

Abstract
Congenital stationary night blindness (CSNB) is a nonprogressive retinal disorder that can be associated with impaired night vision. The last decade has witnessed huge progress in ophthalmic genetics, including the identification of three genes implicated in the pathogenicity of autosomal-recessive CSNB. However, not all patients studied could be associated with mutations in these genes and thus other genes certainly underlie this disorder. Here, we report a large multigeneration family with five affected individuals manifesting symptoms of night blindness. A genome-wide scan localized the disease interval to chromosome 15q, and recombination events in affected individuals refined the critical interval to a 10.41 cM (6.53 Mb) region that harbors SLC24A1, a member of the solute carrier protein superfamily. Sequencing of all the coding exons identified a 2 bp deletion in exon 2: c.1613_1614del, which is predicted to result in a frame shift that leads to premature termination of SLC24A1 (p.F538CfsX23) and segregates with the disorder under an autosomal-recessive model. Expression analysis using mouse ocular tissues shows that Slc24a1 is expressed in the retina around postnatal day 7. In situ and immunohistological studies localized both SLC24A1 and Slc24a1 to the inner segment, outer and inner nuclear layers, and ganglion cells of the retina, respectively. Our data expand the genetic basis of CSNB and highlight the indispensible function of SLC24A1 in retinal function and/or maintenance in humans.
Main Text
Hereditary retinal diseases represent a broad range of retinal dysfunction and/or degeneration, including congenital stationary night blindness (CSNB). CSNB is a clinically and genetically heterogeneous group of nonprogressive retinal disorders that can be characterized by impaired night vision, decreased visual acuity, nystagmus, myopia, and strabismus.1 On the basis of the electroretinographic recordings that exhibit waveforms in response to flashes of light, which correspond to changes in the polarization of the photoreceptor and bipolar cells, CSNB can be classified in two groups.2,3 The Schubert-Bornschein type is characterized by an electronegative electroretinogram (ERG) at the scotopic bright flash, in which the amplitude of the b-wave is smaller than that of the a-wave,2 whereas the Riggs type is defined by proportionally reduced a- and b-waves.3 The Schubert-Bornschein and Riggs types of CSNB patients not only differ electrophysiologically, but manifest different clinical characteristics. Decreased visual acuity, myopia, and nystagmus can be associated with Schubert-Bornschein CSNB. To date, only a few cases of the Riggs type have been described, but patients with this form have visual acuity within a normal range and no symptoms of myopia and/or nystagmus.1
Familial cases of CSNB with autosomal-dominant, autosomal-recessive, or X-linked inheritance have been reported. Mutations in RHO (MIM 180380), PDE6B (MIM 180072), and GNAT1 (MIM 139330) have been associated with autosomal-dominant CSNB, and patients with mutations in these genes show the Riggs or Schubert-Bornschein type of ERG.4–9 On the other hand, all reported autosomal-recessive and X–linked cases manifest a typical Schubert-Bornschein form of ERG, which can be further subdivided into complete and incomplete forms. Clinically complete CSNB is characterized by a drastically reduced rod b-wave response but largely normal 30 Hz flicker cone responses due to solely ON bipolar cell dysfunction, whereas the incomplete type is characterized by both a reduced rod b-wave and substantially reduced 30 Hz flicker cone responses, due to both ON and OFF bipolar cell dysfunction.10 Mutations in GRM6 (MIM 604096) and TRPM1 (MIM 603576) lead to autosomal-recessive complete CSNB, whereas mutations in NYX (MIM 300278) to X-linked complete CSNB.11–17 Mutations in CABP4 (MIM 608965) have been associated with autosomal-recessive incomplete CSNB, and mutations in CACNA1F (MIM 300110) with X-linked incomplete CSNB.18–20
Herein, we report a large consanguineous pedigree named PKRP070 consisting of five affected individuals in four sibships (Figure 1). The family was recruited to participate in a collaborative study between the National Centre of Excellence in Molecular Biology (NCEMB, Pakistan) and the National Eye Institute (NEI, USA) to identify genetic lesions associated with retinal dystrophies. Institutional review board approval was obtained from both institutes, and the participating subjects gave informed written consent consistent with the tenets of the Declaration of Helsinki. Family PKRP070 resides in the southern part of the Punjab province of Pakistan, and altogether 25 individuals from this family agreed to participate in this study. A detailed medical history was obtained by interviewing of family members; patients were queried regarding age at onset, progression, and any other ocular or systemic abnormalities or diseases. All affected individuals complained about night blindness with normal day vision since early childhood. Four of the five affected individuals underwent a detailed ocular examination, and the clinical characteristics of these affected individuals are detailed in Table 1.
Figure 1.
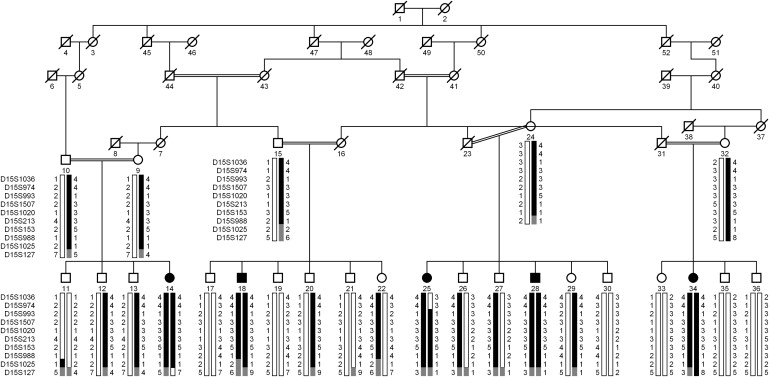
Pedigree of Family PKRP070 with Alleles for Chromosome 15q Markers
Alleles forming the risk haplotypes are shaded black, alleles cosegregating with the risk haplotype without homozygosity are shaded gray, and alleles not cosegregating with CSNB are shown in white.
