

Abstract
Epigenetics, the non-genetic component of how chromatin structure influences gene expression, is amazingly complex, and linking how environmental stimuli can influence epigenetic ‘gene programs’ in specific nerve cells to ultimately control behavior is a seemingly insurmountable puzzle. Cocaine is a highly potent stimulus capable of influencing behavior for the lifetime of an organism. Not surprisingly, psychostimulant-induced epigenetic regulation of gene expression has thus been identified as key to understanding the pathology of addiction. In addition to identifying this essential role of epigenetics in addiction, several important concepts have emerged such as the importance of global, temporal, and spatial control of mRNA expression in considering any given histone modification’s influence on a given gene. Adding to this complexity, one has to account for the cumulative influence of other epigenetic modifications on a gene’s transcription in addition to the interplay between transcription factors and chromatin structure. This review highlights how bioinformatic, molecular, and behavioral studies on addiction provide new insight into these concepts and outlines two distinct psychostimulant-induced patterns of chromatin regulation which are thought to underlie unique programs of gene expression that contribute importantly to the addicted state.
Keywords: addiction, cocaine, nucleus accumbens, chromatin remodeling, epigenetics, histone acetylation, histone methylation, DNA methylation, gene priming, desensitization
Introduction
Drug addiction is a debilitating psychiatric disorder characterized by compulsive drug seeking and taking despite severe adverse consequences (Hyman et al., 2006; Kalivas et al., 2005; Koob and Kreek, 2007). Once a person succumbs to addiction, few effective therapies exist. Even when former addicts remain abstinent for long durations of time, they often find themselves in a lifelong struggle—always vulnerable to drug relapse. Therefore, the two major questions on which basic science research focuses relate to understanding the molecular events that occur during: 1) the transition to the addicted state and 2) the maintenance of the addicted state. A better understanding of these mechanisms would provide insight into how we can block or perhaps reverse the neuroplastic changes that define addiction.
Drug-induced changes in gene expression in key brain reward regions, such as the nucleus accumbens (NAc), prefrontal cortex (PFC), and ventral tegmental area (VTA), represent one mechanism thought to contribute to both of these key questions. A multitude of microarray studies under different experimental conditions have found drug-induced alterations in the expression levels of hundreds of mRNAs in these target regions (Freeman et al., 2010; Heiman et al., 2008; Maze et al., 2010; McClung and Nestler, 2003; McClung et al., 2005; Renthal et al., 2007; Winstanley et al., 2007; Yao et al., 2004). In response to psychostimulants, many genes, such as those encoding c-Fos, FosB, ΔFosB, ATF2 (activating transcription factor 2), ATF3, and ATF4, are rapidly and transiently induced in response to initial drug exposures. Chronic exposure differentially affects the steady state levels of these various mRNA’s as well as their degree of induction upon re-exposure to the same drug dose with some genes showing sensitized responses and others desensitized responses (Alibhai et al., 2007; Green et al., 2008; Hope et al., 1994; Renthal et al., 2008). In contrast, numerous other genes, such as those encoding CDK5 (cyclin-dependent kinase 5), several NFκB (nuclear factor κB) subunits, SIRT2 (sirtuin 2), PSD-95 (postsynaptic density protein of 95 kD), and BDNF (brain-derived neurotrophic factor), are consistently induced only by chronic drug experience, and some even increase further over several weeks of withdrawal after the last drug experience (Alibhai et al., 2007; Bibb et al., 2001; Green et al., 2008; Grimm et al., 2003; Hope et al., 1994; Renthal et al., 2008; Renthal et al., 2009; Russo et al., 2009; Yao et al., 2004).
These complex patterns of transcriptional regulation point to the need to identify the underlying mechanisms responsible for altering a gene’s ‘inducibility’ and those capable of stably influencing transcription for prolonged periods. In focusing on psychostimulant-induced changes in the NAc, recent evidence has suggested that epigenetics—a molecular translator that interprets diverse environmental stimuli into changes in gene expression via the regulation of chromatin structure—contributes to drug-induced transcriptional and behavioral changes (Kumar et al., 2005; Levine et al., 2005; Maze et al., 2010; Renthal et al., 2007; Wang et al., 2010). This review takes an integrative approach in discussing current progress being made toward understanding how epigenetic mechanisms are regulated by cocaine and other psychostimulant drugs of abuse in the NAc to influence specific gene expression programs and how such mechanisms might contribute to addiction-related behaviors.
Epigenetics overview
Historically, the word ‘epigenetic’ refers to a heritable phenotype not coded by DNA itself, but by a cellular process ‘above the genome’. More recently, epigenetics is used to refer to the extremely complex processes of organizing the genome in a manner that allows for regulated gene expression in the appropriate cell type upon appropriate cellular stimuli. On a molecular level, the fundamental unit that accomplishes this feat is chromatin, which is composed of DNA wrapped around histone octomers made up of two copies each of H2A, H2B, H3, and H4. In the past decade, it has been appreciated that the structure of chromatin is highly regulated by post-translational modifications (PTMs) that occur on histones and DNA itself. Importantly, these modifications—via regulation of chromatin structure—profoundly influence gene expression in different ways and, since multiple PTMs can occur on a given histone octomer, it is hypothesized that the combination of these modifications summate to influence gene expression. Also known as the histone code hypothesis (Borrelli et al., 2008; Kouzarides, 2007; Lee and Mahadevan, 2009; Strahl and Allis, 2000). Highlighted in Table 1, are just a few well characterized examples of such PTMs, their associated effect on gene transcription, as well as the enzymes that ‘write’ and ‘erase’ such modifications (reviewed in greater detail in: Bekiranov and Grant, 2010 [this issue of Hormones and Behavior]; Renthal and Nestler, 2008; Strahl and Allis, 2000).
Table 1.
Examples of major chromatin modifications and their function and enzyme mediators.
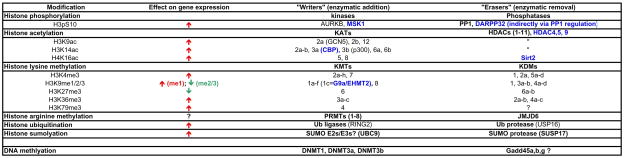 |
List of histone modifications, their known effects on transcription, and examples of enzymes that catalyze their “writing” (enzymatic addition) or “erasing” (enzymatic removal).
 = increased transcription,
= increased transcription,  = decreased transcription;
= decreased transcription;  = examples of enzymes in which cocaine-induced biochemical regulation has been identified and its behavioral significance has been assessed.
