

Abstract
In contrast to endocrine-sensitive and HER2-positive breast cancer, novel agents capable of treating advanced triple negative breast cancer (TNBC) are lacking. PARP (Poly-(adenosine diphosphate [ADP]-ribose) polymerase) inhibitors are emerging as one of the most promising ‘targeted’ therapeutics to treat TNBC, with the intended ‘target’ being DNA repair. PARP's are a family of enzymes involved in multiple cellular processes including DNA repair. TNBC shares multiple clinico-pathologic features with BRCA-mutated breast cancers which harbor dysfunctional DNA repair mechanisms. Investigators hypothesized PARP inhibition, in conjunction with the loss of DNA-repair via BRCA-dependent mechanisms, would result in synthetic lethality and augmented cell death. This hypothesis has borne out in both preclinical models and in clinical trials testing PARP inhibitors in both BRCA-deficient and TNBC. The focus of this review will include an overview of the preclinical rationale for evaluating PARP inhibitors in TNBC, the presumed mechanism of action of this novel therapeutic class, promising results from several influential clinical trials of PARP inhibition in advanced breast cancer (both TNBC and BRCA-deficient), proposed mechanisms of acquired resistance to PARP inhibitors, and, finally, conclude with current challenges and future directions for the development of PARP inhibitors in the treatment of breast cancer.
Introduction
Inhibition of PARP (Poly-(adenosine diphosphate [ADP]-ribose) polymerase) is emerging as one of the most exciting and promising ‘targeted’ therapeutic strategies to treat advanced triple negative breast cancer – the intended ‘target,’ being DNA repair. Diagnosed in an estimated 180,000 women worldwide, triple negative breast cancer (TNBC) is an aggressive subset of breast cancer that lacks expression of the estrogen and progesterone receptors (ER and PR) and the HER2 protein(1). TNBC, classified as basal-like by gene expression 80% of the time, is characterized by distinct risk factors, aggressive and early patterns of metastases, unique molecular characteristics, association with BRCA1 mutations, a relative lack of targeted therapeutics and poor prognosis(2-9).
In contrast to endocrine sensitive and HER2 positive breast cancer, novel agents capable of treating advanced triple negative breast cancer are at present lacking. Currently-available therapies are limited to cytotoxic chemotherapy with or without the addition of the anti-angiogenic agent, bevacizumab (Avastin, Genentech). Despite improvements in progression free survival when combining bevacizumab with paclitaxel among patients with HER2-negative advanced breast cancer (11.8 versus 5.9 months), absolute improvements for the triple negative subset were more limited (9 versus 5 months)(10, 11). Similarly, while there was a progression-free survival benefit of adding ixabepilone (Ixempra, Bristol Meyer Squibb), a newer generation microtubule stabilizing agent, to capecitabine chemotherapy among the triple negative subset (4.1 versus 2.1 months), the benefit was still only modest in part based on poor baseline outcomes among women with advanced breast cancer regardless of subtype (5.8 versus 4.2 months) (11, 12).
Given the poor prognosis and high rate of visceral metastases (including central nervous system recurrence) associated with TNBC, investigators have been actively searching for innovative therapeutic strategies to effectively treat this aggressive disease(13, 14). Building on the observation that TNBC shares several clinical and pathologic characteristics with BRCA-deficient breast cancers known to harbor deficient DNA repair mechanisms, PARP inhibitors have been tested in early phase clinical trials among patients with advanced TNBC. Preliminary results of phase II trials are encouraging and report improvements in response rates, progression free (PFS) and overall survival (OS) when adding PARP inhibition to DNA-damaging chemotherapeutics with minimal additional toxicity(15, 16). We will review the preclinical rationale for evaluating PARP inhibitors in triple negative (and BRCA-deficient) advanced breast cancer, the presumed mechanism of action of this novel therapeutic class, promising results from several influential clinical trials of PARP inhibition in advanced breast cancer (both TNBC and BRCA-deficient), proposed mechanisms of acquired resistance to PARP inhibitors, and, finally, conclude with current challenges and future directions for the development of PARP inhibitors in the treatment of breast cancer.
DNA Repair as a Therapeutic “Target” and Proposed Mechanism of PARP Inhibitors
DNA damage is an ongoing process resulting from both endogenous and exogenous (environmental) assaults to the human genome. Endogenous forms for DNA damage arise from spontaneous base changes, replication errors and oxygen-free radicals, while exogenous forms include chemical mutagens, cytotoxic agents and both ultra-violet and ionizing radiation(17). The genome is armed with multiple DNA repair mechanisms, including but not limited to Mismatch Repair (MMR), Base Excision Repair (BER), Nucleotide Excision Repair (NER) and Double Strand Break Repair (DSR). DNA double strand breaks are highly toxic to cells and two main pathways contribute to their inherent repair – error-prone non-homologous end joining (NHEJ) and error-free homologous recombination (HR, Figure 1a)(17-19). HR is dependent on functional BRCA 1 and 2 pathways and BRCA maintains genome stability, at least in part, by regulating HR according to the type of DNA damage(20-23). As follows, germline mutations in either the BRCA1 or BRCA2 genes are associated with a high risk of developing a number of cancers, including breast, ovarian, and prostate cancer(17, 24, 25). When the BRCA-associated DNA repair pathway – namely HR – is lost or dysfunctional, repair shifts toward alternate DNA repair mechanisms dependent on a unique class of enzymes, Poly-(adenosine diphosphate [ADP]-ribose) polymerase (PARP).
Figure 1.
