

Summary
The composition and activities of microbes from diverse habitats have been the focus of intense research during the past decade with this research being spurred on largely by advances in molecular biology and genomic technologies. In recent years environmental microbiology has entered very firmly into the age of the ‘omics’– (meta)genomics, proteomics, metabolomics, transcriptomics – with probably others on the rise. Microbes are essential participants in all biogeochemical processes on our planet, and the practical applications of what we are learning from the use of molecular approaches has altered how we view biological systems. In addition, there is considerable potential to use information about uncultured microbes in biodiscovery research as microbes provide a rich source of discovery for novel genes, enzymes and metabolic pathways. This review explores the brief history of genomic and metagenomic approaches to study environmental microbial assemblages and describes some of the future challenges involved in broadening our approaches – leading to new insights for understanding environmental problems and enabling biodiscovery research.
A brief history of marine microbiology
Over the past two decades there has been an explosion in understanding of how microbes – bacteria, protists and viruses – critically influence the structure of and function of the oceans. However, this was not always the case. The study of microbial communities in marine environment was a relatively minor aspect of oceanography during most of twentieth century. While epifluorescent microscopy of environmental samples confirmed that bacteria were very abundant (Hobbie et al., 1977), < 1–5% of bacteria could be grown in culture (Staley and Konopka, 1985) and were therefore very difficult to study. Specific filtration methods or the isolation and growth of a few organisms on a variety of different, and often complex, organic substrates were developed mostly for clinical microbiology, but these limited methods were not well suited to studying the vast complexity in natural environmental assemblages. Given this, many marine scientists turned to ‘black box’ ecological approaches (e.g. Redfield, 1958; Carlson et al., 2001) to infer microbial roles in biogeochemical cycles. However, which microbes were responsible for specific biogeochemical processes still remained largely a mystery. While the invention of the microscope over 350 years ago began to open up the richness of the microbial world, it is the development of DNA sequencing tools in the past 30 years that has truly changed how we understand microbial communities and their role in structuring ocean biogeochemical dynamics. We are finding that the extent of marine microbial biodiversity, and consequently natural products potential, seems to be limitless and growing larger as new techniques emerge to measure it.
How diverse is diverse?
The first phylogeny‐based studies of microbial life emerged from evaluation of the small subunit rRNA oligonucleotide studies in the labs of Norman Pace, David Stahl and Carl Woese (Lane et al., 1985; Pace et al., 1985; Stahl et al., 1985; Woese, 1987; Turner et al., 1989). Their advocacy for using rRNA for molecular phylogenetic analysis revolutionized how scientists looked at microbial diversity and evolution.
Initial findings were furthered to study microbial assemblages through the application of the polymerase chain reaction (PCR) (Mullis, 1983) to allow for direct culture‐independent surveys of natural prokaryotic microbial assemblages (e.g. 16S rRNA: Giovannoni et al., 1990; Ward et al., 1990; Britschgi and Giovannoni, 1991; Schmidt et al., 1991). These surveys led to discoveries of extraordinarily unexpected levels of phylogenetic biodiversity in natural systems (e.g. Pace, 1997; Head et al., 1998) and have resulted in a renewed interest in microbial ecology for biodiscovery (Glöckner and Joint, 2010). A case in point is one of the first applications of rRNA sequence analysis to study natural assemblages of bacterial lineages in the oceans (Giovannoni et al., 1990). They found that one group of closely related sequences was very abundant in the Sargasso Sea. This group was code‐named SAR11, and has subsequently been found in almost every marine province that has been sampled. SAR11 accounts for a very significant proportion of bacterioplankton in both surface and deep water (Morris et al., 2002) and has been described as the most abundant bacterium on the planet. Another key outcome of the application of PCR was to show that Archaea, which were originally thought to only inhabit extreme environments such as hot springs or hypersaline ponds, could readily be found in ocean environments (DeLong, 1992; Fuhrman et al., 1992).
There is no question that microbial diversity is vast, but what is the true extent of diversity in the oceans. It has been estimated that there are several hundred to several hundred thousand different phylogenetic species per ml of seawater with total diversity ranging between anything from 100 000 to more than a million (Curtis et al., 2002; Huber et al., 2007). What we have learned about global microbial diversity in the past decade from taxonomic gene surveys far exceeds our previous estimations. Furthermore, we have seen tremendous genome wide diversity within taxonomically identical phylogenetic groups. Even after two decades of concerted efforts to phylogentically characterize microbial diversity, the discovery of novel forms is common. The microbial species definition, despite its eminent practical significance for identification, diagnosis and diversity surveys, remains a very difficult issue to advance (e.g. Hughes et al., 2001; Ward, 2002).
Currently, there are two common approaches to measure diversity. First is a phylotype approach, which is based on assigning taxonomy based on results from an alignment to a sequence in a database that has the most statistically significant similarity a priori. Significant challenges with this approach are that database searches may come back as undefined and that different databases (e.g. ribosomal database project II, SILVA, GreenGenes, Bergey's culture db, NCBI's nt or nr) can yield different taxonomies, many of which are based on ‘copycat’ assignation whereby researchers have blindly accepted the taxonomic assignation of the most homologous sequence in the database. On the other hand, one can define diversity in terms of an ‘unsupervised’ operational taxonomic unit (OTU). In this approach the user defines the taxonomic unit (e.g. peak in ARISA analysis, length and percent similarity criteria), and we allow the sequences to form their own groups during a computational cluster analysis process. Once bins are formed, a representative sequence from each bin is used to determine taxonomy.
With either approach, diversity also depends on which gene or which part of a gene is used in the analysis. For example, a massively parallel tag 454 pyrosequencing approach was used to evaluate the ‘rare biosphere’ diversity of 16S rRNA amplicons (Sogin et al., 2006). The technique uses internal primer sequences to produce restriction‐digest overhanging sequences, which greatly improves the efficiency of serial analysis of ribosomal sequence tags – SARST (Kysela et al., 2005). This produced ∼600 000 unique bacterial OTUs based on actual counts of sequence types from this study rather than from extrapolated estimates (calculations made using the ICoMM database of ∼40 marine microbial 16S rRNA V6454 pyrosequencing projects – Pers. Comm. Sue Huse). Using this methodology, Huber and colleagues (2007) also analysed > 900 000 16S rRNA amplicons to evaluate hydrothermal vent microbial population structure.
A global survey of 18S rRNA marine protistan eukaryotic genes from diverse marine ecosystems was conducted using two fundamentally different high‐throughput sequencing approaches on similar samples: traditional Sanger ABI full length 18S rRNA sequencing (> 12 000 contigs) and 454 pyrosequencing focused on the hyper‐variable V9 region of 18S rRNA genes (> 276 000 short sequence tags). The Sanger approach (∼1000 sequences per sample) yielded non‐parametric OTU diversity estimates (e.g. Chao1, ACE1) that were substantially higher than preliminary diversity analyses that were performed several years earlier on some of the same samples but with many fewer sequences (∼150). The pyrosequenced V9 data revealed a substantial amount of ‘new’ 18S diversity in the samples (D.A. Caron and P.D. Countway, unpubl. data). While pyrosequencing approaches can overestimate taxonomic richness as a result of PCR and sequencing errors (e.g. Quince et al., 2009), these studies show that novel microbial diversity can be found at every taxonomic level. However, even with exciting new technology, we are still massively under‐sampling the diversity in marine ecosystems!
Genome‐based approaches
About the same time that PCR was becoming more commonly used, technology for DNA sequencing of entire genomes was advancing with the Sanger ABI sequencer platform, and the first free‐living bacterium to have its entire genome sequenced was published (Fleischmann et al., 1995). Of course, the ultimate genome project has surely been the 13‐year effort to sequence the 3 billion base pair human genome (Lander et al., 2001). The large investment in resources and people ($2.7 billion 1991) was paired with a period of rapid development in the technology of sequencing. The chemistry involved was basically the same as that originally developed by Sanger and colleagues (1977), but the technology became hugely automated, largely due to the parallel non‐governmental effort led by Celera Corporation (Venter et al., 2001). Similarly, corresponding advances in computational infrastructure and bioinformatics – making sense of sequence data – made it feasible to pair together millions of individual sequence reads by massively parallel pairwise comparison techniques. Applications of these new methodologies helped spur on an explosion in the study of microbes and microbial communities.
