a HIF-1α-dependent, hypoxia-potentiated immunosuppressive circuit active in the tumor microenvironment. These data suggest efforts to develop local HIF-1α inhibition as a cancer treatment modality, alone or in combination with immunotherapy, would address both the pro-survival role of cancer cell HIF-1α while relieving the immunosuppressive action of myeloid cell HIF-1α.
Figure 6. Cartoon depicting a model for myeloid, HIF-dependent suppression of T cell function in hypoxic regions of tumors.
Acknowledgements
We acknowledge the expert assistance and advice of Dennis J. Young of the UCSD Cancer Center Flow Cytometry Core. This work was supported in part by a Susan G. Komen pre-doctoral fellowship granted to A.L.D.
Footnotes
The authors declare no conflicts of interest
References
- 1.Koebel CM, Vermi W, Swann JB, et al. Adaptive immunity maintains occult cancer in an equilibrium state. Nature. 2007;450:903–907. doi: 10.1038/nature06309. [DOI] [PubMed] [Google Scholar]
- 2.Zitvogel L, Apetoh L, Ghiringhelli F, Andre F, Tesniere A, Kroemer G. The anticancer immune response: indispensable for therapeutic success? J Clin Invest. 2008;118:1991–2001. doi: 10.1172/JCI35180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Leen AM, Rooney CM, Foster AE. Improving T cell therapy for cancer. Annu Rev Immunol. 2007;25:243–265. doi: 10.1146/annurev.immunol.25.022106.141527. [DOI] [PubMed] [Google Scholar]
- 4.Talmadge JE. Pathways mediating the expansion and immunosuppressive activity of myeloid-derived suppressor cells and their relevance to cancer therapy. Clin Cancer Res. 2007;13:5243–5248. doi: 10.1158/1078-0432.CCR-07-0182. [DOI] [PubMed] [Google Scholar]
- 5.Vaupel P, Mayer A. Hypoxia in cancer: significance and impact on clinical outcome. Cancer Metastasis Rev. 2007;26:225–239. doi: 10.1007/s10555-007-9055-1. [DOI] [PubMed] [Google Scholar]
- 6.Vaupel P, Hockel M, Mayer A. Detection and characterization of tumor hypoxia using pO2 histography. Antioxid Redox Signal. 2007;9:1221–1235. doi: 10.1089/ars.2007.1628. [DOI] [PubMed] [Google Scholar]
- 7.Wiener CM, Booth G, Semenza GL. In vivo expression of mRNAs encoding hypoxia-inducible factor 1. Biochem Biophys Res Commun. 1996;225:485–488. doi: 10.1006/bbrc.1996.1199. [DOI] [PubMed] [Google Scholar]
- 8.Cramer T, Yamanishi Y, Clausen BE, et al. HIF-1alpha is essential for myeloid cell-mediated inflammation. Cell. 2003;112:645–657. doi: 10.1016/s0092-8674(03)00154-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Takeda N, O'Dea EL, Doedens A, et al. Differential activation and antagonistic function of HIF-{alpha} isoforms in macrophages are essential for NO homeostasis. Genes Dev. 24:491–501. doi: 10.1101/gad.1881410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Peyssonnaux C, Datta V, Cramer T, et al. HIF-1alpha expression regulates the bactericidal capacity of phagocytes. J Clin Invest. 2005;115:1806–1815. doi: 10.1172/JCI23865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bronte V, Serafini P, Apolloni E, Zanovello P. Tumor-induced immune dysfunctions caused by myeloid suppressor cells. J Immunother (1997) 2001;24:431–446. doi: 10.1097/00002371-200111000-00001. [DOI] [PubMed] [Google Scholar]
- 12.Mazzoni A, Bronte V, Visintin A, et al. Myeloid suppressor lines inhibit T cell responses by an NO-dependent mechanism. J Immunol. 2002;168:689–695. doi: 10.4049/jimmunol.168.2.689. [DOI] [PubMed] [Google Scholar]
- 13.Rodriguez PC, Quiceno DG, Zabaleta J, et al. Arginase I production in the tumor microenvironment by mature myeloid cells inhibits T-cell receptor expression and antigen-specific T-cell responses. Cancer Res. 2004;64:5839–5849. doi: 10.1158/0008-5472.CAN-04-0465. [DOI] [PubMed] [Google Scholar]
- 14.Huang B, Pan PY, Li Q, et al. Gr-1+CD115+ immature myeloid suppressor cells mediate the development of tumor-induced T regulatory cells and T-cell anergy in tumor-bearing host. Cancer Res. 2006;66:1123–1131. doi: 10.1158/0008-5472.CAN-05-1299. [DOI] [PubMed] [Google Scholar]
- 15.Liu Y, Van Ginderachter JA, Brys L, De Baetselier P, Raes G, Geldhof AB. Nitric oxide-independent CTL suppression during tumor progression: association with arginase-producing (M2) myeloid cells. J Immunol. 2003;170:5064–5074. doi: 10.4049/jimmunol.170.10.5064. [DOI] [PubMed] [Google Scholar]