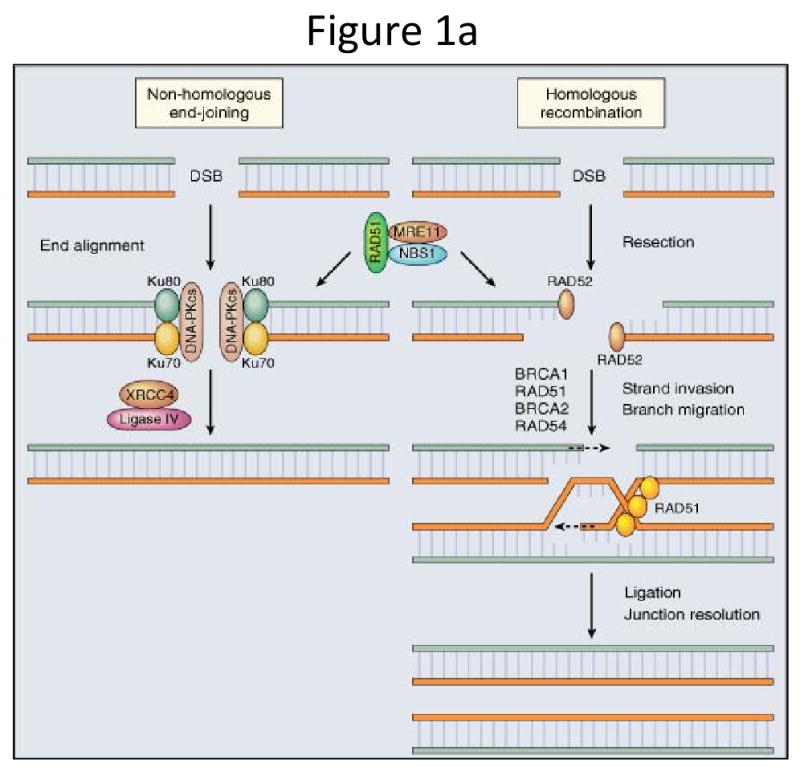
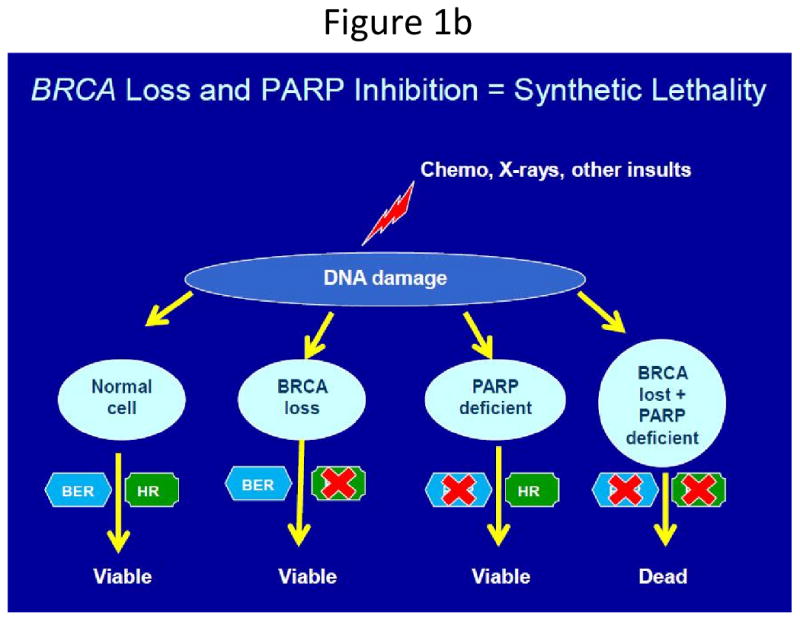
Figure 1a: Two main pathways which contribute to repair of DNA double strand breaks: non-homologous end joining (NHEJ) and homologous recombination (HR). This figure was published in Abeloff's Clinical Oncology, 4th Edition. JM Ford and MB Kastan, Chapter 10: DNA damage response pathways and cancer. p. 149. (Reprinted with permission, Copyright 2008, Churchill Livingstone, an Imprint of Elsevier) (5).
Figure 1b: ‘Synthetic lethality’ and subsequent cell death due to loss of parallel DNA repair pathways. In the presence of one (or both) functional BRCA- or PARP-dependent DNA repair pathways, cells survive. In the absence of both, cell death ensues (Reprinted with permission, from EA Comen and M Robson: ONCOLOGY 24(1):55-68, 2010) (52).
PARP's are a family of enzymes involved in cellular processes such as genomic stability, DNA repair, cell cycle progression, and apoptosis(26, 27). PARP-1, a nuclear, zinc-finger, deoxyribonucleic acid (DNA)-binding protein, localizes to DNA strand breaks as part of the base excision repair (BER) process(28). Cell death from targeting two genes which, alone, do not result in cell death is termed “synthetic lethality.” Investigators hypothesized PARP inhibition, in conjunction with the loss of DNA-repair via BRCA-dependent mechanisms, would result in synthetic lethality and augmented cell death (Figure 1b) – a hypothesis that has borne out in both preclinical models and the clinical trial arena.
It well-recognized that BRCA-deficient, basal-like (as defined by microarray) and triple negative (as defined by immunohistochemistry [IHC]) breast cancers share clinical and pathologic similarities, including high rates of p53 mutation, aneuploidy, high pathological grade, and relative sensitivity to DNA-damaging chemotherapeutics (Table 1)(4, 7, 9, 15, 29, 30). Several mechanisms have been proposed to explain these similarities based on presumed BRCA pathway and subsequent HR dysfunction in sporadic basal-like and TNBC. These mechanisms may include (1) over-expression and copy number gain of ID4, a negative regulator of BRCA1(31, 32), (2) decreased expression of BRCA1 messenger RNA(32), (3) BRCA1 gene promoter methylation(33) and (4) copy number aberrations affecting genes within a ‘BRCA DNA Damage Response’ pathway(32). Each of these observations suggests a role for defective HR DNA repair among sporadic TNBC and basal-like breast tumors providing the basis for the term “BRCAness” of sporadic TNBC(34). Thus, the “BRCAness” of sporadic TNBC has provided the rationale to test PARP inhibitors, not only in advanced BRCA-mutated tumors, but also sporadic TNBC. Results from early phase clinical trials evaluating the efficacy of PARP inhibitors in BRCA-mutated and TNBC, although preliminary, are quite promising (15, 35-37).
Table 1.