= examples of enzymes in which cocaine-induced biochemical regulation has been identified and its behavioral significance has been assessed.
There are several important caveats to consider when studying epigenetics in brain. First, a close look at Table 1 illustrates that many enzymes each have multiple histone substrates and, in fact, although not listed in the table, it is highly likely that many have non-histone substrates as well. For example, in addition to deacetylating H4K16 (histone H4 on Lys16), SIRT2 is well known to also deacetylate tubulin as well as several major transcription factors (Renthal and Nestler, 2009). Therefore, in order to understand the cellular and behavioral role of a given enzyme and histone modification, a multifaceted approach is absolutely crucial. A second major caveat is that most of the discovered PTMs and their ascribed influence on transcription have been derived from in vitro work in non-neuronal cells. Therefore, it is possible that the presumed functions of known PTMs in influencing transcription in cultured cells may not necessarily be the same in brain. Moreover, it is likely that unique PTMs exist in brain. In fact, a recent proteomic study on whole brain tissue identified 196 novel histone modifications (Tweedie-Cullen et al., 2009), and a study analyzing brain DNA identified hydroxy-methylation as a novel brain-specific DNA modification (Kriaucionis and Heintz, 2009; Tweedie-Cullen et al., 2009). A final caveat is that histone PTMs, although the most widely studied, are just one of many epigenetic mechanisms. A few examples that remain virtually unexplored in brain include histone tail clipping, histone substitution, and histone sliding/remodeling, which are reviewed elsewhere (Borrelli et al., 2008; Tsankova et al., 2007).
Epigenetic mechanisms in addiction
One of the most striking findings to date is that, despite the abundance of histone PTMs throughout the genome, drugs of abuse as well as many other types of environmental stimuli are capable of inducing changes in total levels of various histone modifications, for example, as detected by western blotting or immunohistochemistry, in specific brain regions. With respect to cocaine, most global changes observed are consistent with a state of increased gene activation (i.e., a more permissive state) within the NAc. Following a single cocaine injection, total levels of acetylated histone H4 (acH4), phosphoacetylated histone H3 (pacH3), but not acH3 alone, are increased in this brain region within 30 minutes (Kumar et al., 2005). Moreover, in a cocaine self-administration model, both acH4 and acH3 have been found to be increased in the NAc shell, not core, following chronic but not acute self-administration. Interestingly, in these animals, levels of acH3, and not acH4, were found to be positively correlated with motivation for cocaine (Wang et al., 2010). Also, after repeated cocaine injections, a recent study found a global reduction in dimethylation of H3 at Lys9 (H3K9me2), a repressive histone modification, in the NAc (Maze et al., 2010).
In concert with these global epigenetic alterations, cocaine regulates the expression of several chromatin modifying enzymes in the NAc in directions generally consistent with the global modifications (see Table 1). For example, following chronic but not acute cocaine, HDAC5 is shuttled out of the nucleus of NAc neurons and HDAC activity is significantly reduced (Renthal et al., 2007; Romieu et al., 2008). At the same time, phosphorylation of histone H3 at Ser10 (H3pS10) appears to be mediated via direct phosphorylation by MSK1 (mitogen- and stress-activated kinase 1) as well as by nuclear shuttling of DARPP-32, a striatal-enriched protein which indirectly regulates phosphorylation via potent inhibition of PP1 (Brami-Cherrier et al., 2005; Stipanovich et al., 2008). Likewise, the downregulation of H3K9me2 appears to be mediated via transcriptional repression of G9a (also known as KMT1c), a histone methyltransferase that catalyzes H3K9 dimethylation (Maze et al., 2010). Chronic cocaine administration also regulates the expression of DNMT3a, a de-novo DNA methyltransferase, but not other DNMT isoforms, in the NAc (LaPlant et al., 2009; Maze et al., 2010). Thus, chronic cocaine appears to globally induce a more permissive transcriptional state in the NAc (increased acH3, acH4, pacH3; decreased H3K9me2), perhaps via differentially affecting histone modifying enzymes that activate (MSK1, DARPP-32, CBP) and repress (HDAC5, G9a, Dnmt3a) transcription.
Despite the fact that several chromatin modifying enzymes are regulated in the NAc by cocaine, it is still surprising that the absolute levels of PTMs are altered because modifications such as histone acetylation and methylation are known to occur throughout the genome. Thus, in order to have an absolute change, sweeping alterations must occur throughout vast genomic loci despite the fact that many DNA microarray studies of NAc from cocaine-treated animals have found that only a relatively small number (~100’s) of specific genes are differentially expressed. Moreover, many specific gene promoters display changes in histone acetylation and methylation after cocaine that go in the direction opposite of the global modifications (Renthal et al., 2009). Thus, further work is needed to understand the physiological significance of these global changes in epigenetic PTMs.
Insights from genome-wide regulation of gene expression and chromatin structure
Our laboratory recently utilized ChIP-chip (chromatin immunoprecipitation followed by promoter chip) technology to investigate the genome-wide binding profile of chronic cocaine-induced changes in the mouse NAc at two general modifications associated with gene activation and one modification associated with gene repression. The antibodies used in this study referred to as ‘acH3’, ‘acH4’, and ‘meH3’ each recognize multiple PTMs: polyacetylated histone H3 (K9, K14), polyacetylated H4 (K5, K8, K12, K16), and dimethylated H3 at two sites (K9, K27), respectively (Renthal et al., 2009).
This study reported several key findings for epigenetic regulation in brain. An obvious yet often overlooked concept is the importance and complexity of location in analyzing the epigenetic landscape of a gene’s promoter. Here, analysis of the averaged genome-wide spatial binding profile revealed a distinct binding distribution pattern for each histone modification. In general, enrichment of acH3 and acH4 occurs within 500 base pairs (bp’s) of a gene’s transcription start site (TSS), with acH4 having a particularly sharp peak at the TSS (Renthal et al., 2009). Conversely, NAc meH3 enrichment has a broad bimodal distribution with maximal peaks at −1500 bp and +400 bp away from the TSS (Renthal et al., 2009; Wilkinson et al., 2009). Such overt differences in spatial binding profiles have important implications. For example, a ChIP experiment that concludes robust acH3/H4 regulation and the lack of differential binding of meH3 may turn out to be a false negative simply based on the genomic region in which the IP (immunoprecipitation) and PCR reaction are focused. Moreover, based on individual gene promoter plot analyses, several examples exist, such as the CART promoter, in which, depending on the physical location analyzed, it is possible to observe an increase, decrease, or no change in meH3 binding (Fig 1B). This highlights the importance of genome wide studies of chromatin modifications and the need to extensively analyze different locations along the landscape of a given gene promoter.