Table 1.
Clinical Characteristics of Affected Individuals of Family PKRP070 Diagnosed with Autosomal-Recessive CSNB
|
Visual Acuity |
||||||
|---|---|---|---|---|---|---|
| Individual ID | First Symptoms | OD | OS | Disease Progression | Fundus Findings | Electroretinographic Characteristics |
| 14 | night blindness since early childhood | 6/6 | 6/6 | stationary | no macular atrophy, no pigment deposition, and no vascular attenuation | a- and b-waves are absent under the scotopic condition, whereas the cone responses are somewhat reduced under the photopic condition |
| 18 | night blindness since early childhood | 6/6 | 6/24 | stationary | no macular atrophy, no pigment deposition, and no vascular attenuation | a- and b-waves are absent under the scotopic condition, whereas the cone responses are somewhat reduced under the photopic condition |
| 28 | night blindness since early childhood | 6/18 | 6/9 | stationary | no macular atrophy, no pigment deposition, and no vascular attenuation | a- and b-waves are absent under the scotopic condition with normal cone responses under the photopic condition |
| 34 | night blindness since early childhood | 6/6 | 6/6 | stationary | no macular atrophy, no pigment deposition, and no vascular attenuation | a- and b-waves are absent under the scotopic condition with normal cone responses under the photopic condition |
OD, oculus dexter (right eye); OS, oculus sinister (left eye).
Ophthalmological examinations consisting of indirect dilated funduscopy were performed at Layton Rahmatulla Benevolent Trust (LRBT) hospital, Lahore, Pakistan. Fundus photographs were acquired with a Nikon camera (model D70S). Fundus examination revealed no obvious anomalies; the retinal vasculature, optic disc, and maculae were normal (Figure 2). Additionally, no intraretinal pigment migration was visible in the peripheral or central region of the retina (Figure 2). ERG measurements were recorded with equipment manufactured by LKC (Gaithersburg, MD, USA). Full-field ERG was performed on four affected individuals and one normal individual of family PKRP070. For scotopic ERG, patients were dark adapted and rod response was determined through incident flash attenuated by −25 dB, while the rod-cone response was measured at 0 dB. Cone response was recorded at 0 dB with 30 Hz flicker and background illumination of 20 cd/m2. ERG recordings demonstrated the absence of both a- and b-waves under the scotopic condition. Under the photopic condition, cone response was modestly reduced in individuals 14 and 18, whereas for individuals 28 and 34, the response was undistinguishable from the unaffected control (Figure S1, available online). Additionally, the rod response at −25 dB was absent in all four affected individuals and was not recovered 4 hr after dark adaptation (Figure S2). Taken together, the fundus photographs and the ERG results of rod response are strongly suggestive of CSNB with a Riggs type of ERG.
Figure 2.
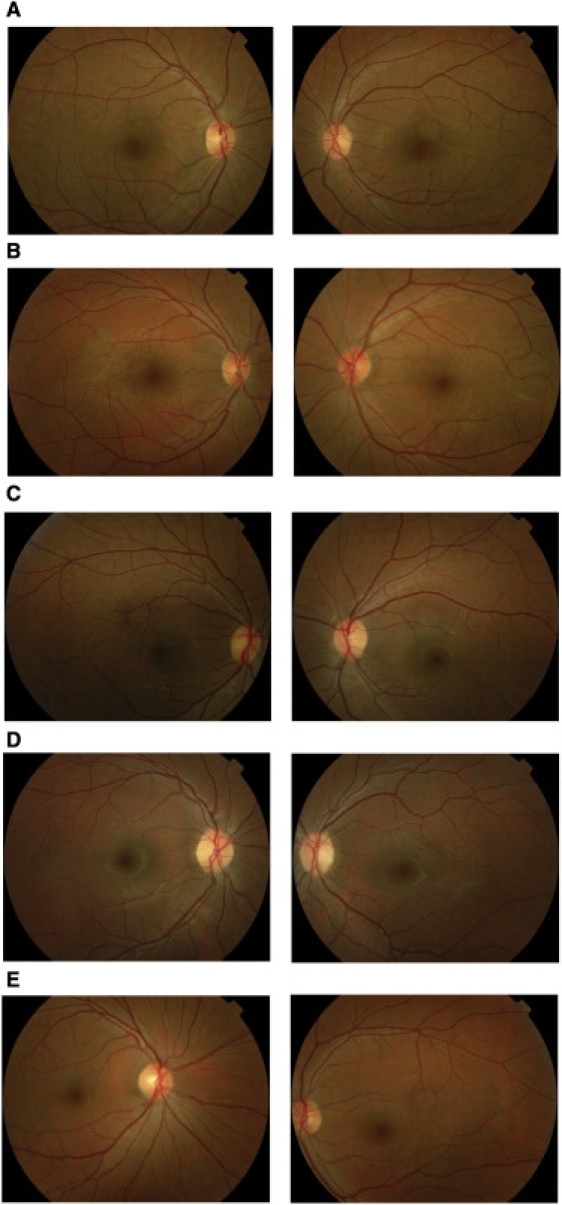
Fundus Photographs of Family PKRP070
(A) OD and OS of affected individual 14.
(B) OD and OS of affected individual 18.
(C) OD and OS of affected individual 28.
(D) OD and OS of affected individual 34.