Shared clinic-pathologic features between BRCA-1 deficient and triple negative/Basal-like breast cancer. Figure adapted from 2009 ASCO Plenary Session(15).
| Clinico-pathologic Characteristics | Hereditary BRCA1 Breast Cancer | Triple Negative/Basal-like Breast Cancer |
|---|---|---|
| ER/PR/HER2 status | Negative | Negative |
| TP53 status | Mutant | Mutant (Up to 80%) |
| BRCA1 status | Mutational Inactivation | Diminished Expression (?) |
| Gene-expression patterns | Basal-like | Basal-like |
| Tumor Histology | Poorly-differentiated (high grade) | Poorly-differentiated (high grade) |
| Chemosensivity to DNA-damaging chemotherapy | Highly sensitive | Likely sensitive |
| Genome-wide Aneuploidy | Yes | Yes |
Preclinical Efficacy of PARP Inhibition in BRCA-deficient and TNBC/Basal-like Breast Cancer Models
Two landmark preclinical studies report (1) feasibility of targeting DNA repair defects inherent to BRCA mutant cell lines as a therapeutic strategy and (2) the acute sensitivity of BRCA-2 deficient embryonic stem cells to PARP inhibition due to deficiencies in HR(38, 39). Farmer et al. conducted a series of experiments illustrating a reduction in clonogenic survival of BRCA-1 and BRCA2-deficient cell lines when transfected with a plasmid expressing a short interfering RNA (siRNA) targeting PARP1. Chemical inhibition of PARP1 was more potent in BRCA-1 and BRCA-2 deficient embryonic stem cells compared to heterozygous mutant and wild-type cells. An in vivo BRCA-2 deficient murine model supported these findings(39). Parallel studies conducted by Bryant et al. also demonstrated cell lines deficient in HR (via defective XRCC2, XRCC3 and BRCA2) were highly sensitive to PARP inhibition. Treatment of BRCA2-deficient cells lines with PARP inhibition induced gamma(γ)-H2AX foci, a marker of DNA double strand breaks, and BRCA2 deficient tumors in a xenograft model were susceptible to PARP inhibition(38). Taken together, results from these studies suggested a new, mechanism-based approach for the treatment of patients with BRCA-1 and BRCA2-deficient tumors. Importantly, models of sensitivity to PARP inhibition suggest that the key ingredient is deficiency in HR, indicating that this approach may be more widely applicable in the treatment of sporadic cancers sharing functional BRCA loss or other impairments of homologus recombination.
As discussed above, BRCA-mutated, basal-like and TNBC share a number of clinico-pathologic characteristics (Table 1). BRCA-1 and BRCA-2 mutation carriers, however, comprise a minority of breast cancer cases(24, 25). The “BRCAness” of TNBC, which is classified as basal-like by cDNA microarray over 80% of the time(9, 40), led investigators to evaluate the sensitivity of PARP inhibition in breast cancer cell lines of varying subtypes (i.e. luminal, basal, etc.) to not only determine subtype-specific sensitivity, but to further elucidate the mechanism of cellular cytotoxicity.
A series of informative preclinical studies evaluating DNA damage response pathways in breast cancer cell lines of different subtypes report selective response of basal-like breast cancer cell lines to PARP inhibition which may, in addition to defective HR, be due to inefficient base excision repair (BER)(41). Oxidative DNA damage (ODD) constitutes a large majority of endogenous DNA damage in human cells. DNA damage from reactive oxygen species usually occurs as a single-base alteration repaired via BER. Investigators hypothesized that basal-like and BRCA-1 mutated breast cancers share defects in maintaining genomic stability via aberrant regulation of ODD. When a variety of breast cancer cell lines of a varying subtypes, including luminal, basal-like and BRCA-1 mutated breast cancers were tested for sensitivity to H202, (as an indicator of response to ODD), basal-like and BRCA1- mutated breast cancer cell lines were most sensitive, thus, least effective in the repair of oxidative damage. (41).
A second set of parallel experiments concluded that the relative inefficiency of basal-like and BRCA-1 mutated breast cancer cell lines to repair ODD was a result of defective BER(41). Briefly, a BER assay involving BER-dependent expression of a GFP [green fluorescent protein] reporter gene showed a relative decrease in GFP expression 24 hours following DNA damage among basal-like and BRCA-1 mutated breast cancer cell lines when compared to luminal breast cancer cell lines. Moreover, transfection of the BRCA-1 mutated cell line SUM149 with wild-type BRCA1 resulted in a 4-fold increase in BER, while shRNA knock-down of BRCA1 resulted in decreased BER. Inefficient BER repair was correlated with sensitivity to PARP inhibition in vitro. In addition, using this same panel of cell lines, it was shown that the basal-like cells were selectively sensitive to platinum and gemcitabine, and that these drugs exhibited synergy when combined with a PARP inhibitor in basal-like but not luminal breast cancer cell lines(42). This series of informative studies continues to shed light on the complicated mechanism of action inherent to PARP inhibitors and provides a plausible mechanism for the selective efficiency of PARP inhibitors in BRCA-mutated and basal-like cell lines – an observation that has borne out in clinical studies.
PARP-inhibitors: Overview of Current Clinical Data
In parallel to strides in the preclinical arena, pharmacologic inhibition of PARP has translated into advances in the clinical management of patients with TNBC and BRCA-deficient advanced breast cancer. Historically, the treatment of advanced TNBC has been fraught with unique challenges. Specifically, in contrast to endocrine sensitive and HER2-enriched breast cancer, TNBC is characterized by a relative lack of “drug-able” targets, rapid recurrence in visceral organs (including the CNS), and inherently poor prognosis(13, 14). Based on elegant preclinical rationale, PARP inhibitors are being tested in BRCA-deficient and TNBC and illustrate clinical efficacy. Although there are several PARP inhibitors in early phase development (Table 2), the lion's share of clinical experience has been with olaparib (AZD2281, AstraZenca/KuDOS) and BSI-201 (BiPAR Sciences/Sanofi Aventis).
Table 2.