Figure 1. Summary of genome wide analysis of cocaine regulation of chromatin structure and gene expression.

A) Chronic cocaine causes extensive genome-wide chromatin modifications marked by little overlap. Venn diagrams from ChIP-chip studies with acetylated H3 (acH3), acetylated H4 (acH4), and methylated H3 (meH3) from nucleus accumbens (NAc) in mice that received chronic cocaine (24 hours after 7 daily cocaine injections at 20 mg/kg dose) (Renthal et al., 2009). Compared to traditional DNA microarray studies that look for genes with altered steady state mRNA levels under similar conditions, a much greater number of gene promoters are significantly regulated by each analyzed histone modification and, yet, a relatively small number of gene promoters share co-regulation of more than one modification suggesting that each analyzed modification has distinct functions. B) Example of promoter plots of two representative genes, Cart and Per1, from the Venn diagram illustrating spatial complexity in chromatin regulation. Y-axis represents fold change in binding relative to saline treated mice and the x-axis represents the physical location of binding on the indicated gene promoter relative to its transcription start site (TSS). The plots illustrate the complex patterns of acH3, acH4, and meH3 on these two representative gene promoters. C) Analysis of cocaine-induced gene expression reveals two patterns of gene induction: desensitized (black line in acute row) and primed genes (purple lines in chronic and withdrawn columns). Scaled heatmaps (*) show the number and pattern of cocaine-upregulated gene expression in NAc at 1 hour after a challenge injection of cocaine (20 mg/kg) in three groups of animals: Acute (no prior cocaine exposure), Chronic (7 daily cocaine injections examined 1 hour after the last injection), or Withdrawn (7 daily cocaine injections followed by 1 week of withdrawal before the challenge injection). The top series of heatmaps show the genes upregulated in the Acute group and how those same genes are regulated in the other two groups. Likewise, the middle and lower series of heatmaps show the genes upregulated in the Chronic and Withdrawal groups, respectively, and how those genes are regulated in the other two groups. Notably, a small but significant number of acutely induced genes are no longer induced in animals with prior chronic cocaine exposure (desensitized genes, black bar***), whereas a larger number of genes that are induced after chronic cocaine lack induction by acute cocaine (primed genes, purple bars***). From Maze et al., 2010.
Another major finding of the Renthal et al., 2009 study was the presence of extensive chromatin modifications found throughout the genome. With rigorous statistical criteria, roughly 1000 gene promoters for acH3, 700 for acH4, and 900 for meH3 were found to have increased promoter binding under cocaine-treated conditions relative to controls (Fig. 1A). These values are consistent with the aforementioned observation that absolute levels of histone PTMs are altered by cocaine. However, these findings raise a paradox: many more gene promoters, almost one order of magnitude more, appear to be cocaine-regulated at the chromatin level compared to the typical number of differentially expressed mRNA transcripts that have been identified under similar cocaine treatment paradigms (Maze et al., 2010; McClung and Nestler, 2003; Renthal et al., 2007; Yao et al., 2004). One explanation for this disparity may lie in differences in the sensitivity of the two techniques. Alternatively, the much higher degree of chromatin regulation may suggest that each histone modification may not directly regulate steady-state mRNA levels, but rather indirectly influences transcription to subsequent stimuli.
An important related discovery is that there is relatively little overlap among the three different histone marks studied at the same gene promoters (Renthal et al., 2009). This is somewhat surprising, since coincident regulation might have been expected: for example, increased acH3, increased acH4, and decreased meH3 at a particular activated gene. However, in this study only about 20% of genes regulated by acH3 were commonly regulated by acH4. Even more striking, only ~1% of hypermethylated ‘repressed’ genes (9 out of 889) show hypoacetylation (Fig 1A). Taken together, these findings suggest that acH4, acH3, and meH3 each have distinct functions that regulate mostly unique transcriptional programs.
In support of these findings, a recent genome wide microarray study, comparing the ability of a challenge dose of cocaine to regulate gene expression in the NAc of cocaine-naïve animals to those treated previously with chronic cocaine, provides large scale evidence for at least two distinct patterns of gene regulation—“desensitized” and “primed” gene expression (Maze et al., 2010). A subset of genes, referred to as desensitized, are induced by acute cocaine and exhibit attenuated induction after chronic cocaine. This blunted induction persists for at least one week, as observed by the lack of effect of a challenge injection in chronic cocaine withdrawn animals (Fig 1C). In contrast, the large majority of genes that are induced by chronic cocaine are not induced by the initial cocaine exposure, represent primed genes. Indeed, approximately 2–3 fold more genes are significantly upregulated in the NAc by chronic cocaine (~275 genes) as well as in cocaine withdrawn animals (~210 genes) compared to those upregulated by initial cocaine exposure (~90 genes) (Maze et al., 2010). From an epigenetic point of view, these findings suggest that throughout the course of acute to chronic cocaine injections, many genomic loci accumulate specific epigenetic ‘hits’ that render them poised for greater induction. Further, the fact that this primed state of gene inducibility remains present after a week of withdrawal from cocaine suggests a prolonged underlying epigenetic influence on transcription. In summary, this microarray study identified distinct patterns of transcriptional regulation which are consistent with our ChIP-chip data, which revealed extensive, mostly non-overlapping patterns of cocaine-induced promoter binding for acH3, acH4, and meH3.
Key concepts in psychostimulant regulation of chromatin structure
Indeed, analysis of the chromatin state and mRNA expression of many genes reveals evidence for distinct epigenetic influence in mediating both gene desensitization and gene priming (Fig. 2).
Figure 2. Distinct patterns of cocaine-induced gene induction correspond to unique patterns of chromatin regulation.