(E) OD and OS of unaffected individual 35.
OD, oculus dexter (right eye); OS, oculus sinister (left eye).
Blood samples were collected from all participating members—both affected individuals and unaffected individuals—and genomic DNA was extracted as described.21,22 The size of the pedigree suggested sufficient power to achieve genome-wide statistical significance under a fully penetrant autosomal-recessive model. We therefore completed a genome-wide scan with 382 polymorphic fluorescent markers with an average resolution of 10 cM, from the ABI PRISM Linkage Mapping Set MD-10 (Applied Biosystems, Foster City, CA, USA). Multiplex PCRs were carried with the GeneAmp PCR System 9700 (Applied Biosystems), resolved on an ABI3100 DNA analyzer, and analyzed with GeneMapper (Applied Biosystems). Two-point linkage analyses were performed with the FASTLINK version of MLINK from the LINKAGE program package, whereas maximum two-point LOD scores were calculated with ILINK.23,24 Autosomal-recessive CSNB was analyzed as a fully penetrant trait with a disease allele frequency of 0.001. The marker order and distances between the markers were obtained from the Marshfield Clinic database (see Web Resources) and the National Center for Biotechnology Information chromosome 15 sequence maps. Equal allele frequencies were assumed for the initial genome scan, whereas for fine mapping, allele frequencies were derived from 96 unrelated and unaffected individuals. During the genome-wide scan, a two-point LOD score of 4.81 at θ = 0 was obtained with marker D15S153 (Table 2). No significant LOD scores (LOD > 3.0) were obtained with markers other than D15S153 during the genome-wide scan (Figure S3). Additional STR markers proximal and distal to D15S153 were selected from the Marshfield database and genotyped. As shown in Table 2, LOD scores of 5.92, 2.55, 5.55, 2.95, and 4.91 at θ = 0 were obtained with D15S993, D15S1507, D15S1020, D15S213, and D15S988, respectively (Table 2). A recombination event in individual 25 at D15S974 defines the proximal boundary, and the distal breakpoint was obtained from individual 14 at D15S127, delimiting the critical linkage interval to a 27.76 cM (28.52 Mb) region on chromosome 15q22.2-q26.1 (Figure 1). Lack of homozygosity in affected individual 18 at D15S1025 further delimits the critical region to a 10.41cM (6.53Mb) interval flanked by markers D15S974 proximally and D15S1025 distally.
Table 2.
Two-Point LOD Scores of Chromosome 15q Markers for Family PKRP070
| Marker | cM | Mb | 0 | 0.01 | 0.05 | 0.09 | 0.1 | 0.2 | 0.3 | Zmax | θmax |
|---|---|---|---|---|---|---|---|---|---|---|---|
| D15S1036 | 57.37 | 62.55 | −∞ | 1.10 | 1.54 | 1.52 | 1.49 | 1.07 | 1.10 | 1.54 | 0.05 |
| D15S974 | 59.05 | 62.87 | −∞ | 2.71 | 2.99 | 2.81 | 2.75 | 1.97 | 2.71 | 2.99 | 0.05 |
| D15S993 | 59.61 | 64.21 | 5.92 | 5.78 | 5.22 | 4.65 | 4.51 | 3.08 | 1.73 | 5.62 | 0.00 |
| D15S1507 | 60.17 | 65.34 | 2.55 | 2.5 | 2.26 | 2.01 | 1.95 | 1.3 | 0.71 | 2.55 | 0.00 |
| D15S1020 | 61.28 | 65.99 | 5.55 | 5.42 | 4.88 | 4.33 | 4.21 | 2.88 | 1.60 | 5.55 | 0.00 |
| D15S213 | 62.40 | 66.45 | 2.95 | 2.86 | 2.49 | 2.13 | 2.04 | 1.15 | 0.43 | 2.95 | 0.00 |
| D15S153a | 62.40 | 66.55 | 4.81 | 4.68 | 4.18 | 3.68 | 3.56 | 2.35 | 1.26 | 5.91 | 0.00 |
| D15S988 | 66.90 | 67.32 | 4.91 | 4.79 | 4.33 | 3.86 | 3.74 | 2.55 | 1.42 | 4.91 | 0.00 |
| D15S1025 | 69.46 | 69.40 | −0.64 | −0.08 | 0.39 | 0.49 | 0.5 | 0.44 | −0.08 | 0.50 | 0.10 |
| D15S127a | 86.81 | 91.39 | −∞ | −4.48 | −1.41 | −0.48 | −0.34 | 0.29 | 0.30 | 0.30 | 0.30 |
LOD scores were calculated at different θ values for each marker.
Marker included in genome scan.
The critical interval on chromosome 15q is a gene-rich region that harbors 109 annotated genes according to the UCSC database. We therefore prioritized candidate genes on the basis of their known function and available expression data in the retina and systematically started sequencing them by using the DNA samples from one affected and one unaffected individual of family PKRP070. SLC24A1 (MIM 603617), a sodium-calcium exchanger, was considered a plausible candidate because of its functionality and expression in the rod outer segment.25–27 The SLC24A1 transcript spans 5.7 kb and consists of ten exons that encode a 1099 amino acid protein. Previously, Sharon and colleagues investigated the involvement of SLC24A1and SLC24A2 (MIM 609838) in retinal diseases and identified 27 previously unreported sequence changes in SLC24A1.28 Of these, 21 variations were not considered sufficient to cause disease because they either did not cosegregate with the disease phenotype or did not affect conserved amino acid residues.28 The remaining two frameshift, three missense, and one likely previously unreported splice-site change were heterozygous variations, and although they were considered plausible, they could not be conclusively associated with the retinal disease phenotype because of the absence of the other pathogenic allele.28 Nonetheless, given the reported expression in the rod outer segment and the presence in the linkage interval, SLC24A1 remained an excellent candidate, and we therefore sequenced the coding exons, and exon-intron boundaries of SLC24A1. Primer pairs for individual exons of SLC24A1 and amplification conditions are available upon request. The PCR primers for each exon were used for bidirectional sequencing with the BigDye Terminator Ready reaction mix, according to manufacturer instructions. Sequencing products were precipitated and resuspended in 10 μl of formamide (Applied Biosystems) and denatured at 95°C for 5 min. Sequencing was performed in an ABI PRISM 3100 Automated Sequencer (Applied Biosystems). Sequencing results were assembled with ABI PRISM sequencing analysis software version 3.7 and analyzed with SeqScape software (Applied Biosystems).