Since 1995, the number of sequenced microbial genomes deposited in public domains has grown tremendously (Fig. 1). Originally sequencing efforts were prioritized to study cultured pathogens, bacteria with specific industrial/bioremediation applications or limited representatives of deep branching organisms. More recently, there has been an exciting expansion to target environmentally important microbes from all domains of life (archaea, bacteria and eukarya). Facilitating programs of particular note are the Gordon and Betty Moore Foundation's Marine Microbial Sequencing Program (http://www.Moore.org/) and the US Department of Energy's Joint Genome Institute (DOE JGI) microbial genomics program, including the Genomic Encyclopedia of Bacteria and Archaea (geba) project (Wu et al., 2009). The expansion of these and other efforts to target environmentally important microbes has not only improved annotation of functional genes but also has resulted in discovery of new processes and functions, possibly those related to bioenergy production, global carbon cycling and bioremediation. For example, Wuchter and colleagues (2006) isolated a culture of a marine Crenarchaota from a seawater aquarium and discovered that it was capable of ammonium oxidation, showing an important unknown nitrification role for Archaea. Previously, it had been assumed that only Bacteria performed the process of nitrification in the oceans. Ammonium oxidizing and nitrite oxidizing bacteria had been well characterized in soils, and similar bacteria were found in the sea. However, Wuchter and colleagues (2006) were able to take their knowledge from the cultured Archaea to target Crenarchaota from the North Sea and demonstrate that its distribution and abundance were correlated with ammonium oxidation to nitrite. Furthermore, the abundance of the gene encoding for the archaeal ammonia monooxygenase A subunit (amoA) was 1–2 orders of magnitude higher than the equivalent gene in nitrifying bacteria. These and other studies are providing important opportunities for comparative genomics, interpretation of environmental genomic read data and better understanding of microbial evolution. At the time of writing (early 2010), there are currently 1128 completed and 4297 ongoing bacterial or archaeal genome projects in the GOLD online database (December 2009; http://www.genomesonline.org/) (Fig. 1).
Figure 1.
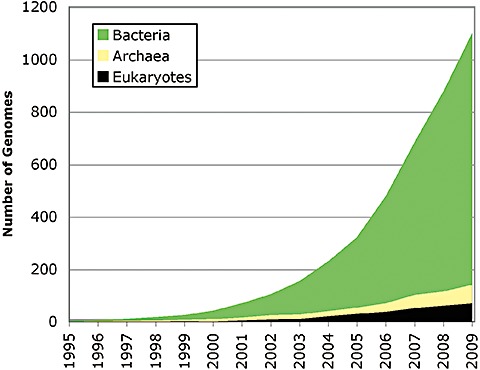
Publicly available completed reference genomes. Data reported by publication date, or if not published, the date that genome data was deposited into NCBI GenBank Data repository (http://www.ncbi.nlm.nih.gov/sites/genome). Data presented represent a total of 1106 genomes (966 Bacteria and 70 Archaea and 70 Eukaryotes). Eukaryotic data is for full or draft genomic data only and does not include mitochondrial or plasmid projects.
Cultured microbes remain an important resource as they provide a mechanism to help pair genetic potential with physiology. However, challenges still remain for culture‐based approaches, and there are efforts underway to improve cultivation and sequencing technologies (e.g. Connon and Giovannoni, 2002; Zengler et al., 2002; Hamaki et al., 2005; Stott et al., 2008; Joint et al., 2010). The most abundant environmental microbes are often the most difficult to grow in the laboratory. For example, it took more than a decade before SAR11 cultures were established. Rappé and colleagues (2002) used a dilution approach to isolate the slow growing SAR11 bacterium and named it ‘Candidatus Pelagibacter ubique’. Even so, the cultures do not grow well, and typically cell densities of only a few million per ml are achievable. Such low biomass in cultures is challenging for complete taxonomic characterization or meaningful physiological studies for this and other hard‐to‐culture organisms.
Linking function to taxonomy in environmental samples
Microbes (used in the largest sense to include archaea, bacteria, single celled eukaryotes, and viruses) are essential participants in virtually all biogeochemical processes on our planet. One of the biggest challenges is the need to integrate the basic science of biodiversity discovery (a listing of genes or organisms within an environment) with an approach to understand functional properties, multi‐scalar responses and interdependencies that connect microbial and abiotic ecosystem processes. One of the first approaches to explore potential function was through the development and sequencing of larger insert clone libraries to link functional genes to known taxonomic markers found within the same insert. Stein and colleagues (1996) cloned a fosmid vector (∼40 kb) of environmental Archaea DNA into Escherichia coli. The library was then screened for clones with phylogentic rRNA markers, and target clones were fully sequenced to search for functional genes flanking the marker. Use of larger‐insert libraries has further been developed for both targeted ecological studies of higher diversity samples (e.g. soils: Rondon et al., 1999; Rondon et al., 2000) and for natural product screening (e.g. Handelsman et al., 1998; Schleper et al., 1998). For example, Schleper and colleagues (1998) demonstrated newly discovered variant heterogeneity within an endosymbiotic Crenarchaeota consistently found within sponges. Béjà and colleagues (2000a) used a bacterial artificial cloning (BAC) insert library (∼80 kb) to isolate metagenomic DNA from marine bacteria. BAC libraries also were found to be particularly effective for obtaining information about the largely uncultured Archaea, the third domain of life. Screened fosmid libraries generated from Antarctic and deep waters of the temperate Pacific Ocean samples, revealed high levels of phylogentic diversity in the Archaea and further characterized the marine Crenarchaota (Béjàet al., 2002a).
Although Kolber and colleagues (2001) analysed the fluorescence properties of natural phytoplankton assemblages and discovered that there were organisms present that contained bacteriochlorophyll, light‐harvesting proteorhodopsin and bacterial rhodopsin genes were not thought to be overly important in marine systems. Subsequently, Béjà and colleagues (2000b; 2002b) discovered that they occurred widely in marine bacteria and archaea in the upper, photic ocean by using a screened large‐insert library. Béjà and colleagues (2002b) analysed the structure of photosynthetic gene content, suggesting that indeed novel phototrophic bacteria in the oceans that did not evolve oxygen during photosynthesis but, unlike most known photosynthetic bacteria, were able to grow aerobically. This pigment was previously believed to be confined to photosynthetic bacteria that could only undertake photosynthesis in anoxic conditions, using sulfide rather than water as electron donor. Since the majority of the oceans are not anoxic, the presence of bacteriochlorophyll was a surprise. A subsequent genomic analysis by Fuchs and colleagues (2007) characterized some of these organisms as the marine gammaproteobacterium, Congergibacter litoralis, a novel phototrophic bacteria, in the oceans that does not evolve oxygen during photosynthesis and could grow aerobically. Proteorhodopsins are not only involved in photosystems but also can fully replace respiration as a cellular energy source in some environmental conditions (Walter et al., 2007). Frigaard and colleagues (2006) later showed that a single lateral transfer event would be enough for other bacteria to acquire the ability to utilize light. Indeed, it has been shown that microbes that maintain proteorhodopsin genes can achieve higher cellular yields when cultured in light than in darkness (Gomez‐Consarnau et al., 2007). This may explain why proteorhodopsin and bacteriorhodopsins are so widely distributed in Bacteria and Archaea in the oceans.
Subsequent studies have used phylogenetic classification of annotated genes within a fosmid insert to infer the taxonomy of the original genome, and additionally to identify gene‐clusters, which occurred from horizontal gene transfer (Nesbøet al., 2005). Fosmid clones from Antarctic coastal waters that had 16S rRNA taxonomic marker were found to have sequences that are not closely related to cultured bacteria. The linked fosmid amino acid sequences also showed variations in protein sequences that are consistent with adaptation to the sub‐zero environment (e.g. Béjàet al., 2002a; Grzymski et al., 2006). A fosmid library was also used to show that there are highly variable regions flanking the 16S rRNA gene that suggested higher diversity in the SAR11 group than would be predicted by analysis of the 16S rRNA gene alone (Gilbert et al., 2008a). This variability may provide a mechanism for adaptability of this highly ubiquitous group to a variety of habitats. These and other studies show that the larger insert library analysis provide useful perspective on the physiological potential of abundant but uncultivated microbes.
Metagenomics: community sequencing approaches
In the early 2000s, shotgun sequencing techniques developed for whole genome sequencing were further adapted to sequencing entire communities of organisms, in a process called metagenomics (reviewed by Handelsman et al., 2007, Hugenholtz and Tysen, 2008). Directly sequencing environmental DNA from communities inhabiting a common environment has provided new opportunities to obtain relatively unbiased views of not only community structure but community metabolic potential. One of the first demonstrations of the viability of this strategy was by Tyson and colleagues (2004), who sequenced a community of DNA from a very low diversity microbial biofilm in an acid mine drainage system. The greatly reduced Bacteria and Archaea diversity provided an excellent test of the approach of high‐throughput community genomics sequencing. Tyson and colleagues were able to reconstruct almost complete genomes of two bacterial groups, Leptospirillum group II and Ferroplasma type II, and were also able to partially describe three other genomes. Their results provided key information on ecosystem function for management and system remediation.