- 16.Albina JE, Henry WL., Jr Suppression of lymphocyte proliferation through the nitric oxide synthesizing pathway. J Surg Res. 1991;50:403–409. doi: 10.1016/0022-4804(91)90210-d. [DOI] [PubMed] [Google Scholar]
- 17.Bingisser RM, Tilbrook PA, Holt PG, Kees UR. Macrophage-derived nitric oxide regulates T cell activation via reversible disruption of the Jak3/STAT5 signaling pathway. J Immunol. 1998;160:5729–5734. [PubMed] [Google Scholar]
- 18.Kropf P, Baud D, Marshall SE, et al. Arginase activity mediates reversible T cell hyporesponsiveness in human pregnancy. Eur J Immunol. 2007;37:935–945. doi: 10.1002/eji.200636542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Atochina O, Daly-Engel T, Piskorska D, McGuire E, Harn DA. A schistosome-expressed immunomodulatory glycoconjugate expands peritoneal Gr1(+) macrophages that suppress naive CD4(+) T cell proliferation via an IFN-gamma and nitric oxide-dependent mechanism. J Immunol. 2001;167:4293–4302. doi: 10.4049/jimmunol.167.8.4293. [DOI] [PubMed] [Google Scholar]
- 20.Guy CT, Cardiff RD, Muller WJ. Induction of mammary tumors by expression of polyomavirus middle T oncogene: a transgenic mouse model for metastatic disease. Mol Cell Biol. 1992;12:954–961. doi: 10.1128/mcb.12.3.954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Clausen BE, Burkhardt C, Reith W, Renkawitz R, Forster I. Conditional gene targeting in macrophages and granulocytes using LysMcre mice. Transgenic Res. 1999;8:265–277. doi: 10.1023/a:1008942828960. [DOI] [PubMed] [Google Scholar]
- 22.Ryan HE, Poloni M, McNulty W, et al. Hypoxia-inducible factor-1alpha is a positive factor in solid tumor growth. Cancer Res. 2000;60:4010–4015. [PubMed] [Google Scholar]
- 23.Corraliza IM, Campo ML, Soler G, Modolell M. Determination of arginase activity in macrophages: a micromethod. J Immunol Methods. 1994;174:231–235. doi: 10.1016/0022-1759(94)90027-2. [DOI] [PubMed] [Google Scholar]
- 24.Lin EY, Jones JG, Li P, et al. Progression to malignancy in the polyoma middle T oncoprotein mouse breast cancer model provides a reliable model for human diseases. Am J Pathol. 2003;163:2113–2126. doi: 10.1016/S0002-9440(10)63568-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lin EY, Nguyen AV, Russell RG, Pollard JW. Colony-stimulating factor 1 promotes progression of mammary tumors to malignancy. J Exp Med. 2001;193:727–740. doi: 10.1084/jem.193.6.727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stewart TJ, Abrams SI. Altered immune function during long-term host-tumor interactions can be modulated to retard autochthonous neoplastic growth. J Immunol. 2007;179:2851–2859. doi: 10.4049/jimmunol.179.5.2851. [DOI] [PubMed] [Google Scholar]
- 27.Forsythe JA, Jiang BH, Iyer NV, et al. Activation of vascular endothelial growth factor gene transcription by hypoxia-inducible factor 1. Mol Cell Biol. 1996;16:4604–4613. doi: 10.1128/mcb.16.9.4604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stockmann C, Doedens A, Weidemann A, et al. Deletion of vascular endothelial growth factor in myeloid cells accelerates tumorigenesis. Nature. 2008 doi: 10.1038/nature07445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Modolell M, Corraliza IM, Link F, Soler G, Eichmann K. Reciprocal regulation of the nitric oxide synthase/arginase balance in mouse bone marrow-derived macrophages by TH1 and TH2 cytokines. Eur J Immunol. 1995;25:1101–1104. doi: 10.1002/eji.1830250436. [DOI] [PubMed] [Google Scholar]
- 30.Bronte V, Serafini P, Mazzoni A, Segal DM, Zanovello P. L-arginine metabolism in myeloid cells controls T-lymphocyte functions. Trends Immunol. 2003;24:302–306. doi: 10.1016/s1471-4906(03)00132-7. [DOI] [PubMed] [Google Scholar]
- 31.Rodriguez PC, Quiceno DG, Ochoa AC. L-arginine availability regulates T-lymphocyte cell-cycle progression. Blood. 2007;109:1568–1573. doi: 10.1182/blood-2006-06-031856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Garvey EP, Oplinger JA, Furfine ES, et al. 1400W is a slow, tight binding, and highly selective inhibitor of inducible nitric-oxide synthase in vitro and in vivo. J Biol Chem. 1997;272:4959–4963. doi: 10.1074/jbc.272.8.4959. [DOI] [PubMed] [Google Scholar]