A summary of representative PARP inhibitors in clinical development by route of administration and current clinical status.
| Agent | Company | Route of Administration | Clinical Status |
|---|---|---|---|
| Olaparib/(AZD2281) | AstraZeneca/KuDOS | Oral | Phase 1 and 2 |
| Veliparib/(ABT-888) | Abbott | Oral | Phase 1 and 2 |
| BS1-201 | BiPar/SanofiAventis | IV | Phase 2 and 3 |
| AG014699 | Pfizer | IV | Phase 1 and 2 |
| MK482 | Merck | Oral | Phase 1 |
| INO-1001 | Inotek | IV | Phase 1 |
| CEP9272 | Cephalon | Oral | Phase 1 |
Olaparib
The efficacy of olaparib, an oral PARP inhibitor, among patients with BRCA-mutated advanced solid tumors was initially reported in the phase I setting(35). In addition to standard dose-escalation and pharmacokinetic/pharmacodyamic analyses, toxicity and efficacy were reported among 60 patients with advanced tumors, a population enriched for BRCA-mutation carriers (n = 22). Pharmacokinetic data indicated rapid absorption and elimination; pharmacodynamic studies confirmed PARP inhibition in surrogate samples (peripheral-blood mononuclear cells and plucked eyebrow-hair follicles) and tumor tissue. There was no obvious increase in adverse effects among BRCA-mutation carriers. Objective anti-tumor activity was reported only in mutation carriers (all heavily pre-treated for breast, ovarian or prostate cancer).
A second phase II, multi-center, single-arm, sequential cohort design study sought to determine the efficacy/tolerability of the olaparib among patients with BRCA1/BRCA2-deficient, advanced breast cancer(43). Fifty-four patients were enrolled in one of two dose-based cohorts (cohort 1 = 27 treated with 400mg orally twice daily; cohort 2 = 27 treated with 100mg orally twice daily). The primary objective was objective tumor response (complete response [CR] plus partial response [PR]). Secondary objectives included progression-free survival (PFS), safety, and tolerability. At the time of progression, patients in cohort 2 were permitted to cross-over to cohort 1. The majority of patients in both cohorts harbored BRCA1 mutations, 67% and 56%, respectively; a smaller proportion had BRCA2 mutations, 33% and 41%, respectively. Over 50% of patients had TNBC and median number of prior therapies was 3. Overall response rates were 41% and 22% among patients in cohort 1 and 2, respectively. PFS was 5.7 (4.6 – 7.4) months in cohort 1 and 3.8 (1.9 – 5.5) months in cohort 2. The most commonly reported grade 3 adverse events were fatigue, nausea and vomiting. Treatment discontinuation due to adverse events was uncommon.
Several Canadian studies are ongoing to define efficacy, mechanisms of resistance, and patient population most likely to respond to olaparib. Canadian 20 study is a phase II study enrolling 4 cohorts of 91 patients with advanced breast or ovarian cancer: A) Ovarian, BRCA negative/unknown, B) TNBC, BRCA negative/unknown, C) Ovarian, BRCA-mutated and D) Breast, BRCA-mutated, ER - or +. Patients received olaparib 400 mg orally twice daily. Assessments at eight weeks include imaging, tumor re-biopsy, blood collection and response assessment. Although responses were seen in Arms A, C and D, Arm B closed early as no responses were seen in 15 enrolled patients with sporadic TNBC. This trial is one of the first to report response assessment for PARP inhibitors as a single agent to treat sporadic TNBC, and with limited and unimpressive results further supports the use of combination PARP inhibitor plus chemotherapy to treat patients with sporadic TNBC(44).
Canadian Study 11, a randomized, double-blind, multi-center study assessing efficacy of olaparib in combination with paclitaxel in the 1st or 2nd line treatment of metastatic TNBC, yielded notable toxicity patterns(45). In the phase I cohort, 8 of 9 patients required paclitaxel dose modifications and 4 of 9 required olaparib dose modifications due to grade 2 – 4 neutropenia prompting the use of G-CSF rescue (granulocyte colony-stimulating factor) in cohort 2. Overall response in cohorts 1 and 2 were 33% (3/9) and 40% (4/10), respectively. Ongoing trials with olaparib and DNA-damaging chemotherapeutics (i.e. phase II, olaparib with carboplatin in advanced BRCA-deficient and TNBC and phase I/II, olaparib with cisplatin in the neoadjuvant treatment of TNBC) will shed light on this toxicity. Moreover, these trials will further define whether or not dose-limiting neutropenia is specific to olaparib in combination with paclitaxel or is more generalizable across the broader class of DNA-damaging chemotherapeutics.
BSI-201
Initial safety, tolerability and efficacy of the intravenous PARP inhibitor, BSI-201, was reported in a series of phase I studies as a single agent and in combination with several DNA-damaging chemotherapeutics, respectively(46, 47). A phase I study of BSI-201 in 23 heavily-pretreated patients with advanced solid tumors report tolerability at doses ranging from 0.5 mg/kg – 8.0 mg/kg. A maximum tolerated dose (MTD) was not reached with the most common side effects being fatigue (56%), nausea (47%), vomiting (39%), and constipation (21%). PARP activity as measured in peripheral blood mononuclear cells (PBMC's) was suppressed by > 50% at the fifth dose level (2.8 mg/kg) and best response of stable disease for > 2 months was reported in 6 of 23 patients(46). A second phase Ib study reported safety and efficacy of BSI-201 in combination with several DNA-damaging chemotherapeutic agents (i.e. gemcitabine, topotecan, temozolomide, or carboplatin/paclitaxel). Among 66 treated patients (24 breast cancer patients), 2 serious adverse events were possibly related to BSI-201 and all patients tolerated doses up to 5.6mg/kg. The addition of BSI-201 did not potentiate expected toxicities of individual cytotoxics. Fifty-three of 66 reported some clinical benefit with partial response observed in 2 advanced breast cancer patients(47).
Both the shared clinico-pathologic features between BRCA-deficient and sporadic TNBC and preclinical evidence illustrating synergy for BSI-201 and gemcitabine/carboplatin provided the rationale to investigate the efficacy of BSI-201 with chemotherapy in advanced TNBC(15, 16). One-hundred and twenty women with metastatic TNBC were enrolled in a multi-center, open-label, randomized, clinical trial in the United States. Enrolled patients who had received 0–2 prior lines of chemotherapy in the metastatic setting were randomized to receive gemcitabine (1000mg mg/m2, IV, days 1, 8) plus carboplatin (AUC 2, IV, days, 1, 8) with or without BSI-201 (5.6mg/kg, IV, days 1, 4, 8, 11) every 21 days. Clinical benefit rate (CR + PR + SD [stable disease]) at 6 months and safety of combination therapy was the primary objective of this trial. Secondary objectives included overall response rates, PFS and overall survival (OS).