Sequencing of SLC24A1 (NM_004727.2) in an affected individual identified a homozygous 2 bp deletion in exon 2, c.1613 _1614del (Figure 3), which is predicted to result in a frameshift leading to either premature termination of the encoded protein, p.F538CfsX23, or nonsense-mediated decay. This mutation segregates with the disease phenotype in the family: all affected individuals are homozygous for this change, whereas unaffected individuals are either heterozygous carriers or homozygous for the wild-type allele. Moreover, we did not find this change in 384 control chromosomes from the Punjab province of Pakistan or in 192 chromosomes of northern European descent. The clinical data of the patient presented herein are suggestive of CSNB with a Riggs type of ERG, and to date this form of CSNB has been described in only few cases with autosomal-dominant CSNB. Although our cohort does not contain any additional CSNB patients or families with a specific Riggs type of ERG, given the fact that many genes associated with retinal dystrophies harbor a wide spectrum of mutations responsible for different retinal phenotypes, we screened 16 different CSNB patients with the following inclusion criteria: patients with an incomplete or complete phenotype were females, and/or their phenotype was excluded in all CSNB-associated genes described to date, and/or consanguinity was noted and thus was indicative of autosomal-recessive inheritance, or their phenotype was not further classified into complete or incomplete CSNB. Additionally, we screened 48 probands from our familial cohort of autosomal-recessive RP. However, no additional pathogenic mutations were identified in SLC24A1.
Figure 3.
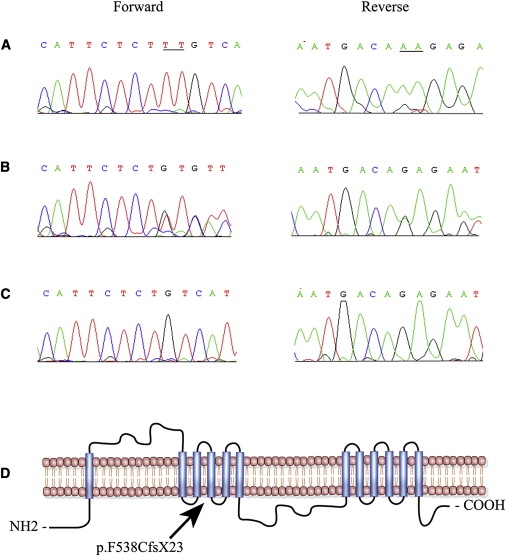
Sequence Chromatogram Illustrating the Causal Lesions and the Topological Model of SLC24A1
(A–C) Individual 11 (A), individual 12 (B), and individual 14 (C) show the wild-type allele, heterozygous, and homozygous 2 bp deletion c.1613_1614del, respectively. The underlined are the two bases deleted in the affected individuals of family PKFP070.
(D) Transmembrane domains of SLC24A1 predicted by Simple Modular Architecture Research Tools (SMART) algorithms. The deletion mutation resides in the fourth transmembrane that constitutes the first of two proposed ion exchanger domains of SLC24A1.
We then examined the expression of Slc24a1 in postnatal eyes (postnatal day 1 [P01] to P30) of C57BL/6 mice. Quantitative RT-PCR (qRT-PCR) was performed on different mouse eye tissues, with forward primer 5′-GGGAAAGAACACTGCGGTAA-3′ and reverse primer 5′-CCCATCCTGGTGCTTACTGT-3′, and the data were analyzed as published.29 The expression of Slc24a1 was normalized against two housekeeping genes, Gapdh (MIM 138400) and RpL19 (MIM 180466), as described previously.30 Expression levels (mean ± SEM) were calculated by analyzing at least three independent samples with replica reactions and presented on an arbitrary scale that represents the expression over the housekeeping genes. Rhodopsin mRNA levels served as a molecular marker for photoreceptor development and were measured in the same set of samples. We observed minimal expression of either Slc24a1 or rhodopsin prior to P05; however, the expression of both genes increased from P07, suggesting that Slc24a1 is present in photoreceptors (Figure 4A). The level of rhodopsin in the retina shows a steady increase after P7, reaching a maximum level at P30 (Figure 4A).31 Although, not surprisingly, the level of expression of the Slc24a1 transcript is much lower than that of rhodopsin, the profile is similar, with a steady increase from P07 to P30 (Figure 4A).
Figure 4.