In contrast, Venter and colleagues (2004) used similar technology applied to a more complex, highly diverse system in an attempt to characterize oceanic microbial assemblages from the Sargasso Sea, near to Bermuda. Venter's team obtained greater than one billion non‐redundant base pairs from Sanger‐based shotgun paired end sequences and used complex bioinformatics approaches to conclude that there were at least 1800 different genomes (i.e. different organisms) with 48 unknown bacterial phylotypes and 1.2 million previously unknown genes. Significantly, their shotgun sequence analysis discovered a vast array of new proteorhodopsin‐like genes found well outside groups of proteobacteria, where they had previously been discovered (Béjàet al., 2000b). These and other metagenomic studies demonstrated conclusively that the techniques that had been developed to sequence individual genomes could also be used with environmental samples. Surprisingly, metagenomic analyses using Sanger ABI sequencer reads, depending on the library or sample, have typically resulted in up to 60% of the sequence reads having no known homologues. With approximately 1 million bacteria per ml of sea water and an estimated average genome size of 2 million bp, the Sargasso Sea project only sequenced 0.05% of the genomic information in a single ml – only a drop in the proverbial ocean. The genomic heterogeneity of the marine ecosystem is so large that there are currently more unknown predicted genes than known sequences, again highlighting the potential of marine microbes as sources of novel genes, enzymes and functions – ideal targets for biodiscovery. This is also clear indication that existing databases contain only a small fraction of the huge quantity of genetic information that resides in the global ecosystem, and additionally, that our comprehension of cellular biochemistry is still in its infancy.
Subsequent to the 2004 Sargasso Sea study, the J. Craig Venter Institute group initiated a global transect study of marine microbial diversity (referred to as the Global Ocean Sampling, GOS Expedition). The first samples were taken from a several thousand mile transect from the North Atlantic to the South Pacific, via the Panama Canal. Sequencing of these samples yielded 6.3 billion base pairs, from 7.7 million sequence reads (Rusch et al., 2007). The massive dataset also required the development of several new bioinformatics analysis tools, which can be divided into two separate analytical techniques. The first was a genome‐orientated approach that helps to define population genetics. Rusch and colleagues (2007) linked observed data to the genomes of known organisms in a process called ‘fragment recruitment’. Sequenced DNA reads were recruited to completed genomes from cultivated organisms and were evaluated for their percentage nucleotide identity to determine the genomic diversity of ecotypes in each ecosystem. This approach makes the analysis tractable with what we know about microbial ecology from sequenced genomes of laboratory cultures and studies on cellular physiology.
The second technique was comparison of richness and composition of functional genes between different samples; this does not rely on sequenced genomes or cultured representatives per se (although invariably, function for a gene is derived from cultured isolates). This approach is loosely a community ecology dynamics approach that relied on a statistical analysis of the frequencies of sequence reads associated with specific functions in specific ecosystems, i.e. how did photosynthetic processes change between ecosystems. To implement this strategy Rusch and colleagues (2007) used a technique called ‘extreme assembly’ enabling the assembly of large non‐clonal segments of abundant organisms. Both approaches relied heavily on the limited databases of sequenced genomes or functional assigned proteins, and so both techniques are heavily biased by previous studies. By ignoring annotation and exploring homologue dynamics in comparative metagenomics can we make full use of the un‐biased datasets to evaluate similarities between ecosystems – although this limits ecological interpretation.
Overall, the GOS survey found that taxonomic diversity and gene diversity were much higher than expected, with a large proportion of the assembled sequences (85%) unique at a level of 98% sequence identity when compared between individual samples. Although primarily a gene‐hunting expedition, which may have been influenced by the potential for biodiscovery, this groundbreaking study has been used to uncover incredible insights into global marine microbial biodiversity (e.g. Raes and Bork, 2008). An analysis of the 16S rRNA and other genes in the GOS shotgun survey showed that there were clear patterns of biogeography for many microbial species and gene variants analysed. For example, Yutin and colleagues (2007) analysed the GOS dataset to assess the abundance and spatial distribution of aerobic anoxygenic photosynthetic bacteria and showed that anoxygenic photosynthetic bacteria are an important component of the bacterioplankton in some regions but less important in others. Prior to 2001, it was also assumed that photosynthesis in the oceans was only carried out by cyanobacteria and eukaryotic phytoplankton, which have chlorophyll a (or in the case of the marine cyanobacterium Prochlorococcus, divinylchlorophyll – a very close derivative) as their primary photosynthetic pigment.
These and other environmental shotgun analyses of DNA have also provided novel information about proteins and protein families from populations of largely uncultured marine microbes. Yooseph and colleagues (2007) used sequence similarity clustering to investigate the putative proteins derived from the GOS sequence database. In a large‐scale bioinformatics undertaking, they compared protein sequences from GOS (6.12 million proteins were predicted from 7.7 million GOS sequences) with databases of nearly all known protein families, by clustering proteins into groups or ‘families’. As with the DNA sequences, a large number of protein clusters were entirely novel. Of a total of 3995 medium‐ and large‐sized clusters from the GOS sequences, 1700 had no homology to known families. Yooseph and colleagues (2007) suggested that some of these proteins might be of viral origin because so little is know about marine viruses and their proteins.
These large metagenomic microbial community datasets clearly offer considerable potential for novel gene biodiscovery research (reviewed by Brady et al., 2009) and holistic comparison of microbial community dynamics. However, it is remarkably difficult to correlate information from metagenomic datasets back to individual cells, species or even small populations of species inhabiting any particular habitat – and to understand the functional context and biogeochemical consequence for their activities. The three biggest limitations of community metagenomic approaches are (i) that the persistently poor sequence coverage means that only the most dominant organisms yield useful sequence assemblies; (ii) that the inability to accurately annotate metagenomic fragments means ecological interpretation becomes problematic; and (iii) that the lack of functional verification for genes that do not have recognizable homology with biochemically characterized proteins means that any analysis lacks reliability. The problem is akin to boiling dinner leftovers in a pot for 24 h, pureeing heavily and then trying to attribute any spice or stew fragment back to the original dish or constituent from which it derived. What we are learning via large‐scale whole‐community metagenomics is important; however, a major problem that remains is in gaining information that can be put into truly meaningful environmental and ecological context that relates to cellular function and activity – the understanding of physiology in an environmental context.
The overlooked microbes
To this point, we have mostly focused on the Bacteria and the Archaea. Some recent studies are also revealing the once‐unimaginable diversity of single cell marine microbial eukaryotes (protists) and fungi, and we would be remiss to not acknowledge these less‐well characterized members of microbial assemblages. Microbial eukaryotes are capable of multiple nutrient and energy acquisition mechanisms and play important roles in ocean food webs and biogeochemical cycling (Caron et al., 2009a). Research during the 20th century established that protists account for most of the conversion of bacterial productivity (Sanders et al., 1992; Sherr and Sherr, 2002). Yet the specific contribution of microbial eukaryotes to environmental systems is often overlooked. The composition, temporal and spatial dynamics, and possible biogeochemical activities of marine eukaryotic communities are now emerging as important, and undercharacterized, topics in marine and environmental sciences and biogeochemistry (Edgcomb et al., 2007; Teske, 2007; Caron et al., 2009b). Applications of molecular techniques to communities of microbial eukaryotes have lagged that of prokaryotic communities attributed to several logical explanations. First, little morphological or physiological data have been obtained for abundant organisms, as similar to the prokaryotes, most fail to grow in culture. Eukaryotes also have larger genomes (10–10 000 times that of prokaryotes) that are less gene‐dense than those of bacteria and archaea, making it more difficult to undertake whole genome sequencing projects or environmental metagenomic sequencing efforts. Eukaryotic gene density is about one gene every 1.3 kb in the smallest free‐living protist to 1 gene per 80 kb in humans. Finally, the complexity of eukaryotic genomes can be daunting when attempting metagenomic approaches, as eukaryotic ribosomal DNA sequences are more often tandomly repeated in genomes making it difficult to acquire meaningful, quantitative information on functional genes (reviewed by Caron et al., 2009a). As a result, most molecular work to date investigating environmental protistan ecology has been limited to 18s rRNA taxonomic diversity surveys. These surveys have revealed unexpected diversity and clarified some of the phylogenetic relationships of protists in the environment (e.g. Díez et al., 2001; Moon‐van der Staay et al., 2001), but this field is ripe for further exploration.
There are far fewer reference genomes available for microbial eukaryotes than prokaryotes (Fig. 1), and this lack of genome data and unidentified sequence data further limits the interpretation of novel environmental genomic databases and results additional uncurrated data in public databases. A better understanding of microbial eukaryotes' specific functional roles and biogeography is vital to our understanding of microbial communities and of evolution of multi‐cellular taxa.
The viral fraction
The smallest entities within microbial communities – viruses – have large potential impacts on microbial communities. While viruses were one of the first communites studied using metagenomics (reviewed by Hugenholtz and Tysen, 2008), in general, viral communities have been less studied despite their significant role in microbial ecology and evolution (e.g. Weinbauer, 2004: Sullivan et al., 2005; Breitbart et al., 2007; 2008). Viruses are the most abundant biological entities on our planet with typical abundances in ocean surface water at 107 ml−1 (Bergh et al., 1989). Metagenomic analysis has confirmed significant interactions between viruses and their hosts that impact several important biological processes in natural systems, such as horizontal gene transfer, microbial diversity and biogeochemical cycling (e.g. Williamson et al., 2008; Banfield and Young 2009). Through cycles of infection, replication and host cell lyses, phages impact multiple pathways and processes involved in host population biology and ecosystem functioning (e.g. Sullivan et al., 2003). Viral diversity projects have been limited as a direct result of the lack of universal marker genes from which total viral communities can be assessed and compared in a single study, although this is being circumnavigated through the use of functional metagenomic studies (reviewed by Edwards and Rohwer, 2005). Understanding the dynamic relationship between microbes and viruses deserves significant attention in the future. The expanding number of environmental viral sequences in public databases through next generation sequencing platforms (see below), paired with information from laboratory challenge experihments, will undoubtedly help this field move forward.