Graphs in the top of A and B display patterns of mRNA expression over time, with the underlying epigenetic binding profile depicted in the bottom tables. Promoter binding profiles are displayed in heatmap style (cocaine relative to saline, with red = increased binding, black = no change, green = reduced binding). The x-axis is divided into 4 sections that highlight mRNA and chromatin changes that occur in response to initial cocaine exposure, in response to a cocaine challenge after prior chronic drug exposure, during drug withdrawal, and in response to a subsequent challenge dose of cocaine. Of note, since chronic injections are given once daily, one can infer that chromatin changes that occur during early withdrawal (24 hours) are presumed to provide insight into events that also occur prior to the last chronic cocaine injection. A) Epigenetic basis of gene priming. Genes that are primed to be induced eventually develop increased acH3 and reduced H3K9me2 binding during a chronic course of cocaine administration. In contrast, during initial cocaine exposure—when there is no mRNA induction—the repressive histone methyltransferase, G9a, is induced and binds to and mediates H3K9me2 binding to nearby promoter regions. B) Epigenetic basis of gene desensitization. Genes that are desensitized exhibit prominent induction of acH4 binding during initial cocaine exposure, which is lost with chronic treatment. This may be due to increased binding of HDAC1. As well, G9a shows increased binding during acute and withdrawal time points. This is correlated with increased H3K9me2 during late withdrawal. Interestingly, the transcription factor, ΔFosB, is bound to both types of genes during cocaine withdrawal, suggesting a dual role in mediating gene repression and gene activation. An important caveat is that these mechanisms of gene priming and desensitization are based on the analysis of a relatively small number of cocaine-regulated genes and must be refined as many more such genes are characterized. N/A, information not available.
Epigenetic basis of gene priming
There are several well established examples of genes that are induced in the NAc by chronic, but not acute, cocaine administration (Maze et al., 2010; McClung and Nestler, 2003). Specific examples for which the chromatin structure has also been extensively studied include the genes encoding BDNF, CDK5, NFκB (p65 subunit), and SIRT2 (Fig 2A). ChIP studies have found that genes which are primed to be induced develop increased acH3 and no change in acH4 in response to chronic and not acute cocaine injections (Kumar et al., 2005; Renthal et al., 2009; Russo et al., 2009; Wang et al., 2010). Interestingly, levels of H3K9me2 and its associated enzyme, G9a, exhibit increased binding at 1 hour after cocaine administration, suggesting a possible role in blockade of acute induction of primed genes (Maze et al., 2010). However, with chronic injections, both H3K9me2 and G9a binding are reduced, consistent with the notion that this loss of methylation on H3K9 may facilitate acetylation on H3K9 and other associated lysine residues and promote gene inducibility (i.e., priming). Priming is also associated with increased recruitment of SWI-SNF chromatin remodeling factors (e.g., BRG1) to the primed gene promoters (Kumar et al., 2005).
Less is known about whether steady state mRNA levels of genes primed for induction remain upregulated or return to baseline in between cocaine exposures, and about what happens to the chromatin state of these genes over this time frame. For two genes known to have prolonged regulation, CDK5 and BDNF, increased acH3 binding was found to persist for at least one week following the last drug injection (Kumar et al., 2005). Presumably, H3K9me2 remains reduced although this remains to be proven. In all likelihood, different subclasses of primed genes exist—those with a prolonged increase in steady-state levels, such as BDNF, and those whose mRNAs return to baseline levels but remain primed for greater induction (sensitization) during re-exposure to drug, as illustrated in Fig. 1C.
Epigenetic basis of gene desensitization
The most well characterized example of gene desensitization is the gene encoding c-Fos, which exhibits desensitized induction in response to repeated cocaine or amphetamine administration, and shows suppressed basal expression levels over several days of withdrawal as well as enhanced desensitization to a challenge dose of the drug (Renthal et al., 2008). Unlike known primed genes, acH4 and not acH3 appears to play a key role in gene desensitization. At the c-Fos promoter, acH4 is dramatically induced by acute but not chronic drug exposure (Kumar et al., 2005). Another interesting observation lies in the temporal dynamics that occur in response to initial drug exposure, when pacH3 precedes both peak mRNA and acH4 levels, an effect that was no longer observed with chronic drug administration (Kumar et al., 2005). This suggests the possible influence of pacH3 on acH4 and subsequent gene activation. Importantly, key chromatin modifying enzymes found to be associated with the c-Fos promoter following chronic drug administration were HDAC1 and G9a, fitting with reduced acetylation (via enhanced deacetylation) and increased H3K9me2 and with gene repression (Maze et al., 2010; Renthal et al., 2008). A major goal of future research is to determine whether other genes that display desensitization exhibit similar chromatin mechanisms as observed for c-Fos.
Interplay between transcription factors and epigenetic mechanisms
Another crucial aspect of chromatin dynamics to consider is how transcription factors and the overall transcriptional machinery interact with epigenetic modifications to ultimately influence transcription. In the addiction field, one of the most well studied examples of such interactions is the transcription factor, ΔFosB (Nestler et al., 1999; Nestler, 2008). Genome wide overexpression studies have suggested that ΔFosB plays a dual role in repressing and activating transcription of cocaine-regulated genes (McClung and Nestler, 2003; Winstanley et al., 2007). As shown in Fig. 2, ChIP studies of NAc from chronically treated animals find that ΔFosB is indeed bound to both primed and desensitized gene promoters and, in each case, ΔFosB is associated with a completely different entourage of modifications and chromatin modifying enzymes. For example, with primed genes, ΔFosB recruits the transcriptional activator, BRG1, whereas with desensitized genes HDAC1 is recruited to the c-Fos gene via ΔFosB (Kumar et al., 2005). Adding to this complexity, it appears that G9a and ΔFosB interact in a complex autoregulatory feedback loop. After acute cocaine, G9a is bound to, and hypermethylates H3, at the FosB promoter. This hypermethylation functionally attenuates cocaine induction of FosB gene expression, as was observed with G9a overexpression. However, due to ΔFosB’s extraordinarily long half life, ΔFosB accumulates in the cell and then represses mRNA levels of G9a and reduces total levels of H3K9me2, both of which ultimately appear to reduce G9a and H3K9me2 binding at the FosB promoter during cocaine withdrawal—thereby allowing ΔFosB to potentiate its own induction (Maze et al., 2010).
Much more work is needed to understand this complex regulation, particularly within the context of the complex behavioral parameters that define an addicted state. For example, it will be critical in future investigations to delineate the role played by these numerous desensitized and primed genes (in the NAc and elsewhere) in different phases of addiction, for example, the decision to take drug initially, the transition from casual drug use to addiction, and the craving and risk for relapse that occur during withdrawal periods.