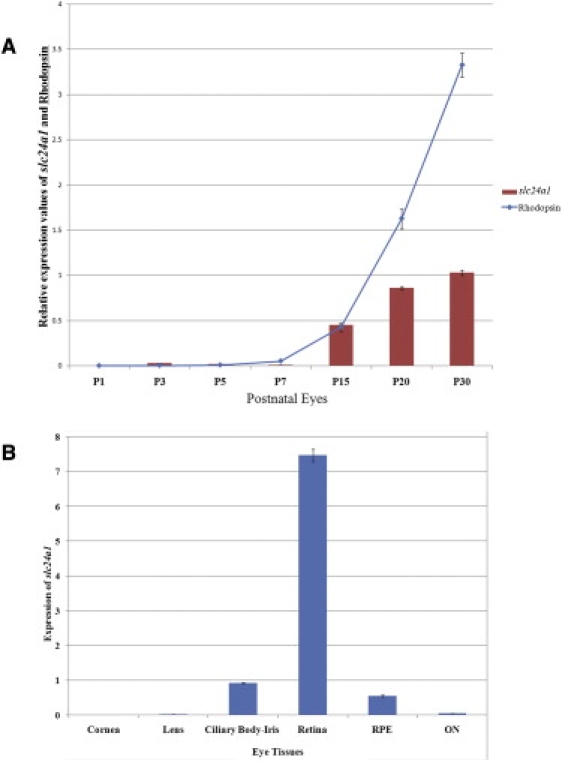
Quantitative Expression Analyses in the Adult Mouse Eye Tissue
(A) Expression of Slc24a1 and Rhodopsin was determined by qRT-PCR in the mouse eyes at P01, P03, P05, P10, P15, and P30. Slc24a1 expression is presented as bars on an arbitrary scale, and Rhodopsin expression is presented as a line. All values are the mean (± SEM) of three independent observations after normalization with the control gene (Rpl-19) expression.
(B) Quantitative expression of Slc24a1 in adult mouse eye tissue (180 days). Slc24a1 expression is presented as bars using an arbitrary scale on the y axis. Values are presented as the mean (± SEM) of three independent observations after normalization with the control gene (Rpl-19) expression. RPE, retinal pigment epithelium; ON, optic nerve.
Once the expression of Slc24a1 in the mouse eye was confirmed, we investigated the expression of Slc24a1 in various eye tissues of 24-week-old C57BL/6 mice. Slc24a1 transcript was detected in the retina, followed by low levels of expression in the iris-cilliary body and retinal pigment epithelium (Figure 4B). Slc24a1 is either not expressed or expressed at low levels in the mouse cornea, lens, and optic nerve (Figure 4B). The levels of Slc24a1 expressed in the mouse retina prompted us to further investigate the transcript levels in different retinal layers. We generated RNA probes (size ranging from 200 bp to 1200 bp) from a cDNA clone of SLC24A1 and performed in situ hybridization on 7 μm cryosections from the posterior segment of the human eye according to published protocols.32 In situ hybridization detected SLC24A1 message in the inner segment, the outer and inner nuclear layers, and ganglion cells (Figure 5).
Figure 5.
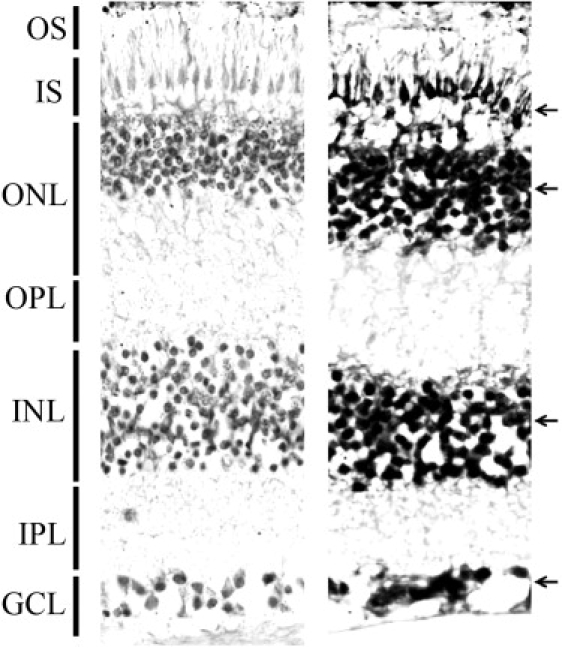
Localization of SLC24A1 in Human Retina with In Situ Hybridization Using Digoxigenin-Labeled Sense and Antisense Probes
Hybridization with sense probe as a control (left) and antisense probe (right) show strong labeling (black arrows) in the inner segment, outer and inner nuclear layers, and ganglion cell layer. OS, outer segment; IS, inner segment; ONL, outer nuclear layer; OPL, outer plexiform layer; IPL, inner plexiform layer; GCL, ganglion cell layer.
Previously, Reid and colleagues identified a sodium-calcium exchanger protein in the bovine rod outer segments and localized it exclusively to the plasma membrane of rod outer segments.25 Because our in situ results suggested a broader localization pattern for SLC24A1, we sought additional evidence and used a commercially available anti-SLC24A1 antibody (SAB2102180, Sigma-Aldrich, St. Louis, MO, USA) to detect Slc24a1 in different mouse retinal layers. P13 and adult CD1 mouse eyes were dissected and fixed overnight in 4% paraformaldehyde (Electron Microscopy Services) in 1× PBS. After cryoprotection using a sucrose gradient, mouse eyes were embedded in optimal cutting temperature compound medium. The retinal distribution of Slc24a1 was visualized by incubating 12-μm-thick sections of the retina with rabbit polyclonal anti-SLC24A1 antibody at 1:250 dilution overnight at 4°C, followed by goat anti-rabbit IgG conjugated with AlexaFluor 594 (Invitrogen). Slides were costained for F-actin with AlexaFluor488-conjugated phalloidin (Invitrogen, A12379) and nuclei with the use of Hoechst 33342 (1:2000 dilutions of each). Apotome-sectioned images were captured with a Zeiss Imager Z1 microscope equipped with AxioVS40 software version 4.8.1.0. Slc24a1 was detected in the inner segment, the outer and inner nuclear layers, and ganglion cells in both the P13 and adult mouse eyes, consistent with the results of the in situ hybridization experiment (Figure 6).