Scientific discoveries through technological advancement
High‐throughput sequencing has historically been undertaken using Sanger ABI sequencing platforms, but more recently there has been a fundamental shift away from this platform to less expensive, ‘next generation’ sequencing (NGS) platforms. Despite some cloning related biases (Béjàet al., 2000; Temperton et al., 2009), Sanger dideoxy sequencing is still the standard for read length and sequence accuracy (Bonetta, 2006); however costs make large‐scale sequencing on this platform prohibitively expensive. In 2005, two new sequencing platforms were introduced: sequencing‐by‐synthesis or pyrosequencing, developed by 454 Life Sciences (Margulies et al., 2005) and the multiplex polony sequencing protocol of George Church's lab (Shendure et al., 2005). Both NGS sequencing technologies utilize less expensive massively parallel sequencing approaches and yield far more read volume sequence output at a lower cost than Sanger‐based sequencing or microarray technology (Table 1).
Table 1.
Summary of Roche 454, Illumina GA and ABI SOLiD seqeuncing capabilities.
| Roche 454 FLX Titanium | Illumina Solexa GA | ABI SOLiD 3 | |
|---|---|---|---|
| Reads per run (M) | 1.25 | 250 | 320 |
| Average read length (bp)a | 330 | 75 or 100 | 50 |
| Usable reads that pass quality filtersb | > 99.5% | 55% | 35% |
| Raw accuracy readsc | 96.0–97.0% | 96.2–99.7% | 99.0 to > 99.9%d |
| Primary bias | Homopolymer read errors | short read length | short read length |
| Biases in eukaryotic sequencingb | Minimal low coverage of AT‐rich regions | Low coverage in AT‐rich repetitive regions | Low coverage in AT‐rich repetitive regions |
| Amplicon overrepresentation in 50 bp end regionsb | 5% | 56% | 11% (after amplicon end removal) |
| Saturating level of redundant sequence coverageb | 43× | 188× | 841× |
Reviewed by Metzker (2010).
Reviewed by Harismendy and colleagues (2009).
Reviewed by Chan (2009).
Higher accuracy achieved by reading each base twice in a two‐base encoding scheme.
The NGS platforms have revolutionized how we obtain genetic information from microbial communities. Previously unaffordable targeted studies evaluating composition and functional potential of microbial communities are now quite common. Currently, the three most used NGS platforms are the 454 pyrosequencer (Roche Diagnostics Corporation, Branford, CT, USA), Illumina (Illumina, San Diego, CA, USA) and SOLiD (Life Technologies Corporation, Carlsbad, CA, USA). While these platforms have enormous breadth in applications, including the newer approaches of ChIP‐seq, transcriptome (Craft et al., 2010), microRNA discovery (Shi et al., 2009; Yanmei et al., 2009) and whole genome resequencing (e.g. Blazewicz et al., 2009), usability of the outputted reads varies significantly when compared with a Sanger ABI sequencing read length of ∼800 bp and accuracy rate of 99.0% to 99.999% (Table 1). A detailed review is provided by Metzker (2010).
Each of the newer platforms also has specific biases, which must be considered when evaluating the appropriateness of the technology for a given study (reviewed by Harismendy et al., 2009). For example, 454 pyrosequencers do not resolve homopolymer DNA segments well, while other NGS platforms output very short read lengths. Strategies combining platform data analysis and advances in technologies are circumventing some of these challenges for many approaches (e.g. 800 bp read lengths are expected on the 454 platform later in 2010).
An increasing number of publications have used 454‐Pyrosequencing for analysis of environmental DNA and RNA (> 600 as of January 2010, for example, Angly et al., 2006; Edwards et al., 2006; Leininger et al., 2006; Liu et al., 2007; Roesch et al., 2007; Wegley et al., 2007; Desnues et al., 2008; Dinsdale et al., 2008; Frias‐Lopez et al., 2008). Increasingly, 454‐pyrosequencing technology has also been used to evaluate changes in gene transcription (expression) of a community through mass‐sequencing of the metatranscriptome (e.g. Leininger et al., 2006; Frias‐Lopez et al., 2008; Gilbert et al., 2008b; Urich et al., 2008; Hewson et al., 2009; Poretsky et al., 2009; Shi et al., 2009).
Shortly, third‐generation sequencing technologies, such as Heliscope (Helicos Bioscience Corporation, Cambridge, MA, USA), Complete Genomics (Complete Genomics, Mountain View, CA, USA), SMRT (Pacific Biosciences, Menlo Park, CA, USA), Ion Torrent Semiconductor Sequencing (San Francisco, CA, USA) and Roche GS‐FLX Junior, will further increase access and usher in a new dawn in our capability – including the potential to completely sequence all genomes from all organisms in a community. With this toolbox at our disposal it is entirely possible that the dream of understanding the genomic potential and transcriptional response of entire communities will become a reality.
Much of the recent gain provided by access to NGS platforms is being offset by increased costs and efforts on the bioinformatics front (Fig. 2). Dropping sequencing costs and better access to sequencing platforms by scientists working in a variety of fields is now producing data at a prodigious rate, and the volume of sequence data entering public domains is staggering. Genomic datasets are taxing the computational infrastructure and computational community, and significant limitations now lie with bioinformatic tools – the programs and the computer processing power necessary to deal with massive datasets. For a given analysis, a general first step is to use a basic local alignment search tool (blast) to compare partial gene reads with all existing sequences in the database. blast has processing times that scale linerally with input size, and the amount of sequence data being generated is doubling at a much faster rate than computational infrastructure. This is one of the most significant issues facing our field today, and improvements in infrastructures are needed to better proceed from (meta)genomic sequence information to biochemical and physiological function prediction and ecosystem understanding (Wilkening et al., 2009). Another major bioinformatics hurdle lies is the current difficulty in assembling short genomic reads into larger contiguous elements. If this could be done more reliably, it would provide greater understanding of the genetic context of sequences and help to recreate the diverse genomic elements found in natural ecosystems. Cloud computing and other newer technologies are emerging to address many of these computational needs for NGS data analysis.
Figure 2.
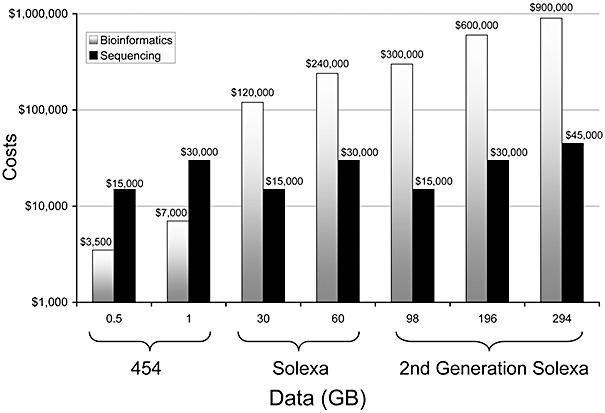
Challenges for processing genomic sequence data and Moore's law. Moore's law describes a relationship in the rate that computational infrastructure increases – basically a doubling approximately every two years. Genomic data in public domains is growing faster than the computational technological capacity for processing. Costs for blast analysis are presented in Amazon EC2 units and do not include storage or transfer costs [Figure modified from F. Meyer (IGSB, Argonne National Lab), with data from Wilkening et al., 2009].
A return to the basics to move forward
While there is still a need to expand metagenomic and other ‘‐omic’ efforts for exploring marine habitats (e.g. especially underexplored habitats like brines, deep sea and high latitude ecosystems) and organisms (e.g. viruses, protists and marine microbial symbionts), there is also a need to pair environmental work with carefully controlled laboratory studies (reviewed by Heidelberg et al., 2008). In some regards, large‐scale environmental sequencing programs have resulted in a regression back to treating communities of microbes as a black box (as previously discussed). Fine scale detail has been lost in a sea of sequence information that focuses not on the individual organisms but on the presence or absence of potential genes involved in a given function. There is now a strong need for coordination of disparate fields and the merging of the traditional methodologies to begin to evaluate microbial function under differing environmental conditions (e.g. Zehr and Ward, 2002).