Role of epigenetic mechanisms in drug-related behaviors
As stated previously, biochemical and genome-wide evidence suggests that chronic cocaine administration causes chromatin structure to be in a generally more permissive state. This is evidenced by a reduction in total levels of the repressive histone mark, H3K9me2, as well as an increase in total levels of acH3 and acH4. On a genome-wide scale, in addition to each of the aforementioned modifications being heavily regulated by chronic cocaine at literally thousands of gene promoters, 2–3 fold more genes are activated by chronic cocaine compared to acute cocaine. Behaviorally, increasing evidence suggests that the permissive chromatin structure induced by chronic cocaine is a pathological adaptation to the rewarding effects of the drug (Table 2). For example, mimicking this ‘permissive state’ by systemic administration of trichostatin A (TSA) or intra-NAc delivery of suberoylanilide hydroxamic acid (SAHA), which are both selective class I and class II HDAC inhibitors, increase cocaine conditioned place preference, which provides an indirect measure of cocaine reward (Kumar et al., 2005; Renthal et al., 2007). In addition, intra-NAc pharmacological blockade as well as intra-NAc conditional knock-out of two other repressive modifications, G9a (H3K9me2) and DNMT3a (DNA methylation), also increase cocaine place conditioning (LaPlant et al., 2009; Maze et al., 2010). Conversely, experimental manipulations that promote gene repression, such as intra-NAc overexpression of HDAC4, HDAC5, G9a, or DNMT3a, reduce cocaine place conditioning. These effects appear to depend specifically on enzymatic activity of these enzymes, since catalytically dead mutants of HDAC5 or G9a (G9a-H1093K) have no effect on cocaine reward, and since overexpression of wildtype HDAC5 co-administered with TSA completely blocks HDAC5’s effect on behavioral responses to cocaine. Furthermore, these effects on cocaine reward are specific to certain enzymes and are not generalizable, since, for example, NAc-specific overexpression of HDAC9 and NAc-specific knock-out of DNMT1—two enzymes that are not cocaine regulated, but are expressed in the NAc—do not have any effect on cocaine reward (LaPlant et al., 2009; Renthal et al., 2007).
Table 2.
Effect of chromatin modifications on behavioral responses to cocaine.
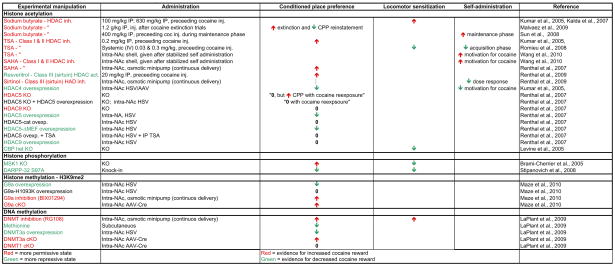 |
inh., inhibitor; act., activator; TSA, tichostatin A; KO, knockout; cKO, conditional KO; het KO, heterozygous KO; G9a-H1093K, catalytically dead G9a mutant; HDAC5- MEF, HDAC5 with mutated MEF binding domain.
It is important to note that this general notion that promoting a more permissive chromatin state does not always produce increased cocaine reward. For example, resvertitrol, a sirtuin (class III HDAC) agonist, and sirtintol, a sirtuin antagonist, unexpectedly increase and decrease cocaine reward, respectively (Renthal et al., 2009). This is surprising since SIRT1, a key target of these agents, is a major component of transcriptional repression complexes (Finkel et al., 2009). Also, despite both manipulations reducing H3S10 phosphorylation, MSK1 knockout increases cocaine reward whereas DARPP-32 S97A knock-in decreases cocaine reward (Brami-Cherrier et al., 2005; Stipanovich et al., 2008). However, while sirtuins, MSK1, and nuclear DARPP-32 have effects on histones, they also have a multitude of non-histone substrates, which makes it difficult to establish a solid link between their effects on chromatin and their resultant behavioral actions—an important concept.
From a theoretical standpoint, since chromatin modifications such as histone acetylation are affected differently by acute compared to chronic cocaine, one would expect that experiments that manipulate chromatin dynamics at different times of drug administration (i.e., before receiving initial cocaine, after receiving chronic cocaine, or during cocaine extinction) may have different results. A limited number of experiments have specifically addressed this question and support this concept (Table 2). All of the aforementioned experiments have analyzed baseline manipulations that occur during behavioral training and testing. However, in one experiment, it was found that loss of HDAC5 and not HDAC9 results in lasting sensitization to cocaine reward: HDAC5 knock-out had no effect on cocaine place conditioning at baseline but sensitized the ability of prior cocaine exposure to enhance place conditioning (Renthal et al., 2007). Furthermore, a series of key cocaine self-administration studies have found that the timing of HDAC inhibition is likely to be crucial. For example, Romieu et al (2008) found that the daily systemic (IV) administration of TSA during the initial acquisition phase of cocaine self-administration reduced instrumental responding including reduced break point in a progressive ratio test, whereas another study found that a single systemic injection of sodium butyrate (a broadly acting and non-specific HDAC inhibitor among many other actions) during the maintenance phase significantly enhanced administration behavior (Sun et al., 2008). Similarly, a recent study found that TSA and SAHA infusions in the NAc shell, but not core, greatly enhanced motivation for cocaine during the chronic phase of cocaine self-administration (long-term reinforcement of cocaine self-administration for more than 30 days) (Wang et al., 2010). Finally, Malvaez and colleagues found that systemic sodium butyrate injections given after CPP extinction and reinstatement training resulted in an enhancement of extinction and reduced reinstatement (Malvaez et al., 2010). Based on this range of interesting results, it is clear that, similar to ChIP and mRNA studies, acute exposure to cocaine imposes different effects compared to chronic cocaine, and presumably the environmental context also imposes unique effects—all of which are simultaneously affected by these manipulations.
Future directions and summary
Regulation of chromatin structure has greatly advanced our basic understanding of the pathophysiology of the addicted state, and offers a fundamentally new approach for the development of more effective treatments for drug addiction. Behavioral studies to date have left open major avenues of investigation as well as leaving key mechanistic questions unanswered. The hypothesis that ‘blocking cocaine’s ability to create a more permissive chromatin state serves as a potential therapeutic treatment for addiction’ needs further investigation. A promising line of research would be to assess the behavioral function of enzymes that mediate transcriptional activation such as H3K4, H3K36, or H3K79 methyltransferases. To date, the majority of studies have focused on the enzymes that mediate transcriptional repression—such as HDACs, H3K9 methyltransferases, and DNA methyltransferases. Moreover, despite having similar behavioral outcomes, the gene targets of HDACs, G9a, or DNMT inhibitors remain unknown. It is not known if these manipulations are affecting primarily cocaine-expressed genes (primed or desensitized genes) (Renthal et al., 2007) or completely novel genes—as was found in a recent study comparing target genes affected in the NAc by the HDAC inhibitor MS-275 or by an antidepressant medication (Covington et al., 2009). Furthermore, the degree of cross-talk and gene target overlap of each manipulation is not known. For example, since class III HDACs (sirtuins) regulate excitability of NAc neurons (Renthal et al., 2009), does this extend to other chromatin modifying enzymes as well? Similarly, do manipulations that inhibit DNA methylation also affect histone acetylation, and vice versa, as has been shown in the learning and memory field (Miller et al., 2008)?