Figure 6.
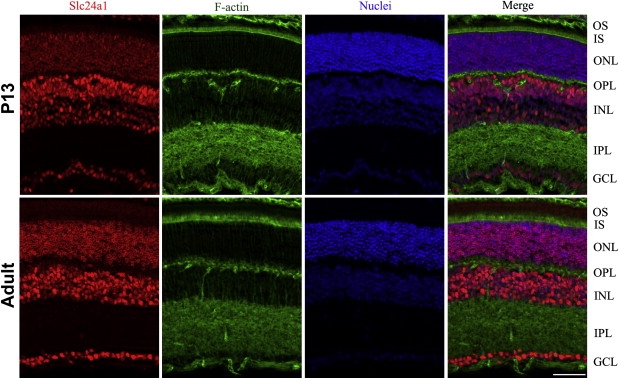
Immunolocalization of Slc24a1 Expression in P13 and Adult Mouse Retinal Layers
The slides were stained for Slc24a1 with goat anti-rabbit IgG conjugated with AlexaFluor 594 (red), F-actin using AlexaFluor488 conjugated phalloidin (green), and nuclei using Hoechst 33342 (blue). Merge represents an overlay of images from the first three columns in P13 and adult mouse retinal layers, respectively. The localization pattern illustrates a strong signal in the inner segment, outer and inner nuclear layers, and ganglion cell layer. The scale bar represents 50 um. OS, outer segment; IS, inner segment; ONL, outer nuclear layer; OPL, outer plexiform layer; IPL, inner plexiform layer; GCL, ganglion cell layer.
The genomic structure of this locus predicts that the pathogenic frameshift mutation will likely trigger nonsense-mediated decay (NMD). Even if some of the mutant mRNA escapes NMD, the topological models predicted by SMART algorithms suggest that the mutation resides early in the first of the two Na+/Ca2+- K+ ion exchanger domains and as such the resulting protein will lack part of the first and the entire second Na+/Ca2+- K+ ion exchanger domains that are involved in binding and transport of these physiologically important cations. Thus, either way the mutation is expected to have deleterious effect on the transport activity of SLC24A1. Calcium and sodium ions enter the rod outer segment through cGMP-gated channels,26 and Ca2+ balance is thought to be maintained by SLC24A1, where extrusion of Ca2+ is coupled with an inward flux of Na+. The complete or even partial loss of the ion exchange function would result in abnormal levels of intracellular Ca2+ concentrations that could potentially interfere with the proper functioning of the rod photoreceptors, resulting in the disease phenotype. In that regard, it is perhaps surprising that the homozygous frameshift results in a CSNB, but not an RP, phenotype. It is potentially significant that Sharon and colleagues reported several variations in SLC24A1 in RP patients but none of them could be causally associated with a retinal phenotype.28 As such, it is possible that this locus is sufficient to cause CSNB but also contributes modifying alleles to more severe, progressive retinal degenerations.
It is also unclear why the loss of SLC24A1 transporter protein that is expressed in different retinal layers manifests its phenotype only in the rod photoreceptor cells. One possible explanation is that a secondary transport mechanism for Ca2+ rescues the loss of SLC24A1 disease phenotype in other retinal layers. Development of an animal model will potentially help us understand the mechanism and the molecular players that help rescue the abnormal Ca2+ concentrations in inner segment, inner nuclear layers, and ganglion cells and will further elucidate the physiological significance of SLC24A1 and its role in the pathology of CSNB.
In conclusion, we report mapping of a locus for autosomal-recessive CSNB to 15q in a family with a Riggs type of ERG and subsequent identification of a pathogenic mutation in SLC24A1, segregating in an autosomal-recessive fashion in a consanguineous multigenerational family. Implication of SLC24A1 in the etiology of CSNB reaffirms the genetic heterogeneity of the disease. Further functional analysis will aid in clarifying the intricate details of retinal cellular machinery and rod outer segment physiology.