- 33.Gallina G, Dolcetti L, Serafini P, et al. Tumors induce a subset of inflammatory monocytes with immunosuppressive activity on CD8+ T cells. J Clin Invest. 2006;116:2777–2790. doi: 10.1172/JCI28828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mantovani A, Sozzani S, Locati M, Allavena P, Sica A. Macrophage polarization: tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. 2002;23:549–555. doi: 10.1016/s1471-4906(02)02302-5. [DOI] [PubMed] [Google Scholar]
- 35.Bronte V, Serafini P, De Santo C, et al. IL-4-induced arginase 1 suppresses alloreactive T cells in tumor-bearing mice. J Immunol. 2003;170:270–278. doi: 10.4049/jimmunol.170.1.270. [DOI] [PubMed] [Google Scholar]
- 36.Munder M, Schneider H, Luckner C, et al. Suppression of T-cell functions by human granulocyte arginase. Blood. 2006;108:1627–1634. doi: 10.1182/blood-2006-11-010389. [DOI] [PubMed] [Google Scholar]
- 37.Matlack R, Yeh K, Rosini L, et al. Peritoneal macrophages suppress T-cell activation by amino acid catabolism. Immunology. 2006;117:386–395. doi: 10.1111/j.1365-2567.2005.02312.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Aruga A, Shu S, Chang AE. Tumor-specific granulocyte/macrophage colony-stimulating factor and interferon gamma secretion is associated with in vivo therapeutic efficacy of activated tumor-draining lymph node cells. Cancer Immunol Immunother. 1995;41:317–324. doi: 10.1007/BF01517220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Barth RJ, Jr, Mule JJ, Spiess PJ, Rosenberg SA. Interferon gamma and tumor necrosis factor have a role in tumor regressions mediated by murine CD8+ tumor-infiltrating lymphocytes. J Exp Med. 1991;173:647–658. doi: 10.1084/jem.173.3.647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shankaran V, Ikeda H, Bruce AT, et al. IFNgamma and lymphocytes prevent primary tumour development and shape tumour immunogenicity. Nature. 2001;410:1107–1111. doi: 10.1038/35074122. [DOI] [PubMed] [Google Scholar]
- 41.Bronte V, Zanovello P. Regulation of immune responses by L-arginine metabolism. Nat Rev Immunol. 2005;5:641–654. doi: 10.1038/nri1668. [DOI] [PubMed] [Google Scholar]
- 42.Grunstein J, Roberts WG, Mathieu-Costello O, Hanahan D, Johnson RS. Tumor-derived expression of vascular endothelial growth factor is a critical factor in tumor expansion and vascular function. Cancer Res. 1999;59:1592–1598. [PubMed] [Google Scholar]
- 43.Dinapoli MR, Calderon CL, Lopez DM. The altered tumoricidal capacity of macrophages isolated from tumor-bearing mice is related to reduce expression of the inducible nitric oxide synthase gene. J Exp Med. 1996;183:1323–1329. doi: 10.1084/jem.183.4.1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sotomayor EM, DiNapoli MR, Calderon C, Colsky A, Fu YX, Lopez DM. Decreased macrophage-mediated cytotoxicity in mammary-tumor-bearing mice is related to alteration of nitric-oxide production and/or release. Int J Cancer. 1995;60:660–667. doi: 10.1002/ijc.2910600516. [DOI] [PubMed] [Google Scholar]
- 45.Kakuda DK, Sweet MJ, Mac Leod CL, Hume DA, Markovich D. CAT2-mediated L-arginine transport and nitric oxide production in activated macrophages. Biochem J. 1999;340(Pt 2):549–553. [PMC free article] [PubMed] [Google Scholar]
- 46.Nicholson B, Manner CK, Kleeman J, MacLeod CL. Sustained nitric oxide production in macrophages requires the arginine transporter CAT2. J Biol Chem. 2001;276:15881–15885. doi: 10.1074/jbc.M010030200. [DOI] [PubMed] [Google Scholar]
- 47.Rothenberg ME, Doepker MP, Lewkowich IP, et al. Cationic amino acid transporter 2 regulates inflammatory homeostasis in the lung. Proc Natl Acad Sci U S A. 2006;103:14895–14900. doi: 10.1073/pnas.0605478103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Vig M, Srivastava S, Kandpal U, et al. Inducible nitric oxide synthase in T cells regulates T cell death and immune memory. J Clin Invest. 2004;113:1734–1742. doi: 10.1172/JCI20225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kahn DA, Archer DC, Gold DP, Kelly CJ. Adjuvant immunotherapy is dependent on inducible nitric oxide synthase. J Exp Med. 2001;193:1261–1268. doi: 10.1084/jem.193.11.1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Qin HY, Sadelain MW, Hitchon C, Lauzon J, Singh B. Complete Freund's adjuvant-induced T cells prevent the development and adoptive transfer of diabetes in nonobese diabetic mice. J Immunol. 1993;150:2072–2080. [PubMed] [Google Scholar]