First, environmental levels of microbial biodiversity is still not reflected in bacterial culture collections, and considerable challenges remain to bring into culture those bacteria that are abundant in natural environments (Joint et al., 2010). Even after 15 years of genome sequencing, the lack of completed ‘reference’ genomes poses an obstacle in interpreting environmental sequence data, determining ecological roles of the uncultured microorganisms and in deconstructing microbial metabolic pathways. Complete genomes provide information on the full biochemical potential of an organism (genome, plasmids, etc.) and also provide a template to allow directed biochemical analysis of specific gene targets, which will hopefully accelerate our future understanding of the cell systems. The inability to culture the vast majority of bacteria pose significant challenges for microbial ecologists and our ability to understand how microbes shape the natural environments. Rappé and Giovannoni (2003) reviewed microbial biodiversity based on comparisons of 16S rRNA genes with what was known from cultured bacteria whose taxonomy was fully characterized. At that time, molecular approaches had identified 52 major lineages. The number of major bacterial groups has continued to increase – at time of writing there are 61 recognized bacterial phyla comprising over 100 proposed unique lineages – with only 40% having a cultured representative (Fig. 1). Hence, laboratory cultures still remain an important tool to understand physiology of a microbe (Joint et al., 2010), and consequently to investigate the physiology and mode of infection for associated viruses. For organism that we still cannot culture, pairing existing sequencing technology with newly developing sophisticated techniques provides new opportunities for genome analysis (Fig. 3). For example, fluorescent‐activated cell sorting (FACS) (Brehm‐Stecher and Johnson, 2004) can be used to enrich samples and can sometimes provide enough biomass for direct DNA or RNA sequencing. FACS or other techniques can also be used to obtain single cells for whole‐genome amplification by multiple strand displacement amplification (MDA) (e.g. Hosono et al., 2003; Woyke et al., 2009). The challenges with this process include overcoming issues of differential cell lysis, problems with contamination DNA from another cell or from reagents, MDA bias, and potential issues with chimaric sequences. Preliminary tests of this method have shown that 80–95% of the genome can be recovered, when measured against a reference genome assembly (Woyke et al., 2009). Consequently, this procedure works best when paired with another genome sequencing process. When further developed, enhancing basic metagenomic approaches with complimentary technologies, such as single cell sorting or targeted enrichment for a specific feature of the population, provides tremendous future promise for increasing our abilities to study ecology and potential function of uncultured microorganisms (Ishoey et al., 2008; reviewed by Warnecke and Hugenholtz, 2007).
Figure 3.
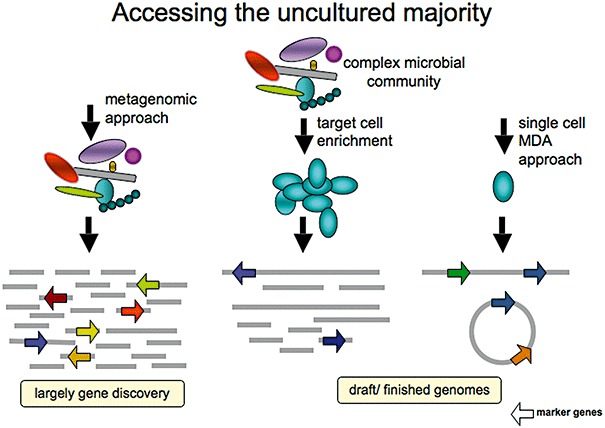
Schematic representation of sample handling options for metagenomic studies. One strategy is to concentrated (often after pre‐filtration to remove larger eukaryotic organisms), and the total DNA or RNA extracted for shotgun or large‐insert library construction. A second strategy is to enrich the sample for a particular community member through specific culturing techniques or through FACS. Most recently, single cell approaches are being attempted to clone and sequence the uncultured majority (Figure courtesy of Tanja Woyke, DOE Joint Genome Institute).
Second, while the application of genomic and metagenomic approaches is still valuable for evaluating diversity for within and between ecosystem comparisons, our challenge is now to use these new technologies to frame questions in environmentally relevant scales and across important gradients of space and time. Exploding databases holds enormous promise for refining concepts of microbial biodiversity co‐evolution of life and understanding environmental function when used in well‐planed research programs and combined approaches. Just as high‐throughput DNA sequencing has brought big changes in genomic and biodiscovery research, other high‐throughput technologies will do the same for areas such as gene expression, protein and metabolic characterization (reviewed by Warnecke and Hugenholtz, 2007).
The potential to now be able to pair (meta)genomic analysis with other approaches to evaluate gene expression and protein production offer a great opportunity for future ecological studies. Such observations may yield unprecedented discoveries, and change our perception of community structure and function in environmental systems. Paired approaches will also offer an unprecedented opportunity for gene discovery and biotechnological advance. Approaches like these will allow for refining understanding of basic relationships between community diversity and ecosystem function and provide important opportunities to gain a predictive understanding of the response of ecosystems in the face of environmental change.
Acknowledgments
This work is supported by National Science Foundation (NSF) grants MCB 0732066 and EF 0626526 to K.B.H. and by a Natural Environment Research Council (NE/C507902/1) award to I.J. The topic relates to a part of the core research programme of the Plymouth Marine Laboratory, a collaborative centre of NERC. K.B.H. and I.J. also acknowledge funding from the European Commission and NSF to attend the Joint EC‐US CIESM Workshop on Marine Genomics: At the Interface of Marine Microbial Ecology and Biotechnological Applications in October 2008 in Monaco. This meeting and the resulting report provided the stimulation for the development of this manuscript (http://ec.europa.eu/research/biotechnology/ec‐us/docs/monaco.pdf).
References
- Angly F.E., Felts B., Breitbart M., Salamon P., Edwards R.A., Carlson C. The marine viromes of four oceanic regions. PLoS Biol. 2006;4:2121–2131. doi: 10.1371/journal.pbio.0040368. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banfield J.F., Young M. Microbiology. Variety the spice of life – in microbial communities. Science. 2009;326:1198–1199. doi: 10.1126/science.1181501. [DOI] [PubMed] [Google Scholar]
- Béjà O., Suzuki M.T., Koonin E.V., Aravind L., Hadd A., Nguyen L.P. Construction and analysis of bacterial artificial chromosome libraries from a marine microbial assemblage. Environ Microbiol. 2000a;2:516–529. doi: 10.1046/j.1462-2920.2000.00133.x. et al. [DOI] [PubMed] [Google Scholar]
- Béjà O., Aravind L., Koonin E.V., Suzuki M.T., Hadd A. Bacterial Rhodopsin: Evidence for a new type of phototrophy in the sea. Science. 2000b;289:1902–1906. doi: 10.1126/science.289.5486.1902. et al. [DOI] [PubMed] [Google Scholar]
- Béjà O., Koonin E.V., Aravind L., Taylor L.T., Seitz H., Stein J.L. Comparative genomic analysis of archaeal genotypic variants in a single population and in two different oceanic provinces. Appl Environ Microbiol. 2002a;68:335–345. doi: 10.1128/AEM.68.1.335-345.2002. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Béjà O., Suzuki M.T., Heidelberg J.F., Nelson W.C., Preston C.M., Hamada T. Unsuspected diversity among marine aerobic anoxygenic phototrophs. Nature. 2002b;415:630–633. doi: 10.1038/415630a. et al. [DOI] [PubMed] [Google Scholar]
- Bergh O., Borsheim K.Y., Bratbak G., Heldal M. High abundance of viruses found in aquatic environments. Nature. 1989;340:476–468. doi: 10.1038/340467a0. [DOI] [PubMed] [Google Scholar]
- Blazewicz J., Bryja M., Figlerowicz M., Gawron P., Kasprzak M. Whole genome assembly from 454 sequencing output via modified DNA graph concept. Comput Biol Chem. 2009;33:224–230. doi: 10.1016/j.compbiolchem.2009.04.005. et al. [DOI] [PubMed] [Google Scholar]
- Bonetta L. Genome sequencing in the fast lane. Nat Methods. 2006;3:141–147. [Google Scholar]