Similar to behavior manipulations, a more thorough analysis of cocaine’s effects on chromatin structure and function demand attention. Two key weaknesses of the previously described genome-wide ChIP-chip study are that 1) the immunoprecipitation utilized antibodies that were not specific to individual PTMs, and 2) analysis was localized only to gene promoter regions (an inherent limitation of ChIP-chip). For the next step in gaining insight into why, for example, cocaine globally reduces H3K9me2, it will be crucial to immunoprecipitate with a specific anti-H3K9me2 antibody in a completely unbiased genome-wide setting using ChIP-seq (ChIP-sequencing) technology.
In addition to more thorough genomic analysis and more thorough characterization of novel modifications, two crucial technical challenges exist in linking chromatin function with gene expression. First is differentiating causality from correlation. At best, the presence of a given PTM at a given gene promoter can only be associated with a gene’s expression. Such correlation is even further confounded in examples such as the CART gene promoter, which has a bidirectional methylated H3 profile (see Fig. 1B). Even with pharmacological or overexpression studies, one cannot conclusively determine whether overexpression of G9a, for example, directly vs. indirectly influences transcription of the CART gene because such overexpression has global effects. To address this question, one must specifically target a PTM to the CART gene promoter. One possible route of accomplishing this very challenging task could be fusing a chromatin modifying enzyme to a protein that specifically binds to the CART gene promoter. This could be accomplished by knocking in specific ‘targetable’ sequences at a promoter, or perhaps by designing zinc-finger peptides to target specific sequences of DNA. This approach holds promise since it has proven successful in cell culture using a DNA methyltransferase fused to a zinc finger protein (Smith et al., 2008). If successfully applied in vivo, this could prove to be an extremely powerful tool in causally establishing the functional influence on transcription as well as providing insight in the in vivo half-life of a given PTM.
A second major technical challenge lies in further refining cell specific tools to analyze chromatin state within a given brain region of interest like the NAc. Because brain tissue is highly heterogeneous and contains many different classes of neurons and glia, data generated from ChIP studies may be a poor reflection of what is actually occurring in specific neurons. Furthermore, because ChIP analyses are carred out on tissue homogenates, it is difficult to ascertain whether differences reflect large binding changes that occur in absolute levels in a minority of cells, such as neuronal ensembles, or incremental changes that are occurring in a majority of cells in that region. Again referring to the CART promoter example (Fig. 1B), could this bidirectional profile be due to differences occurring in different populations of cells? This scenario is entirely plausible since NAc neurons which express Gs-coupled dopamine D1 receptors have vastly different baseline and cocaine-induced mRNA expression profiles compared to Gi-coupled D2 receptor containing neurons (Heiman et al., 2008; Lobo et al., 2006). Theoretically, since ChIP has been successfully combined with FACS (Flourescence Activated Cell Sorting) of brain tissue (by sorting Neun+ cells to isolate neurons specifically), it should be possible to ChIP even further purified populations of cells (Jiang et al., 2008).
In summary, we have outlined an emerging concept that chronic exposure to cocaine induces at least two distinct patterns of gene induction and that these patterns can be thought of as transcriptional states (gene desensitization vs. gene priming) that likely have an epigenetic basis. Desensitized genes, such as c-fos, are less induced with repeated cocaine use and this is thought to be due to a loss of acH4 and persistent gain of H3K9me2 at these promoters. At the same time, many examples exist where genes are “primed” to be induced by chronic cocaine. Primed genes appear to be largely characterized by a gain of promoter acH3 and a loss of H3K9me2. Moreover, microarray and ChIP-chip analyses suggest that these transcriptional states (desensitized and primed gene promoters) exist at hundreds if not thousands of loci in the genome of NAc neurons. The aforementioned modifications represent just a minute fraction of the types of histone modifications known, and genome-wide promoter analyses represent just a small fraction of the total genome, and all of these data come from only a single time point (24 hrs) after cocaine administration. Clearly, we are at the very beginning stages in understanding the epigenetic basis of cocaine-induced gene expression. Overlaid on this genomic complexity lies the need to understand the detailed behavioral significance of epigenetic modifications. Currently, the data suggest that blocking cocaine’s ability to create a more permissive chromatin state blunts the rewarding effects of cocaine, but we do not yet know whether promoting or blunting such reward represents the best therapeutic approach for addiction. Finally, going one step beyond general manipulations of histone modifications, creative work is needed to understand whether different states of gene desensitization or priming may be related to different motivational states or stages of addiction. Altogether, such studies of chromatin open a new window on the molecular basis of drug addiction. The hope is that as novel insight into addiction is achieved, it will be possible one day to take advantage of this information to develop better diagnostic tests for addiction and to develop improved treatments and ultimately preventive measures.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Bibliography