Patients' baseline characteristics were well-balanced between treatments arms. Notably, > 50% of patients in both arms had received prior taxane and anthracycline-based chemotherapy and 10%, prior bevacizumab. Clinical benefit rates were superior among patients who received BSI-201 in combination with chemotherapy compared to those who received chemotherapy alone (62% versus 21%, respectively; p = 0.0002). In addition, PFS and OS was superior among patients who received BSI-201 (6.9 versus 3.3 months; Hazard Ratio [HR] = 0.342, p <0.0001 and 9.2 versus 5.7 months; HR = 0.348, p = 0.0005, respectively)(15). An updated analysis continues to confirm an OS advantage among patients who received BSI-201 in combination with chemotherapy compared to chemotherapy alone (12.2 versus 7.7 months, p = 0.005; Figure 2)(36). The updated safety analysis indicates the addition of BSI-201 does not potentiate side effects of chemotherapy. Preliminary efficacy results prompted the design of an ongoing multicenter, randomized, phase III trial to confirm efficacy of BSI-201 in combination with gemcitabine/carboplatin chemotherapy, results of which are eagerly awaited.
Figure 2.
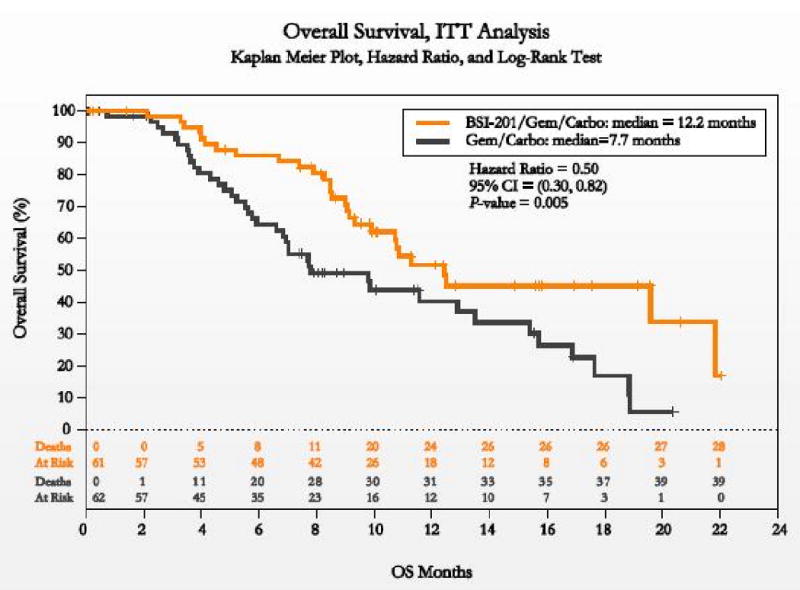
Kaplan Meier curves illustrating an overall survival advantage for patients with triple negative metastastic breast cancer treated with the PARP inhibitor, BSI-201 (BiPAR Sciences/Sanofi Aventis) plus carboplatin/gemcitabine chemotherapy compared to chemotherapy alone (12.2 versus 7.2 months, p = 0.005)(36).
Proposed Mechanisms for Resistance to PARP inhibition
PARP inhibitors have illustrated clinical efficacy in historically-challenging, often treatment-refractory malignancies, however disease control rarely exceeds one year. Although not entirely understood, resistance to PARP inhibitors is hypothesized to result from one of several mechanisms including, but not limited to up-regulation of the multi-drug resistance (MDR 1,2) efflux pumps(48) and reversion of BRCA mutation with restoration of BRCA function(49). A series of several studies support the proposed mechanisms of PARP resistance.
Long-term treatment of a BRCA-1 deficient genetically engineered mouse model (GEMM) treated with the PARP inhibitor, AZD2281, caused up-regulation of the Abcb1a/b genes encoding for the P-glycoprotein efflux pumps, and occasionally up-regulation of the drug target, PARP1. Moreover, resistance in all cases was reversed by co-administration of tariquidar, a P-glycoprotein inhibitor(48). A separate series of experiments employed a PARP-inhibitor-resistant clone from a human pancreatic cell line homozygous for the BRCA2 protein-truncating c.6174delT frameshift mutation. Resistant cell lines were found to express new BRCA2 isoforms as a result of intragenic deletion spanning the c.6174delT mutation, resulting in restoration of the open reading frame, creating a novel BRCA2 allele functional for homologous recombination and causing decreased sensitivity to PARP inhibition(49). Finally, based on the knowledge that PARP inhibitors and platinum chemotherapeutics have overlapping targets, platinum resistance may inform mechanisms of PARP inhibitor resistance. Among a series of recurrent BRCA1 mutated ovarian cancers, 4 of 6 recurrent, platinum-resistant tumors had developed secondary genetic changes in BRCA1 that restored the open reading frame of the BRCA1 protein, thus conferring diminished platinum sensitivity(50). Although complex, rational strategies to circumvent the two flavors of PARP inhibitor resistance, up-regulation of MDR efflux pumps and reversion mutations, may exist. Such strategies will be challenging to translate clinically and underscore the need for tissue collection from the numerous early phase clinical trials evaluating PARP inhibition in both BRCA-mutated and non-BRCA-mutated advanced malignancies to study mechanisms of resistance.