- Brady S.F., Simmons L., Kima J.H., Schmidt E.W. Metagenomic approaches to natural products from free‐living and symbiotic organisms. Nat Prod Rep. 2009;26:1488–1503. doi: 10.1039/b817078a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brehm‐Stecher B.F., Johnson E.A. Single‐cell microbiology: tools, technologies, and applications. Microbiol Mol Biol Rev. 2004;68:538–559. doi: 10.1128/MMBR.68.3.538-559.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breitbart M., Middelboe M., Rohwer F. Marine viruses: community dynamics, diversity, and impact on microbial processes. In: Kirchman D., editor. 2nd. John Wiley and Sons; 2008. pp. 443–479. [Google Scholar]
- Breitbart M., Thompson L.R., Suttle C.A., Sullivan M.B. Exploring the vast diversity of marine viruses. Oceanogr. 2007;20:135–139. [Google Scholar]
- Britschgi T.B., Giovannoni S.J. Phylogenetic analysis of a natural marine bacterioplankton population by rRNA gene cloning and sequencing. Appl Environ Microbiol. 1991;57:1707–1713. doi: 10.1128/aem.57.6.1707-1713.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlson C.A., Bates N.R., Hansell D.A., Steinberg D.K. Nutrient cycling: the carbon cycle. In: Steele J., Thorpe S., Turekian K., editors. Academic Press; 2001. pp. 390–400. [Google Scholar]
- Caron D.A., Worden A.Z., Countway P.D., Demir E., Heidelberg K.B. Protists are microbes too: a perspective. ISME J. 2009a;3:4–12. doi: 10.1038/ismej.2008.101. [DOI] [PubMed] [Google Scholar]
- Caron D.A., Gast R.J., Countway P.D., Heidelberg K.B. Microbial eukaryote ecology: questions of diversity and biogeography. Microbe. 2009b;4:71–77. [Google Scholar]
- Chan E.Y. Next‐generation sequencing methods: impact of sequencing accuracy on SNP discovery. Methods Mol Biol. 2009;578:95–111. doi: 10.1007/978-1-60327-411-1_5. [DOI] [PubMed] [Google Scholar]
- Connon S.A., Giovannoni S.J. High‐throughput methods for culturing microorganisms in very low nutrient media yield diverse new marine isolates. Appl environ Microbiol. 2002;68:3878–3885. doi: 10.1128/AEM.68.8.3878-3885.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craft J.A., Gilbert J.A., Temperton B., Dempsey K.E., Ashelford K., Tiwari B. Pyrosequencing of Mytilus galloprovincialis cDNAs: tissue‐specific expression patterns. PLoS ONE. 2010;5:e8875. doi: 10.1371/journal.pone.0008875. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis T.P., Sloan W.T., Scannell J.W. Estimating prokaryotic diversity and its limits. Proc Natl Acad Sci USA. 2002;99:10494–10499. doi: 10.1073/pnas.142680199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLong E.F. Archaea in coastal marine environments. Proc Natl Acad Sci USA. 1992;89:5685–5689. doi: 10.1073/pnas.89.12.5685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desnues C., Rodriguez‐Brito B., Rayhawk S., Kelley S., Tran T., Haynes M. Biodiversity and biogeography of phages in modern stromatolites and thrombolites. Nature. 2008;452:340–343. doi: 10.1038/nature06735. et al. [DOI] [PubMed] [Google Scholar]
- Díez B., Pedrós‐Alió C., Massana R. Study of genetic diversity of eukaryotic picoplankton in different oceanic regions by small‐subunit rRNA gene cloning and sequencing. Appl Environ Microbiol. 2001;67:2932–2941. doi: 10.1128/AEM.67.7.2932-2941.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinsdale E.A., Edwards R.A., Hall D., Angly F., Breitbart M., Brulc J.M. Functional metagenomic profiling of nine biomes. Nature. 2008;452:629–631. doi: 10.1038/nature06810. et al. [DOI] [PubMed] [Google Scholar]
- Edgcomb V.P., Bernhard J., Jeon S. Deep‐sea microbial eukaryotes in anoxic, microoxic, and sulfidic environments. In: Seckbach J., editor. Springer; 2007. pp. 713–734. , and . In Cellular Origins, Life in Extreme Habitats, and Astrobiology (COLE (Series Editor). Berlin, Germany: , pp. [Google Scholar]
- Edwards R.A., Rohwer F. Viral Metaegnomics. Nat Rev Microbiol. 2005;3:504–510. doi: 10.1038/nrmicro1163. [DOI] [PubMed] [Google Scholar]
- Edwards R.A., Rodriguez‐Brito B., Wegley L., Haynes M., Breitbart M., Peterson D.M. Using pyrosequencing to shed light on deep mine microbial ecology. BMC Genomics. 2006;7:e57. doi: 10.1186/1471-2164-7-57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleischmann R.D., Adams M.D., White O., Clayton R.A., Kirkness E.F., Kerlavage A.R. Whole‐genome random sequencing and assembly of Haemophilus influenzae Rd. Science. 1995;269:496–512. doi: 10.1126/science.7542800. et al. [DOI] [PubMed] [Google Scholar]
- Frias‐Lopez J., Shi Y., Tyson G.W., Coleman M.L., Schuster S.C., Chisholm S.W., DeLong E.F. Microbial community gene expression in ocean surface waters. Proc Natl Acad Sci USA. 2008;105:3805–3810. doi: 10.1073/pnas.0708897105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frigaard N.‐U., Martinez A., Mincer T.J., DeLong E.F. Proteorhodopsin lateral gene transfer between marine planktonic Bacteria and Archaea. Nature. 2006;439:847–850. doi: 10.1038/nature04435. [DOI] [PubMed] [Google Scholar]
- Fuchs B.M., Spring S., Teeling H., Quast C., Wulf J. Characterization of a marine gammaproteobacterium capable of aerobic anoxygenic photosynthesis. Proc Natl Acad Sci USA. 2007;104:2891–2896. doi: 10.1073/pnas.0608046104. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuhrman J.A., McCallum K., Davis A.A. Novel major Archaebacterial group from marine plankton. Nature. 1992;356:148–149. doi: 10.1038/356148a0. [DOI] [PubMed] [Google Scholar]
- Gilbert J.A., Mühling M., Joint I. A rare SAR11 fosmid clone confirming genetic variability in the ‘Candidatus Pelagibacterubique’ genome. ISME J. 2008a;2:790–793. doi: 10.1038/ismej.2008.49. [DOI] [PubMed] [Google Scholar]
- Gilbert J.A., Field D., Huang Y., Edwards R., Li W., Gilna P., Joint I. Detection of large numbers of novel sequences in the metatranscriptomes of complex marine microbial communities. PLoS ONE. 2008b;3:e3042. doi: 10.1371/journal.pone.0003042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giovannoni S.J., Britschgi T.B., Moyer C.L., Field K.G. Genetic diversity in Sargasso Sea bacterioplankton. Nature. 1990;345:60–63. doi: 10.1038/345060a0. [DOI] [PubMed] [Google Scholar]
- Glöckner F.O., Joint I. Marine microbial genomics in Europe: current status and perspectives. Microb Biotechnol. 2010;3:523–530. doi: 10.1111/j.1751-7915.2010.00169.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez‐Consarnau L., González J.M., Coll‐Lladó M., Gourdon P., Pascher T., Neutze R. Light stimulates growth of proteorhodopsin‐containing marine Flavobacteria. Nature. 2007;445:210–213. doi: 10.1038/nature05381. et al. [DOI] [PubMed] [Google Scholar]
- Grzymski J.J., Carter B.J., DeLong E.F., Feldman R.A., Ghadiri A., Murray A.E. Comparative genomics of DNA fragments from six Antarctic marine planktonic bacteria. Appl Environ Microbiol. 2006;72:1532–1541. doi: 10.1128/AEM.72.2.1532-1541.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamaki T., Suzuki M., Fudou R., Jojima Y., Kajiura T., Tabuchi A. Isolation of novel bacteria and actinomycetes using soil‐extract agar medium. J Biosci Bioengin. 2005;99:485–492. doi: 10.1263/jbb.99.485. et al. [DOI] [PubMed] [Google Scholar]
- Handelsman J., Rondon M.R., Brady S.F., Clardy J., Goodman R.M. Molecular biological access to the chemistry of unknown soil microbes: a new frontier for natural products. Chem Biol. 1998;5:245–249. doi: 10.1016/s1074-5521(98)90108-9. [DOI] [PubMed] [Google Scholar]
- Handelsman J., Tiedge J., Alvarez‐Cohen L., Ashburner M., Cann I.K.O. The National Academies Press; 2007. et al) The New Science of Metagenomics: Revealing the Secrets of Our Microbial Planet. Washington DC, USA: [Google Scholar]
- Harismendy O., Ng P.C., Strausberg R.L., Wang X., Stockwell T.B., Beeson K.Y. Evaluation of next generation sequencing platforms for population targeted sequencing studies. Genome Biol. 2009;10:R32. doi: 10.1186/gb-2009-10-3-r32. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Head I.M., Saunders J.R., Pickup R.W. Microbial evolution, diversity, and ecology: A decade of ribosomal RNA analysis of uncultivated microorganisms. Microb Ecol. 1998;35:1–21. doi: 10.1007/s002489900056. [DOI] [PubMed] [Google Scholar]
- Heidelberg K.B., Allen A., Stepanauskas R., Yildiz F., Murray A., Sullivan M., Yakimiv M. 2008.