Acknowledgments
The authors are grateful to all family members for their participation in this study. None of the contributing authors have any financial interest related to this work. This work was supported in part by Higher Education Commission (H.E.C.), Islamabad, Pakistan; Ministry of Science and Technology, Islamabad, Pakistan; NIH-R00-DC009287-03 (Z.A.); Research to Prevent Blindness (Z.A.); Foundation Fighting Blindness, USA (R.A.), Research to prevent blindness (R.A.) and NIH-EY013198 (R.A.). N.K. is a Distinguished George W. Brumley Professor.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Marshfield Clinic Research Foundation, http://research.marshfieldclinic.org
National Center for Biotechnology Information (NCBI), http://www.ncbi.nlm.nih.gov/
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/
Primer3, http://frodo.wi.mit.edu/primer3
Retinal Information Network (RetNet), http://www.retnet.org
Simple Modular Architecture Research Tools (SMART), http://smart.embl-heidelberg.de
UCSC Genome Bioinformatics, http://genome.ucsc.edu
References
- 1.Zeitz C. Molecular genetics and protein function involved in nocturnal vision. Expert Rev. Ophthalmol. 2007;2:467–485. [Google Scholar]
- 2.Schubert G., Bornschein H. [Analysis of the human electroretinogram.] Ophthalmologica. 1952;123:396–413. doi: 10.1159/000301211. [DOI] [PubMed] [Google Scholar]
- 3.Riggs L.A. Electroretinography in cases of night blindness. Am. J. Ophthalmol. 1954;38:70–78. doi: 10.1016/0002-9394(54)90011-2. [DOI] [PubMed] [Google Scholar]
- 4.Dryja T.P., Berson E.L., Rao V.R., Oprian D.D. Heterozygous missense mutation in the rhodopsin gene as a cause of congenital stationary night blindness. Nat. Genet. 1993;4:280–283. doi: 10.1038/ng0793-280. [DOI] [PubMed] [Google Scholar]
- 5.Gal A., Orth U., Baehr W., Schwinger E., Rosenberg T. Heterozygous missense mutation in the rod cGMP phosphodiesterase beta-subunit gene in autosomal dominant stationary night blindness. Nat. Genet. 1994;7:551. doi: 10.1038/ng0894-551a. [DOI] [PubMed] [Google Scholar]
- 6.Dryja T.P., Hahn L.B., Reboul T., Arnaud B. Missense mutation in the gene encoding the alpha subunit of rod transducin in the Nougaret form of congenital stationary night blindness. Nat. Genet. 1996;13:358–360. doi: 10.1038/ng0796-358. [DOI] [PubMed] [Google Scholar]
- 7.Sandberg M.A., Weigel-DiFranco C., Dryja T.P., Berson E.L. Clinical expression correlates with location of rhodopsin mutation in dominant retinitis pigmentosa. Invest. Ophthalmol. Vis. Sci. 1995;36:1934–1942. [PubMed] [Google Scholar]
- 8.Rosenberg T., Haim M., Piczenik Y., Simonsen S.E. Autosomal dominant stationary night-blindness. A large family rediscovered. Acta Ophthalmol. (Copenh.) 1991;69:694–702. doi: 10.1111/j.1755-3768.1991.tb02046.x. [DOI] [PubMed] [Google Scholar]
- 9.Zeitz C., Gross A.K., Leifert D., Kloeckener-Gruissem B., McAlear S.D., Lemke J., Neidhardt J., Berger W. Identification and functional characterization of a novel rhodopsin mutation associated with autosomal dominant CSNB. Invest. Ophthalmol. Vis. Sci. 2008;49:4105–4114. doi: 10.1167/iovs.08-1717. [DOI] [PubMed] [Google Scholar]
- 10.Audo I., Robson A.G., Holder G.E., Moore A.T. The negative ERG: clinical phenotypes and disease mechanisms of inner retinal dysfunction. Surv. Ophthalmol. 2008;53:16–40. doi: 10.1016/j.survophthal.2007.10.010. [DOI] [PubMed] [Google Scholar]
- 11.Bech-Hansen N.T., Naylor M.J., Maybaum T.A., Sparkes R.L., Koop B., Birch D.G., Bergen A.A., Prinsen C.F., Polomeno R.C., Gal A. Mutations in NYX, encoding the leucine-rich proteoglycan nyctalopin, cause X-linked complete congenital stationary night blindness. Nat. Genet. 2000;26:319–323. doi: 10.1038/81619. [DOI] [PubMed] [Google Scholar]
- 12.Pusch C.M., Zeitz C., Brandau O., Pesch K., Achatz H., Feil S., Scharfe C., Maurer J., Jacobi F.K., Pinckers A. The complete form of X-linked congenital stationary night blindness is caused by mutations in a gene encoding a leucine-rich repeat protein. Nat. Genet. 2000;26:324–327. doi: 10.1038/81627. [DOI] [PubMed] [Google Scholar]
- 13.Dryja T.P., McGee T.L., Berson E.L., Fishman G.A., Sandberg M.A., Alexander K.R., Derlacki D.J., Rajagopalan A.S. Night blindness and abnormal cone electroretinogram ON responses in patients with mutations in the GRM6 gene encoding mGluR6. Proc. Natl. Acad. Sci. USA. 2005;102:4884–4889. doi: 10.1073/pnas.0501233102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zeitz C., van Genderen M., Neidhardt J., Luhmann U.F., Hoeben F., Forster U., Wycisk K., Mátyás G., Hoyng C.B., Riemslag F. Mutations in GRM6 cause autosomal recessive congenital stationary night blindness with a distinctive scotopic 15-Hz flicker electroretinogram. Invest. Ophthalmol. Vis. Sci. 2005;46:4328–4335. doi: 10.1167/iovs.05-0526. [DOI] [PubMed] [Google Scholar]