Conclusions, Challenges and Future Directions
Results from elegant preclinical studies and promising clinical trials continue to highlight PARP inhibitors as one of the most promising “targeted therapies” for aggressive TNBC. As we as a medical community move forward, we are challenged with several tasks including (1) more precisely defining the patient population (even within TNBC patient population) most likely to respond to the PARP inhibition, (2) discovering and validating candidate biomarkers to predict responders (i.e. (γ) H2AX, RAD51 [as a marker of intact HR], germline DNA studies, etc.), (3) determining the optimal chemotherapy back-bone to combine with PARP inhibitors, (4) defining the most effective schedule of administration (ie. continuous versus intermittent dosing with chemotherapy), and finally, (5) moving PARP inhibitors into “niche” settings (i.e. brain metastases arising from TNBC). In this era of personalized medicine, investigators are actively working to further define TNBC patients most likely to respond to PARP inhibitors. This is particularly important as these drugs move to the adjuvant setting since long-term toxicities to normal tissues as a result of prolonged suppression of DNA repair have yet to be defined. As an example, an as of yet unvalidated, but intriguing 25-gene assay was developed to identify BRCA-like sporadic TNBC; an approach such as this may help us select patients most likely to respond to this class of drugs (51). The role of PARP inhibitors in non-TNBC phenotypes is also of interest since the mechanism of PARP inhibition may serve to sensitize endocrine sensitive and/or HER2-positive breast tumors to DNA-damaging chemotherapy. Each of the aforementioned challenges are areas deserving of further study and are the subject of ongoing clinical investigation. Although challenging at first glance, thoughtful planning and coordinated efforts between collaborating pre-clinical, translational, and clinical investigators will continue to move the development of PARP inhibitors forward and hold the potential to improve the lives of the hundreds and thousands of women diagnosed with breast cancer worldwide each year.
Statement of Translational Relevance
The treatment of triple negative breast cancer with PARP inhibitors is a perfect example of translational medicine. Close to a decade ago, investigators hypothesized that preventing DNA repair via PARP inhibition, in conjunction with the loss of DNA-repair via BRCA-dependent mechanisms, would result in synthetic lethality and augmented cell death. This hypothesis has borne out in preclinical studies evaluating the effect of PARP inhibition on BRCA-deficient cell lines and BRCA-deficient preclinical tumor models. Profound efficacy observed in the preclinical arena was readily-transferrable to the clinical setting. Recognizing the clinico-pathologic similarities between BRCA-deficient and triple negative breast tumors (often termed “BRCA-ness”), PARP inhibitors have been tested in both patient populations and illustrate apparent clinical efficacy. Future studies to define PARP inhibitor mechanisms of resistance and response biomarkers to define optimal patient populations will involve returning back to the bench, such that strides at the bedside continue – the essence of translational medicine.
Acknowledgments
Funding Sources: 5K12CA120780-03 (CA), NCI CA058223 (UNC SPORE, CA and LC), National Institute of Health R01 CA108794 (JF), Breast Cancer Research Foundation (JF), Susan G. Komen for the Cure (JF); National Cancer Institute Program of Research Excellence (SPORE) in BreastCancer at the Dana- Farber/Harvard Cancer Center CA089393 (DS), and the Cogan Family Foundation(DS); and Canadian Breast Cancer Research Foundation (RD).
On December 9, 2009, the Triple Negative Breast Cancer Foundation and Susan G. Komen for the Cure convened a meeting of clinicians, investigators, and advocates to review the state of current clinical and translational research on TNBC, specifically as it relates to novel therapies and the direction of future research opportunities in this subset of breast cancer. This is a report derived from that meeting. We would like to acknowledge and sincerely thank the Triple Negative Breast Cancer Foundation and the Susan G. Komen for the Cure for their support of this symposium.
Members attending this symposium include: Marianne Alciati, PhD; Carey K. Anders, MD; Allison Axenrod; Sunil Badve, MD; Judith Balmaña, MD; Powel Brown, MD, PhD; Lisa A. Carey, MD; Jennifer De Los Santos, MD; Rebecca Dent, MD; Hayley Dinerman; James Ford, MD; Andres Forero, MD; William D. Foulkes, MB, PhD; Liz Frank; Sharon Fredman; Shridar Ganesan, MD, PhD; Judy E. Garber, MD, MPH; Montserrat Garcia-Closas, MD, Dr.P.H.; Vincent L. Giranda, MD, PhD; Cindy Geoghegan; Lyndsay N. Harris, MD; Gabriel N. Hortobagyi, MD, FACP; Clifford A. Hudis, MD; Steven Jay Isakoff, MD, PhD; Gilbert Jirau-Lucca, MS; Helen Krontiras, MD; Gabriella L. Mariani, MD; Olufunmilayo I. Olopade, MD, FACP; Charles M. Perou, PhD; Peggy L. Porter, MD; Lajos Pusztai, MD, PhD; Andrea Richardson, MD, PhD; Hope Rugo, MD; Daniel Silver, MD, PhD; George W. Sledge, Jr. MD; Sandra M. Swain, MD; Melinda L. Telli, MD; Elizabeth Thompson; Andrew Tutt, MRCP, FRCR, PhD; Eric P. Winer, MD; Antonio C. Wolff, MD; Jo Anne Zujewski, MD.