- Hewson I., Poretsky R.S., Beinart R.A., White A., Shi T., Bench S.R. In situ transcriptomic analysis of the globally important keystone N‐2‐fixing taxon Crocosphaera watsonii. ISME J. 2009;3:618–631. doi: 10.1038/ismej.2009.8. et al. [DOI] [PubMed] [Google Scholar]
- Hobbie J.E., Daley R.J., Jasper S. Use of Nuclepore filters for counting bacteria by fluorescence microscopy. Appl Environ Microbiol. 1977;33:1225–1228. doi: 10.1128/aem.33.5.1225-1228.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosono S., Faruqi A.F., Dean F.B., Du Y., Sun Z., Wu X. Unbiased whole‐genome amplification directly from clinical samples. Genome Res. 2003;13:954–964. doi: 10.1101/gr.816903. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber J.A., Welch D.B.M., Morrison H.G., Huse S.M., Neal P.R., Butterfield D.A., Sogin M.L. Microbial population structures in the deep marine biosphere. Science. 2007;318:97–100. doi: 10.1126/science.1146689. [DOI] [PubMed] [Google Scholar]
- Hugenholtz P., Tysen G.W. Metagenomics. Nature. 2008;455:481–483. doi: 10.1038/455481a. [DOI] [PubMed] [Google Scholar]
- Hughes J.B., Hellmann J.J., Ricketts T.H., Bohannan B.J.M. Counting the uncountable: statistical approaches to estimating microbial diversity. Appl Environ Microbiol. 2001;67:4399–4406. doi: 10.1128/AEM.67.10.4399-4406.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishoey T., Woyke T., Stepanauskas R., Novotny M., Lasken R.S. Genomic sequencing of single microbial cells from environmental samples. Curr Opin Microbiol. 2008;11:198–204. doi: 10.1016/j.mib.2008.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joint I., Mühling M., Querellou J. Culturing marine bacteria – an essential prerequisite for biodiscovery. Microb Biotechnol. 2010;3:564–575. doi: 10.1111/j.1751-7915.2010.00188.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolber Z.S., Plumley F.G., Lang A.S., Beatty J.T., Blankenship R.E., VanDover C.L. Contribution of aerobic photoheterotrophic bacteria to the carbon cycle in the ocean. Science. 2001;292:2492–2495. doi: 10.1126/science.1059707. et al. [DOI] [PubMed] [Google Scholar]
- Kysela D.T., Palacios C., Sogin M.L. Serial analysis of V6 ribosomal sequence tags (SARST‐V6): a method for efficient, high‐throughput analysis of microbial community composition. Environ Microbiol. 2005;7:356–364. doi: 10.1111/j.1462-2920.2004.00712.x. [DOI] [PubMed] [Google Scholar]
- Lander E.S., Linton L.M., Birren B., Nusbaum C., Zody M.C., Baldwin J. Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. doi: 10.1038/35057062. et al. [DOI] [PubMed] [Google Scholar]
- Lane D.J., Stahl D.A., Olsen G.J., Pace N.R. Analysis of hydrothermal vent‐associated symbionts by ribosomal RNA sequences. Proc Biol Soc Wash. 1985;6:389–400. doi: 10.1126/science.224.4647.409. [DOI] [PubMed] [Google Scholar]
- Leininger S., Urich T., Schloter M., Schwark L., Qi J., Nicol G.W. Archaea predominate among ammonia‐oxidizing prokaryotes in soils. Nature. 2006;442:806–809. doi: 10.1038/nature04983. et al. [DOI] [PubMed] [Google Scholar]
- Liu Z.Z., Lozupone C., Hamady M., Bushman F.D., Knight R. Short pyrosequencing reads suffice for accurate microbial community analysis. Nucleic Acids Res. 2007;35:e120. doi: 10.1093/nar/gkm541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margulies M., Egholm M., Altman W.E., Attiya S., Bader J.S. Genome sequencing in microfabricated high‐density picolitre reactors. Nature. 2005;437:376–380. doi: 10.1038/nature03959. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metzker M.L. Sequencing technologies‐ the next generation. Nat Rev Genet. 2010;11:31–46. doi: 10.1038/nrg2626. [DOI] [PubMed] [Google Scholar]
- Moon‐van der Staay S.Y., De Wachter R., Vaulot D. Oceanic 18S rDNA sequences from picoplankton reveal unsuspected eukaryotic diversity. Nature. 2001;409:607–610. doi: 10.1038/35054541. [DOI] [PubMed] [Google Scholar]
- Morris R.M., Rappé M.S., Connon S.A., Vergin K.L., Siebold W.A., Carlson C.A. SAR11 clade dominates ocean surface bacterioplankton communities. Nature. 2002;420:806–810. doi: 10.1038/nature01240. et al. [DOI] [PubMed] [Google Scholar]
- Mullis K.B. The unusual origin of the polymerase chain reaction. Sci Am. 1983;262:56–65. doi: 10.1038/scientificamerican0490-56. [DOI] [PubMed] [Google Scholar]
- Nesbø C.L., Boucher Y., Dlutek M., Doolittle W.F. Lateral gene transfer and phylogenetic assignment of environmental fosmid clones. Environ Microbiol. 2005;7:2011–2026. doi: 10.1111/j.1462-2920.2005.00918.x. [DOI] [PubMed] [Google Scholar]
- Pace N.R. A molecular view of microbial diversity and the biosphere. Science. 1997;276:734–740. doi: 10.1126/science.276.5313.734. [DOI] [PubMed] [Google Scholar]
- Pace N.R., Stahl D.A., Lane D.J., Olsen G.J. The analysis of natural microbial populations by ribosomal RNA sequences. Am Soc Microbiol News. 1985;51:4–12. [Google Scholar]
- Poretsky R.S., Gifford S., Rinta‐Kanto J., Vila‐Costa M., Moran M.A. Analyzing gene expression from marine microbial communities using environmental transcriptomics. J Vis Exp. 2009;18:1086. doi: 10.3791/1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quince C., Lanzén A., Curtis T.P., Davenport R.J., Hall N., Head I.M. Accurate determination of microbial diversity from 454 pyrosequencing data. Nature Methods. 2009;6:639–641. doi: 10.1038/nmeth.1361. et al. [DOI] [PubMed] [Google Scholar]
- Raes J., Bork P. Molecular eco‐systems biology: towards an understanding of community function. Nat Rev Microbiol. 2008;6:693–699. doi: 10.1038/nrmicro1935. [DOI] [PubMed] [Google Scholar]
- Rappé M.S., Giovannoni S.J. The uncultured microbial majority. Annu Rev Microbiol. 2003;57:369–394. doi: 10.1146/annurev.micro.57.030502.090759. [DOI] [PubMed] [Google Scholar]
- Rappé M.S., Connon S.A., Vergin K.L., Giovannoni S.J. Cultivation of the ubiquitous SAR11 marine bacterioplankton clade. Nature. 2002;418:630–633. doi: 10.1038/nature00917. [DOI] [PubMed] [Google Scholar]
- Redfield A.C. The biological control of chemical factors in the environment. Am Sci. 1958;46:205–221. [PubMed] [Google Scholar]
- Roesch L.F., Fulthorpe R.R., Riva A., Casella G., Hadwin A.K.M., Kent A.D. Pyrosequencing enumerates and contrasts soil microbial diversity. ISME J. 2007;1:283–290. doi: 10.1038/ismej.2007.53. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rondon M.R., Raffel S.J., Goodman R.M., Handelsman J. Toward functional genomics in bacteria: Analysis of gene expression in Escherichia coli from a bacterial artificial chromosome library of Bacillus cereus. Proc Natl Acad Sci USA. 1999;96:6451–6455. doi: 10.1073/pnas.96.11.6451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rondon M.R., August P.R., Betterman A.D., Brady S.F., Grossman T.H., Liles M.R. Cloning the soil metagenome: a strategy for accessing the genetic and functional diversity of uncultured microorganisms. Appl Environ Microbiol. 2000;66:2541–2547. doi: 10.1128/aem.66.6.2541-2547.2000. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rusch D.B., Halpern A.L., Sutton G., Heidelberg K.B., Williamson S., Shibu Y. The sorcerer II global ocean sampling expedition: Northwest Atlantic through eastern tropical Pacific. PLoS Biol. 2007;5:e77. doi: 10.1371/journal.pbio.0050077. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders R.W., Caron D.A., Berninger U.‐G. Relationships between bacteria and heterotrophic nanoplankton in marine and fresh water: an inter‐ecosystem comparison. Mar Ecol Prog Ser. 1992;86:1–14. [Google Scholar]
- Sanger F., Nicklen S., Coulson A.R. DNA sequencing with chain‐terminating inhibitors. Proc Natl Acad Sci USA. 1977;74:5463–5467. doi: 10.1073/pnas.74.12.5463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schleper C., DeLong E.F., Preston C.M., Feldman R.A., Wu K.‐Y., Swanson R.V. Genomic analysis reveals chromosomal variation in natural populations of the uncultured psychrophilic archaeon Cenarchaeum symbiosum. J Bacteriol. 1998;180:5003–5009. doi: 10.1128/jb.180.19.5003-5009.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt T.M., DeLong E.F., Pace N.R. Analysis of a marine picoplankton community by 16S rRNA gene cloning and sequencing. J Bacteriol. 1991;173:4371–4378. doi: 10.1128/jb.173.14.4371-4378.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shendure J., Porreca G.J., Nikos B.R., Xiaoxia L., McCutcheon J.P., Rosenbaum A.M. Accurate multiplex polony sequencing of an evolved bacterial genome. Science. 2005;309:1728–1732. doi: 10.1126/science.1117389. et al. [DOI] [PubMed] [Google Scholar]