- 15.van Genderen M.M., Bijveld M.M., Claassen Y.B., Florijn R.J., Pearring J.N., Meire F.M., McCall M.A., Riemslag F.C., Gregg R.G., Bergen A.A., Kamermans M. Mutations in TRPM1 are a common cause of complete congenital stationary night blindness. Am. J. Hum. Genet. 2009;85:730–736. doi: 10.1016/j.ajhg.2009.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Audo I., Kohl S., Leroy B.P., Munier F.L., Guillonneau X., Mohand-Saïd S., Bujakowska K., Nandrot E.F., Lorenz B., Preising M. TRPM1 is mutated in patients with autosomal-recessive complete congenital stationary night blindness. Am. J. Hum. Genet. 2009;85:720–729. doi: 10.1016/j.ajhg.2009.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li Z., Sergouniotis P.I., Michaelides M., Mackay D.S., Wright G.A., Devery S., Moore A.T., Holder G.E., Robson A.G., Webster A.R. Recessive mutations of the gene TRPM1 abrogate ON bipolar cell function and cause complete congenital stationary night blindness in humans. Am. J. Hum. Genet. 2009;85:711–719. doi: 10.1016/j.ajhg.2009.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bech-Hansen N.T., Naylor M.J., Maybaum T.A., Pearce W.G., Koop B., Fishman G.A., Mets M., Musarella M.A., Boycott K.M. Loss-of-function mutations in a calcium-channel alpha1-subunit gene in Xp11.23 cause incomplete X-linked congenital stationary night blindness. Nat. Genet. 1998;19:264–267. doi: 10.1038/947. [DOI] [PubMed] [Google Scholar]
- 19.Strom T.M., Nyakatura G., Apfelstedt-Sylla E., Hellebrand H., Lorenz B., Weber B.H., Wutz K., Gutwillinger N., Rüther K., Drescher B. An L-type calcium-channel gene mutated in incomplete X-linked congenital stationary night blindness. Nat. Genet. 1998;19:260–263. doi: 10.1038/940. [DOI] [PubMed] [Google Scholar]
- 20.Zeitz C., Kloeckener-Gruissem B., Forster U., Kohl S., Magyar I., Wissinger B., Mátyás G., Borruat F.X., Schorderet D.F., Zrenner E. Mutations in CABP4, the gene encoding the Ca2+-binding protein 4, cause autosomal recessive night blindness. Am. J. Hum. Genet. 2006;79:657–667. doi: 10.1086/508067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kaul H., Riazuddin S.A., Yasmeen A., Mohsin S., Khan M., Nasir I.A., Khan S.N., Husnain T., Akram J., Hejtmancik J.F., Riazuddin S. A new locus for autosomal recessive congenital cataract identified in a Pakistani family. Mol. Vis. 2010;16:240–245. [PMC free article] [PubMed] [Google Scholar]
- 22.Kaul H., Riazuddin S.A., Shahid M., Kousar S., Butt N.H., Zafar A.U., Khan S.N., Husnain T., Akram J., Hejtmancik J.F., Riazuddin S. Autosomal recessive congenital cataract linked to EPHA2 in a consanguineous Pakistani family. Mol. Vis. 2010;16:511–517. [PMC free article] [PubMed] [Google Scholar]
- 23.Lathrop G.M., Lalouel J.M. Easy calculations of lod scores and genetic risks on small computers. Am. J. Hum. Genet. 1984;36:460–465. [PMC free article] [PubMed] [Google Scholar]
- 24.Schäffer A.A., Gupta S.K., Shriram K., Cottingham R.W., Jr. Avoiding recomputation in linkage analysis. Hum. Hered. 1994;44:225–237. doi: 10.1159/000154222. [DOI] [PubMed] [Google Scholar]
- 25.Reid D.M., Friedel U., Molday R.S., Cook N.J. Identification of the sodium-calcium exchanger as the major ricin-binding glycoprotein of bovine rod outer segments and its localization to the plasma membrane. Biochemistry. 1990;29:1601–1607. doi: 10.1021/bi00458a035. [DOI] [PubMed] [Google Scholar]
- 26.Schnetkamp P.P. The SLC24 Na+/Ca2+-K+ exchanger family: vision and beyond. Pflugers Arch. 2004;447:683–688. doi: 10.1007/s00424-003-1069-0. [DOI] [PubMed] [Google Scholar]
- 27.Kimura M., Jeanclos E.M., Donnelly R.J., Lytton J., Reeves J.P., Aviv A. Physiological and molecular characterization of the Na+/Ca2+ exchanger in human platelets. Am. J. Physiol. 1999;277:H911–H917. doi: 10.1152/ajpheart.1999.277.3.H911. [DOI] [PubMed] [Google Scholar]
- 28.Sharon D., Yamamoto H., McGee T.L., Rabe V., Szerencsei R.T., Winkfein R.J., Prinsen C.F., Barnes C.S., Andreasson S., Fishman G.A. Mutated alleles of the rod and cone Na-Ca+K-exchanger genes in patients with retinal diseases. Invest. Ophthalmol. Vis. Sci. 2002;43:1971–1979. [PubMed] [Google Scholar]
- 29.Mandal M.N., Vasireddy V., Reddy G.B., Wang X., Moroi S.E., Pattnaik B.R., Hughes B.A., Heckenlively J.R., Hitchcock P.F., Jablonski M.M., Ayyagari R. CTRP5 is a membrane-associated and secretory protein in the RPE and ciliary body and the S163R mutation of CTRP5 impairs its secretion. Invest. Ophthalmol. Vis. Sci. 2006;47:5505–5513. doi: 10.1167/iovs.06-0312. [DOI] [PubMed] [Google Scholar]
- 30.Ayyagari R., Mandal M.N., Karoukis A.J., Chen L., McLaren N.C., Lichter M., Wong D.T., Hitchcock P.F., Caruso R.C., Moroi S.E. Late-onset macular degeneration and long anterior lens zonules result from a CTRP5 gene mutation. Invest. Ophthalmol. Vis. Sci. 2005;46:3363–3371. doi: 10.1167/iovs.05-0159. [DOI] [PubMed] [Google Scholar]
- 31.He L., Campbell M.L., Srivastava D., Blocker Y.S., Harris J.R., Swaroop A., Fox D.A. Spatial and temporal expression of AP-1 responsive rod photoreceptor genes and bZIP transcription factors during development of the rat retina. Mol. Vis. 1998;4:32. [PubMed] [Google Scholar]
- 32.Nieto M.A., Patel K., Wilkinson D.G. In situ hybridization analysis of chick embryos in whole mount and tissue sections. Methods Cell Biol. 1996;51:219–235. doi: 10.1016/s0091-679x(08)60630-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.