- Alibhai IN, Green TA, Potashkin JA, Nestler EJ. Regulation of fosB and DeltafosB mRNA expression: in vivo and in vitro studies. Brain Res. 2007;1143:22–33. doi: 10.1016/j.brainres.2007.01.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bibb JA, Chen J, Taylor JR, Svenningsson P, Nishi A, Snyder GL, Yan Z, Sagawa ZK, Ouimet CC, Nairn AC, Nestler EJ, Greengard P. Effects of chronic exposure to cocaine are regulated by the neuronal protein Cdk5. Nature. 2001;410:376–80. doi: 10.1038/35066591. [DOI] [PubMed] [Google Scholar]
- Borrelli E, Nestler EJ, Allis CD, Sassone-Corsi P. Decoding the epigenetic language of neuronal plasticity. Neuron. 2008;60:961–74. doi: 10.1016/j.neuron.2008.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brami-Cherrier K, Valjent E, Herve D, Darragh J, Corvol JC, Pages C, Arthur SJ, Girault JA, Caboche J. Parsing molecular and behavioral effects of cocaine in mitogen- and stress-activated protein kinase-1-deficient mice. J Neurosci. 2005;25:11444–54. doi: 10.1523/JNEUROSCI.1711-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Covington HE, 3rd, Maze I, LaPlant QC, Vialou VF, Ohnishi YN, Berton O, Fass DM, Renthal W, Rush AJ, 3rd, Wu EY, Ghose S, Krishnan V, Russo SJ, Tamminga C, Haggarty SJ, Nestler EJ. Antidepressant actions of histone deacetylase inhibitors. J Neurosci. 2009;29:11451–60. doi: 10.1523/JNEUROSCI.1758-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finkel T, Deng CX, Mostoslavsky R. Recent progress in the biology and physiology of sirtuins. Nature. 2009;460:587–91. doi: 10.1038/nature08197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeman WM, Lull ME, Patel KM, Brucklacher RM, Morgan D, Roberts DC, Vrana KE. Gene expression changes in the medial prefrontal cortex and nucleus accumbens following abstinence from cocaine self-administration. BMC Neurosci. 2010;11:29. doi: 10.1186/1471-2202-11-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green TA, Alibhai IN, Unterberg S, Neve RL, Ghose S, Tamminga CA, Nestler EJ. Induction of activating transcription factors (ATFs) ATF2, ATF3, and ATF4 in the nucleus accumbens and their regulation of emotional behavior. J Neurosci. 2008;28:2025–32. doi: 10.1523/JNEUROSCI.5273-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimm JW, Lu L, Hayashi T, Hope BT, Su TP, Shaham Y. Time-dependent increases in brain-derived neurotrophic factor protein levels within the mesolimbic dopamine system after withdrawal from cocaine: implications for incubation of cocaine craving. J Neurosci. 2003;23:742–7. doi: 10.1523/JNEUROSCI.23-03-00742.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heiman M, Schaefer A, Gong S, Peterson JD, Day M, Ramsey KE, Suarez-Farinas M, Schwarz C, Stephan DA, Surmeier DJ, Greengard P, Heintz N. A translational profiling approach for the molecular characterization of CNS cell types. Cell. 2008;135:738–48. doi: 10.1016/j.cell.2008.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hope BT, Nye HE, Kelz MB, Self DW, Iadarola MJ, Nakabeppu Y, Duman RS, Nestler EJ. Induction of a long-lasting AP-1 complex composed of altered Fos-like proteins in brain by chronic cocaine and other chronic treatments. Neuron. 1994;13:1235–44. doi: 10.1016/0896-6273(94)90061-2. [DOI] [PubMed] [Google Scholar]
- Hyman SE, Malenka RC, Nestler EJ. Neural mechanisms of addiction: the role of reward-related learning and memory. Annu Rev Neurosci. 2006;29:565–98. doi: 10.1146/annurev.neuro.29.051605.113009. [DOI] [PubMed] [Google Scholar]
- Jiang Y, Matevossian A, Huang HS, Straubhaar J, Akbarian S. Isolation of neuronal chromatin from brain tissue. BMC Neurosci. 2008;9:42. doi: 10.1186/1471-2202-9-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalivas PW, Volkow N, Seamans J. Unmanageable motivation in addiction: a pathology in prefrontal-accumbens glutamate transmission. Neuron. 2005;45:647–50. doi: 10.1016/j.neuron.2005.02.005. [DOI] [PubMed] [Google Scholar]
- Koob G, Kreek MJ. Stress, dysregulation of drug reward pathways, and the transition to drug dependence. Am J Psychiatry. 2007;164:1149–59. doi: 10.1176/appi.ajp.2007.05030503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouzarides T. Chromatin modifications and their function. Cell. 2007;128:693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- Kriaucionis S, Heintz N. The nuclear DNA base 5-hydroxymethylcytosine is present in Purkinje neurons and the brain. Science. 2009;324:929–30. doi: 10.1126/science.1169786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A, Choi KH, Renthal W, Tsankova NM, Theobald DE, Truong HT, Russo SJ, Laplant Q, Sasaki TS, Whistler KN, Neve RL, Self DW, Nestler EJ. Chromatin remodeling is a key mechanism underlying cocaine-induced plasticity in striatum. Neuron. 2005;48:303–14. doi: 10.1016/j.neuron.2005.09.023. [DOI] [PubMed] [Google Scholar]
- LaPlant Q, Vialou V, Covington HE, III, Maze I, Dietz DM, Renthal W, Oosting RS, Fan G, Nestler EJ. Role of DNA methylation in persistent cocaine-induced plasticity in the nucleus accumbens. Soc Nerosci Abs. 2009:254.2. [Google Scholar]
- Lee BM, Mahadevan LC. Stability of histone modifications across mammalian genomes: implications for ‘epigenetic’ marking. J Cell Biochem. 2009;108:22–34. doi: 10.1002/jcb.22250. [DOI] [PubMed] [Google Scholar]
- Levine AA, Guan Z, Barco A, Xu S, Kandel ER, Schwartz JH. CREB-binding protein controls response to cocaine by acetylating histones at the fosB promoter in the mouse striatum. Proc Natl Acad Sci U S A. 2005;102:19186–91. doi: 10.1073/pnas.0509735102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobo MK, Karsten SL, Gray M, Geschwind DH, Yang XW. FACS-array profiling of striatal projection neuron subtypes in juvenile and adult mouse brains. Nat Neurosci. 2006;9:443–52. doi: 10.1038/nn1654. [DOI] [PubMed] [Google Scholar]
- Malvaez M, Sanchis-Segura C, Vo D, Lattal KM, Wood MA. Modulation of chromatin modification facilitates extinction of cocaine-induced conditioned place preference. Biol Psychiatry. 2010;67:36–43. doi: 10.1016/j.biopsych.2009.07.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maze I, Covington HE, 3rd, Dietz DM, LaPlant Q, Renthal W, Russo SJ, Mechanic M, Mouzon E, Neve RL, Haggarty SJ, Ren Y, Sampath SC, Hurd YL, Greengard P, Tarakhovsky A, Schaefer A, Nestler EJ. Essential role of the histone methyltransferase G9a in cocaine-induced plasticity. Science. 2010;327:213–6. doi: 10.1126/science.1179438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClung CA, Nestler EJ. Regulation of gene expression and cocaine reward by CREB and DeltaFosB. Nat Neurosci. 2003;6:1208–15. doi: 10.1038/nn1143. [DOI] [PubMed] [Google Scholar]