References
- 1.Swain S. Triple-Negative Breast Cancer. Metastatic Risk and Role of Platinum Agents 2008 ASCO Clinical Science Symposium 2008; June 3, 2008. [Google Scholar]
- 2.Carey L, Perou C, Livasy C, et al. Race, breast cancer subtypes, and survival in the Carolina Breast Cancer Study. JAMA. 2006;295:2492–502. doi: 10.1001/jama.295.21.2492. [DOI] [PubMed] [Google Scholar]
- 3.Dent R, Trudeau M, Pritchard K, et al. Triple-negative breast cancer: clinical features and patterns of recurrence. Clin Cancer Res. 2007;13:4429–34. doi: 10.1158/1078-0432.CCR-06-3045. [DOI] [PubMed] [Google Scholar]
- 4.Livasy C, Karaca G, Nanda R, et al. Phenotypic evaluation of the basal-like subtype of invasive breast carcinoma. Mod Pathol. 2006;19:264–71. doi: 10.1038/modpathol.3800528. [DOI] [PubMed] [Google Scholar]
- 5.Millikan RC, Newman B, Tse CK, et al. Epidemiology of basal-like breast cancer. Breast Cancer Research and Treatment. 2008;109:123–39. doi: 10.1007/s10549-007-9632-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nielsen T, Hsu F, Jensen K, et al. Immunohistochemical and clinical characterization of the basal-like subtype of invasive breast carcinoma. Clin Cancer Res. 2004;10:5367–74. doi: 10.1158/1078-0432.CCR-04-0220. [DOI] [PubMed] [Google Scholar]
- 7.Perou CM, Sorlie T, Eisen MB, et al. Molecular portraits of human breast tumours. Nature. 2000;406:747–52. doi: 10.1038/35021093. [DOI] [PubMed] [Google Scholar]
- 8.Smid M, Wang Y, Zhang Y, et al. Subtypes of breast cancer show preferential site of relapse. Cancer Research. 2008;68:3108–14. doi: 10.1158/0008-5472.CAN-07-5644. [DOI] [PubMed] [Google Scholar]
- 9.Sorlie T, Tibshirani R, Parker J, et al. Repeated observation of breast tumor subtypes in independent gene expression data sets. Proc Natl Acad Sci U S A. 2003;100:8418–23. doi: 10.1073/pnas.0932692100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Miller K, Wang M, Gralow J, et al. Paclitaxel plus bevacizumab versus paclitaxel alone for metastatic breast cancer. New England Journal of Medicine. 2007;357:2666–76. doi: 10.1056/NEJMoa072113. [DOI] [PubMed] [Google Scholar]
- 11.Burstein HJ. Therapeutic Options for Treatment Refractory Breast Cancer. Discussion 2010 Metastatic Breast Cancer Oral Abstract Presentation 2010; June 8, 2010. [Google Scholar]
- 12.Thomas ES, Gomez HL, Li RK, et al. Ixabepilone Plus Capecitabine for Metastatic Breast Cancer Progressing After Anthracycline and Taxane Treatment. Journal of Clinical Oncology. 2007;25:5210–7. doi: 10.1200/JCO.2007.12.6557. [DOI] [PubMed] [Google Scholar]
- 13.Dent RTM, Pritchard KI, Hanna WM, et al. Triple-negative breast cancer: clinical features and patterns of recurrence. Clin Cancer Res. 2007;13:4429–34. doi: 10.1158/1078-0432.CCR-06-3045. [DOI] [PubMed] [Google Scholar]
- 14.Lin N, Claus E, Sohl J, et al. Sites of Distant Recurrence and Clinical Outcomes in Patients With Metastatic Triple-negative Breast Cancer High Incidence of Central Nervous System Metastases. Cancer. 2008;113:2638–45. doi: 10.1002/cncr.23930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.O'Shaughnessy J, Osborne C, Pippen J, et al. Efficacy of BSI-201, a poly (ADP-ribose) polymerase-1 (PARP1) inhibitor, in combination with gemcitabine/carboplatin (G/C) in patients with triple-negative breast cancer (TNBC): Results of a randomized phase II trial. J Clin Oncol. 2009;27 Abstract 3. [Google Scholar]
- 16.O'Shaughnessy J, Yoffe M, Osborne C, et al. Triple negative breast cancer: a phase 2, multi-center, open-label, randomized trial of gemcitabine/carboplatin (G/C), with or without BSI-201, a PARP inhibitor. San Antonio Breast Cancer Symposium; 2008. Abstract 2120. [Google Scholar]
- 17.Ford J. DNA Damage Response Pathways and Cancer. In: Abeloff MD, editor. Clinical Oncology. 3rd. Churchill Livingstone; 2004. [Google Scholar]
- 18.Hartman A, Ford J. BRCA1 induces DNA damage recognition factors and enhances nucleotide excision repair. Nature Genetics. 2002;32:180–4. doi: 10.1038/ng953. [DOI] [PubMed] [Google Scholar]
- 19.Zhang J, Powell S. The role of the BRCA1 tumor suppressor in DNA double-strand break repair. Mol Cancer Res. 2005;3:531–9. doi: 10.1158/1541-7786.MCR-05-0192. [DOI] [PubMed] [Google Scholar]
- 20.Moynahan ME, Pierce AJ, Jasin M. BRCA2 is required for homology-directed repair of chromosomal breaks. Molecular Cell. 2001;7:263–72. doi: 10.1016/s1097-2765(01)00174-5. [DOI] [PubMed] [Google Scholar]
- 21.Cousineau I, Belmaaza A. BRCA1 haploinsufficiency, but not heterozygosity for a BRCA1-truncating mutation, deregulates homologous recombination. Cell Cycle. 2007;6:962–71. doi: 10.4161/cc.6.8.4105. [DOI] [PubMed] [Google Scholar]
- 22.Powell SN, Kachnic LA. Roles of BRCA1 and BRCA2 in homologous recombination, DNA replication fidelity and the cellular response to ionizing radiation. Oncogene. 2003;22:5784–91. doi: 10.1038/sj.onc.1206678. [DOI] [PubMed] [Google Scholar]
- 23.Snouwaert JN, Gowen LC, Latour AM, et al. BRCA1 deficient embryonic stem cells display a decreased homologous recombination frequency and an increased frequency of non-homologous recombination that is corrected by expression of a brca1 transgene. Oncogene. 1999;18:7900–7. doi: 10.1038/sj.onc.1203334. [DOI] [PubMed] [Google Scholar]
- 24.Narod S. Modifiers of risk of hereditary breast and ovarian cancer. Nat Rev Cancer. 2002;2:113–23. doi: 10.1038/nrc726. [DOI] [PubMed] [Google Scholar]
- 25.Narod S, Foulkes W. BRCA1 and BRCA2: 1994 and beyond. Nat Rev Cancer. 2004;4:665–76. doi: 10.1038/nrc1431. [DOI] [PubMed] [Google Scholar]
- 26.Tentori L, Graziani G. Chemopotentiation by PARP inhibitors in cancer therapy. Pharmacol Res. 2005;52:25–33. doi: 10.1016/j.phrs.2005.02.010. [DOI] [PubMed] [Google Scholar]
- 27.Tentori L, Porarena I, Graziani G. Potential Clinical Applications of Poly (ADP-ribose) Polymerase Inhibitors. Pharmacological Research. 2002;45:73–85. doi: 10.1006/phrs.2001.0935. [DOI] [PubMed] [Google Scholar]