- Sherr E.B., Sherr B.F. Significance of predation by protists in aquatic microbial food webs. Antonie Van Leeuwenhoek. 2002;81:293–308. doi: 10.1023/a:1020591307260. [DOI] [PubMed] [Google Scholar]
- Shi Y., Tyson G.W., DeLong E.F. Metatranscriptomics reveals unique microbial small RNAs in the ocean's water column. Nature. 2009;459:266–269. doi: 10.1038/nature08055. [DOI] [PubMed] [Google Scholar]
- Sogin M.L., Morrison H.G., Huber J.A., Welch D.M., Huse S.M., Neal P.R. Microbial diversity in the deep sea and the underexplored ‘rare biosphere’. Proc Natl Acad Sci USA. 2006;103:12115–12120. doi: 10.1073/pnas.0605127103. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stahl D.A., Lane D.J., Olsen G.J., Pace N.R. Characterization of a Yellowstone hot spring microbial community by 5S ribosomal RNA sequences. Appl Environ Microbiol. 1985;49:1379–1384. doi: 10.1128/aem.49.6.1379-1384.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staley J.T., Konopka A. Measurements of in situ activities of nonphotosynthetic microorganisms in aquatic and terrestrial habitats. Annu Rev Microbiol. 1985;39:321–346. doi: 10.1146/annurev.mi.39.100185.001541. [DOI] [PubMed] [Google Scholar]
- Stein J.L., Marsh T.L., Wu K.Y., Shizuya H., DeLong E.F. Characterization of uncultivated prokaryotes: Isolation and analysis of a 40‐kilobase‐pair genome fragment from a planktonic marine archaeon. J Bacteriol. 1996;178:591–599. doi: 10.1128/jb.178.3.591-599.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stott M.B., Crowe M.A., Mountain B.W., Smirnova A.V., Hou S., Alam M., Dunfield P.F. Isolation of novel bacteria, including a candidate division, from geothermal soils in New Zealand. Environ Microbiol. 2008;10:2030–2041. doi: 10.1111/j.1462-2920.2008.01621.x. [DOI] [PubMed] [Google Scholar]
- Sullivan M.B., Waterbury J.B., Chisholm S.W. Cyanophages infecting the oceanic cyanobacterium Prochlorococcus. Nature. 2003;426:548–584. doi: 10.1038/nature01929. [DOI] [PubMed] [Google Scholar]
- Sullivan M.B., Coleman M.L., Weigele P., Rohwer F., Chisholm S.W. Three Prochlorococcus cyanophage genomes: signature features and ecological interpretations. PLoS Biol. 2005;3:e144. doi: 10.1371/journal.pbio.0030144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Temperton B., Field D., Oliver A., Tiwari B., Mühling M., Joint I., Gilbert J.A. Bias in assessments of marine microbial biodiversity in fosmid libraries as evaluated by pyrosequencing. ISME J. 2009;3:792–796. doi: 10.1038/ismej.2009.32. [DOI] [PubMed] [Google Scholar]
- Teske A. Enigmatic archaeal and eukaryotic life at hydrothermal vents and in marine subsurface sediments. In: Seckbach J., editor. Springer; 2007. pp. 521–533. In Cellular Origins, Life in Extreme Habitats, and Astrobiology (COLE (Series Editor). Berlin, Germany: , pp. [Google Scholar]
- Turner S., Burger‐Wiersma T., Giovannoni S.J., Mur L.R., Pace N.R. The relationship of a prochlorophyte, Prochlorothrix hollandica, to green chloroplasts. Nature. 1989;337:380–382. doi: 10.1038/337380a0. [DOI] [PubMed] [Google Scholar]
- Tyson G.W., Chapman J., Hugenholtz P., Allen E.E., Ram R.J., Richardson P.M. Community structure and metabolism through reconstruction of microbial genomes from the environment. Nature. 2004;428:37–43. doi: 10.1038/nature02340. et al. [DOI] [PubMed] [Google Scholar]
- Urich T., Lanzén A., Qi J., Huson D.H., Schleper C., Schuster S.C. Simultaneous assessment of soil microbial community structure and function through analysis of the meta‐transcriptome. PLoS ONE. 2008;3:e2527. doi: 10.1371/journal.pone.0002527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venter J.C., Adams M.D., Myers E.W., Li P.W., Mural R.J., Sutton G.G. The sequence of the human genome. Science. 2001;291:1304–1351. doi: 10.1126/science.1058040. et al. [DOI] [PubMed] [Google Scholar]
- Venter J.C., Remington K., Heidelberg J.F., Halpern A.L., Rusch D., Eisen J.A. Environmental genome shotgun sequencing of the Sargasso Sea. Science. 2004;304:66–74. doi: 10.1126/science.1093857. et al. [DOI] [PubMed] [Google Scholar]
- Walter J.M., Greenfield D., Bustamante C., Liphardt J. Light‐powering Escherichia coli with proteorhodopsin. Proc Natl Acad Sci USA. 2007;104:2408–2412. doi: 10.1073/pnas.0611035104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward B.B. How many species of prokaryotes are there? Proc Natl Acad Sci USA. 2002;99:10234–10236. doi: 10.1073/pnas.162359199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward D.M., Weller R., Bateson M.M. 16S rRNA sequences reveal numerous uncultured microorganisms in a natural community. Nature. 1990;345:63–65. doi: 10.1038/345063a0. [DOI] [PubMed] [Google Scholar]
- Warnecke F., Hugenholtz P. Building on metagenomics with complimentary technologies. Genome Biol. 2007;8:231.0–231.5. doi: 10.1186/gb-2007-8-12-231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wegley L., Edwards R., Rodriguez‐Brito B., Liu H., Rohwer F. Metagenomic analysis of the microbial community associated with the coral Porites astreoides. Environ Microbiol. 2007;9:2707–2719. doi: 10.1111/j.1462-2920.2007.01383.x. [DOI] [PubMed] [Google Scholar]
- Weinbauer M.G. Ecology of prokaryotic viruses. FEMS Microbiol Rev. 2004;28:127–186. doi: 10.1016/j.femsre.2003.08.001. [DOI] [PubMed] [Google Scholar]
- Wilkening J., Wilke A., Desai N., Meyer F. Using clouds for metagenomics: a case study. IEEE Cluster. 2009;2009:1–6. [Google Scholar]
- Williamson S.J., Rusch D.B., Yooseph S., Halpern A., Heidelberg K.B., Glass J.I. The sorcerer II global ocean sampling expedition: Metagenomic characterization of viruses within aquatic microbial samples. PLoS One. 2008;1:e1456. doi: 10.1371/journal.pone.0001456. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woese C.R. Bacterial evolution. Microbiol Rev. 1987;51:221–271. doi: 10.1128/mr.51.2.221-271.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woyke T., Xie G., Copeland A., Gonzalez J.M., Han C., Kiss H. Assembling the marine metagenome, one cell at a time. PLoS ONE. 2009;4:e5299. doi: 10.1371/journal.pone.0005299. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu D., Hugenholtz P., Mavromatis K., Pukall R., Dalin E. A phylogeny‐driven genomic encyclopaedia of Bacteria and Archaea. Nature. 2009;462:1056–1060. doi: 10.1038/nature08656. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wuchter C., Abbas B., Coolen M.J.L., Herfort L., Van Bleijswijk J., Timmers P. Archaeal nitrification in the ocean. Proc Natl Acad Sci USA. 2006;103:12317–12322. doi: 10.1073/pnas.0600756103. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanmei S., Tyson G.W., DeLong E.F. Metatranscriptomics reveals unique microbial small RNAs in the ocean's water column. Nature. 2009;459:266–269. doi: 10.1038/nature08055. [DOI] [PubMed] [Google Scholar]
- Yooseph S., Sutton G., Rusch D.B., Halpern A.L., Williamson S.J., Remington K. The sorcerer II global ocean sampling expedition: expanding the universe of protein families. PLoS Biol. 2007;5:432–466. doi: 10.1371/journal.pbio.0050016. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yutin N., Suzuki M.T., Teeling H., Weber M., Venter J.C., Rusch D.B., Béjà O. Assessing diversity and biogeography of aerobic anoxygenic phototrophic bacteria in surface waters of the Atlantic and Pacific Oceans using the Global Ocean Sampling expedition metagenomes. Environ Microbiol. 2007;9:1464–1475. doi: 10.1111/j.1462-2920.2007.01265.x. [DOI] [PubMed] [Google Scholar]
- Zehr J.P., Ward B.B. Nitrogen cycling in the ocean: New perspectives on processes and paradigms. Appl Environ Microbiol. 2002;68:1015–1024. doi: 10.1128/AEM.68.3.1015-1024.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zengler K., Toledo G., Rappé M., Elkins J., Mathur E.J., Short J.M., Keller M. Cultivating the uncultured. Proc Natl Acad Sci USA. 2002;99:15681–15686. doi: 10.1073/pnas.252630999. [DOI] [PMC free article] [PubMed] [Google Scholar]