- McClung CA, Nestler EJ, Zachariou V. Regulation of gene expression by chronic morphine and morphine withdrawal in the locus ceruleus and ventral tegmental area. J Neurosci. 2005;25:6005–15. doi: 10.1523/JNEUROSCI.0062-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller CA, Campbell SL, Sweatt JD. DNA methylation and histone acetylation work in concert to regulate memory formation and synaptic plasticity. Neurobiol Learn Mem. 2008;89:599–603. doi: 10.1016/j.nlm.2007.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nestler EJ, Kelz MB, Chen J. DeltaFosB: a molecular mediator of long-term neural and behavioral plasticity. Brain Res. 1999;835:10–7. doi: 10.1016/s0006-8993(98)01191-3. [DOI] [PubMed] [Google Scholar]
- Nestler EJ. Review. Transcriptional mechanisms of addiction: role of DeltaFosB. Philos Trans R Soc Lond B Biol Sci. 2008;363:3245–55. doi: 10.1098/rstb.2008.0067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renthal W, Maze I, Krishnan V, Covington HE, 3rd, Xiao G, Kumar A, Russo SJ, Graham A, Tsankova N, Kippin TE, Kerstetter KA, Neve RL, Haggarty SJ, McKinsey TA, Bassel-Duby R, Olson EN, Nestler EJ. Histone deacetylase 5 epigenetically controls behavioral adaptations to chronic emotional stimuli. Neuron. 2007;56:517–29. doi: 10.1016/j.neuron.2007.09.032. [DOI] [PubMed] [Google Scholar]
- Renthal W, Carle TL, Maze I, Covington HE, 3rd, Truong HT, Alibhai I, Kumar A, Montgomery RL, Olson EN, Nestler EJ. Delta FosB mediates epigenetic desensitization of the c-fos gene after chronic amphetamine exposure. J Neurosci. 2008;28:7344–9. doi: 10.1523/JNEUROSCI.1043-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renthal W, Nestler EJ. Epigenetic mechanisms in drug addiction. Trends Mol Med. 2008;14:341–50. doi: 10.1016/j.molmed.2008.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renthal W, Kumar A, Xiao G, Wilkinson M, Covington HE, 3rd, Maze I, Sikder D, Robison AJ, LaPlant Q, Dietz DM, Russo SJ, Vialou V, Chakravarty S, Kodadek TJ, Stack A, Kabbaj M, Nestler EJ. Genome-wide analysis of chromatin regulation by cocaine reveals a role for sirtuins. Neuron. 2009;62:335–48. doi: 10.1016/j.neuron.2009.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renthal W, Nestler EJ. Histone acetylation in drug addiction. Semin Cell Dev Biol. 2009;20:387–94. doi: 10.1016/j.semcdb.2009.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romieu P, Host L, Gobaille S, Sandner G, Aunis D, Zwiller J. Histone deacetylase inhibitors decrease cocaine but not sucrose self-administration in rats. J Neurosci. 2008;28:9342–8. doi: 10.1523/JNEUROSCI.0379-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russo SJ, Wilkinson MB, Mazei-Robison MS, Dietz DM, Maze I, Krishnan V, Renthal W, Graham A, Birnbaum SG, Green TA, Robison B, Lesselyong A, Perrotti LI, Bolanos CA, Kumar A, Clark MS, Neumaier JF, Neve RL, Bhakar AL, Barker PA, Nestler EJ. Nuclear factor kappa B signaling regulates neuronal morphology and cocaine reward. J Neurosci. 2009;29:3529–37. doi: 10.1523/JNEUROSCI.6173-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith AE, Hurd PJ, Bannister AJ, Kouzarides T, Ford KG. Heritable gene repression through the action of a directed DNA methyltransferase at a chromosomal locus. J Biol Chem. 2008;283:9878–85. doi: 10.1074/jbc.M710393200. [DOI] [PubMed] [Google Scholar]
- Stipanovich A, Valjent E, Matamales M, Nishi A, Ahn JH, Maroteaux M, Bertran-Gonzalez J, Brami-Cherrier K, Enslen H, Corbille AG, Filhol O, Nairn AC, Greengard P, Herve D, Girault JA. A phosphatase cascade by which rewarding stimuli control nucleosomal response. Nature. 2008;453:879–84. doi: 10.1038/nature06994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strahl BD, Allis CD. The language of covalent histone modifications. Nature. 2000;403:41–5. doi: 10.1038/47412. [DOI] [PubMed] [Google Scholar]
- Sun J, Wang L, Jiang B, Hui B, Lv Z, Ma L. The effects of sodium butyrate, an inhibitor of histone deacetylase, on the cocaine- and sucrose-maintained self-administration in rats. Neurosci Lett. 2008;441:72–6. doi: 10.1016/j.neulet.2008.05.010. [DOI] [PubMed] [Google Scholar]
- Tsankova N, Renthal W, Kumar A, Nestler EJ. Epigenetic regulation in psychiatric disorders. Nat Rev Neurosci. 2007;8:355–67. doi: 10.1038/nrn2132. [DOI] [PubMed] [Google Scholar]
- Tweedie-Cullen RY, Reck JM, Mansuy IM. Comprehensive mapping of post-translational modifications on synaptic, nuclear, and histone proteins in the adult mouse brain. J Proteome Res. 2009;8:4966–82. doi: 10.1021/pr9003739. [DOI] [PubMed] [Google Scholar]
- Wang L, Lv Z, Hu Z, Sheng J, Hui B, Sun J, Ma L. Chronic cocaine-induced H3 acetylation and transcriptional activation of CaMKIIalpha in the nucleus accumbens is critical for motivation for drug reinforcement. Neuropsychopharmacology. 2010;35:913–28. doi: 10.1038/npp.2009.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkinson MB, Xiao G, Kumar A, LaPlant Q, Renthal W, Sikder D, Kodadek TJ, Nestler EJ. Imipramine treatment and resiliency exhibit similar chromatin regulation in the mouse nucleus accumbens in depression models. J Neurosci. 2009;29:7820–32. doi: 10.1523/JNEUROSCI.0932-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winstanley CA, LaPlant Q, Theobald DE, Green TA, Bachtell RK, Perrotti LI, DiLeone RJ, Russo SJ, Garth WJ, Self DW, Nestler EJ. DeltaFosB induction in orbitofrontal cortex mediates tolerance to cocaine-induced cognitive dysfunction. J Neurosci. 2007;27:10497–507. doi: 10.1523/JNEUROSCI.2566-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao WD, Gainetdinov RR, Arbuckle MI, Sotnikova TD, Cyr M, Beaulieu JM, Torres GE, Grant SG, Caron MG. Identification of PSD-95 as a regulator of dopamine-mediated synaptic and behavioral plasticity. Neuron. 2004;41:625–38. doi: 10.1016/s0896-6273(04)00048-0. [DOI] [PubMed] [Google Scholar]