- 28.Plummer E. Inhibition of poly(ADP-ribose) polymerase in cancer. Current Opinion in Pharmacology. 2006;6 doi: 10.1016/j.coph.2006.02.004. [DOI] [PubMed] [Google Scholar]
- 29.Cleator S, Heller W, Coombes R. Triple-negative breast cancer: therapeutic options. Lancet Oncol. 2007;3:235–44. doi: 10.1016/S1470-2045(07)70074-8. [DOI] [PubMed] [Google Scholar]
- 30.Sorlie T, Perou C, Tibshirani R, et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci U S A. 2001;98:10869–74. doi: 10.1073/pnas.191367098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Turner N, Reis-Filho J, Russell A, et al. BRCA1 dysfunction in sporadic basal-like breast cancer. Oncogene. 2007;26:2126–32. doi: 10.1038/sj.onc.1210014. [DOI] [PubMed] [Google Scholar]
- 32.Natrajan R, Weigelt B, Mackay A, et al. An integrative genomic and transcriptomic analysis reveals molecular pathways and networks regulated by copy number aberrations in basal-like, HER2 and luminal cancers. Breast Cancer Research and Treatment. 2009;121:575–89. doi: 10.1007/s10549-009-0501-3. [DOI] [PubMed] [Google Scholar]
- 33.Grushko T, Nwachukwu C, Caroenthammaraksa S, et al. Evaluation of BRCA1 inactivation by promoter methylation as a marker of triple-negative and basal-like breast cancer. Journal of Clinical Oncology. 2010;28:7S. Abstract 10510. [Google Scholar]
- 34.Turner N, Tutt A, Ashworth A. Hallmarks of ‘BRCAness’ in sporadic cancers. Nat Rev Cancer. 2004;4:814–9. doi: 10.1038/nrc1457. [DOI] [PubMed] [Google Scholar]
- 35.Fong P, Boss D, Yap T, Tutt A, Wu P, Mergui-Roelvink M. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. New England Journal of Medicine. 2009;361:123–34. doi: 10.1056/NEJMoa0900212. [DOI] [PubMed] [Google Scholar]
- 36.O'Shaughnessy J, Osborne C, Pippen J, et al. Updated results of a randomized phase II study demonstrating efficacy and safety of BSI-201, a PARP inhibitor, in combination with gemcitabine/carboplatin in metastastic triple-negative breast cancer. Cancer Research. 2009;69 Abstract 3122. [Google Scholar]
- 37.Tutt A, Robson M, Garber JE, et al. Phase II trial of the oral PARP inhibitor olaparib in BRCA-deficient advanced breast cancer. Journal of Clinical Oncology. 2009;27 Abstract CRA501. [Google Scholar]
- 38.Bryant H, Schultz N, Thomas H, et al. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature. 2005;434:913–7. doi: 10.1038/nature03443. [DOI] [PubMed] [Google Scholar]
- 39.Farmer H, McCabe N, Lord CJ, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434:917–21. doi: 10.1038/nature03445. [DOI] [PubMed] [Google Scholar]
- 40.Silver D, Richardson A, Eklund A, et al. Efficacy of neoadjuvant Cisplatin in triple-negative breast cancer. Journal of Clinical Oncology. 2010;28:1145–53. doi: 10.1200/JCO.2009.22.4725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Alli E, Sharma V, Sunderesakumar P, Ford J. Defective Repair of Oxidative DNA Damage in Triple Negative Breast Cancer Confers Sensitivity to Inhibition of Poly(ADP-Ribose) Polymerase. Cancer Research. 2009;69:3589–96. doi: 10.1158/0008-5472.CAN-08-4016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hastak K, Alli E, Ford J. Synergistic chemosensitivity of triple-negative breast cancer cell lines to a PARP inhibitor, cisplatin and gemcitabine. Proc AACR. 2009;50 doi: 10.1158/0008-5472.CAN-09-4521. Abstract 5642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tutt A, Robson M, Garber JE, et al. Phase II trial of the oral PARP inhibitor olaparib in BRCA-deficient advanced breast cancer. J Clin Oncol. 2009;27:CRA501. [Google Scholar]
- 44.Gelmon K, Hirte H, Robidoux A, et al. Can we define tumors that respond to PARP inhibitors? A phase II correlative study of olaparib in advanced serous ovarian cancer and triple negative breast cancer. Journal of Clinical Oncology. 2010;28 Abstract 3002. [Google Scholar]
- 45.Dent R, Lindeman G, Clemons M, et al. Safety and efficacy of the oral PARP inhibitor olaparib (AZD2281) in combination with paclitaxel for the first- or second-line treatment of patients with metastatic triple-negative breast cancer: Results from the safety cohort of a phase I/II multicenter trial. Journal of Clinical Oncology. 2010;28:1018. [Google Scholar]
- 46.Kopetz S, Mita MM, Mok I, et al. First in human phase I study of BSI-201, a small molecule inhibitor of poly ADP-ribose polymerase (PARP) in subjects with advanced solid tumors. J Clin Oncol. 2008;26 Abstract 3577. [Google Scholar]
- 47.Mahany J, Lewis N, Heath E, et al. A phase IB study evaluating BSI-201 in combination with chemotherapy in subjects with advanced solid tumors. J Clin Oncology. 2008;26 Abstract 3579. [Google Scholar]
- 48.Rottenberga S, Jaspersa JE, Kersbergena A, et al. High sensitivity of BRCA1-deficient mammary tumors to the PARP inhibitor AZD2281 alone and in combination with platinum drugs. Proc Natl Acad Sci USA. 2008;105:17079–84. doi: 10.1073/pnas.0806092105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Edwards S, Brough R, Lord C, et al. Resistance to therapy caused by intragenic deletion in BRCA2. Nature. 2008;451:1111–6. doi: 10.1038/nature06548. [DOI] [PubMed] [Google Scholar]
- 50.Swisher E, Sakai W, Karlan B, Wurz K, Urban N, Toshiyasu T. Secondary BRCA1 Mutations in BRCA1-mutated Ovarian Carcinomas with Platinum Resistance. Cancer Research. 2008;68:2581–6. doi: 10.1158/0008-5472.CAN-08-0088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rodriguez A, Rimawi M, Wu M, et al. A BRCA1-Like, 25-gene Assay Predicts for Anthracycline-chemosenstivity in Sporadic Triple-Negative Breast Cance. 2009 San Antonio Breast Cancer Symposium; 2009. Abstract 110. [Google Scholar]
- 52.Comen E, Robson M. Inhibitionof Poly(ADP)-Ribose Polymerase as a Therapeutic Strategy for Breast Cancer. Oncology. 2010;24:55–68. [PubMed] [Google Scholar]