

Abstract
Diaphanospondylodysostosis (DSD) is a rare, recessively inherited, perinatal lethal skeletal disorder. The low frequency and perinatal lethality of DSD makes assembling a large set of families for traditional linkage-based genetic approaches challenging. By searching for evidence of unknown ancestral consanguinity, we identified two autozygous intervals, comprising 34 Mbps, unique to a single case of DSD. Empirically testing for ancestral consanguinity was effective in localizing the causative variant, thereby reducing the genomic space within which the mutation resides. High-throughput sequence analysis of exons captured from these intervals demonstrated that the affected individual was homozygous for a null mutation in BMPER, which encodes the bone morphogenetic protein-binding endothelial cell precursor-derived regulator. Mutations in BMPER were subsequently found in three additional DSD cases, confirming that defects in BMPER produce DSD. Phenotypic similarities between DSD and Bmper null mice indicate that BMPER-mediated signaling plays an essential role in vertebral segmentation early in human development.
Main Text
Homozygosity mapping in consanguineous families can be an efficient way to define loci for recessively inherited disorders.1 In families with closely related parents, such as first-cousin matings, a large number of autozygous intervals are typically found in their affected offspring,2 which can make progress from locus to causative mutation difficult. Fewer and smaller intervals are predicted when the parents have more distant familial relationships, but knowledge of these relationships is frequently unknown. However, detection of autozygosity in sporadic cases of recessive disorders can be used to identify unknown ancestral consanguinity, reflecting more distant genetic relationships and thereby reducing the candidate intervals in which the mutated gene is likely to be localized to a more tractable size, simplifying the identification of the causative mutations.
Diaphanospondylodysostosis (DSD, MIM 608022) is a rare, recessively inherited, perinatal lethal skeletal disorder.3–6 The primary skeletal characteristics of the phenotype include a small chest, abnormal vertebral segmentation, and posterior rib gaps containing incompletely differentiated mesenchymal tissue.6 Consistent craniofacial features include ocular hypertelorism, epicanthal folds, a depressed nasal bridge with a short nose, and low-set ears. The most commonly described extraskeletal finding is nephroblastomatosis with cystic kidneys, but other visceral findings have been described in some cases.3
The low frequency and perinatal lethality of DSD made it impossible to assemble a set of families for linkage studies. We therefore tested four genetically independent DSD cases for autozygosity with whole-genome SNP data. All clinical data, radiographs, and tissue were obtained through the International Skeletal Dysplasia Registry (ISDR) and with informed consent under an institutional review board-approved protocol. The SNP array genotyping and identity-by-descent (IBD) analysis were performed as described in Tompson et al., 2008.7 Briefly, DNA was processed and hybridized to GeneChip Human Mapping 250K arrays (NspI) as per manufacturer's recommendations (Affymetrix). Genotypes were called with GeneChip DNA analysis software (version 2.0, Affymetrix) and then analyzed in Mathematica via a sliding-window approach with a maximum of five errors allowed in 100 SNPs, as described previously.7
In one case, ISDR reference number R05-062A (II-2 in Figures 1A and 1B), there were three blocks of homozygosity, each larger than 5 Mbps (Figure 1C), providing strong evidence of a consanguineous mating. Homozygosity for the same haplotype for one of these intervals, on chromosome 6, was shared with an unaffected sibling (II-1), leaving the two intervals on chromosomes 1 and 7 as candidate intervals uniquely associated with DSD. These two regions together contained 34 Mbps, comprising 166 known genes (see Table S1 available online).
Figure 1.

Radiographic Phenotype and Homozygosity Mapping in Diaphanospondylodysostosis Family R05-062
(A) Pedigree of the family.
(B) Anteroposterior radiograph of individual II-2. Note the abnormalities of the ribs, including rib gaps, and the defects in vertebral segmentation and ossification.
(C) Homozygosity mapping in II-2 showing the sizes of the blocks across the genome in cM. The block on chromosome 6 was also present in the unaffected sibling, II-2 (see Table S1).
To determine whether one of the intervals contained a DSD mutation, we employed a targeted capture-and-resequencing approach for individual II-2. The genomic library was prepared as described in Illumina library generation protocol version 2.3, and the array-based targeting enrichment protocol was performed with custom 244K microarrays (Agilent) via a protocol modified from Lee et al., 2009.8 The arrays were designed to target every exon in RefSeq in the autozygous intervals identified. Specifically, the probes included all validated comparative genomic hybridization probes for all exons in the intervals, at least two probes were used for each exon, and there was 30 bp spacing between adjacent probes. The hybridization of the DNA library to the arrays and all washing steps were performed as recommended in the Agilent CGH array protocol. However, after hybridization, stripping was performed with a low salt buffer at 5° above the hybridization temperature. The eluted sample was then enriched by PCR, and the hybridization-and-washing process was repeated with the same array for further enrichment. The sample was loaded onto one channel of an eight channel Illumina GAIIx flow cell, and 75 bp paired-end sequencing was performed. The image data were processed with the Genome Analyzer Pipeline version 1.0, and six million sequences derived from the captured exons were aligned to the human reference genome, hg18 (University of California, Santa Cruz), by Burrows-Wheeler Alignment. Variants were further filtered, analyzed, and visualized with SAMtools9 and the UCSC Genome Browser (Figure 2).10 Single-nucleotide variants were selected for those observed in the homozygous state in exons but not present in dbSNP130, with a minimum coverage of four reads at the variant position, and the variant call represented greater than 90% of the reads (summarized in Table S2).
Figure 2.
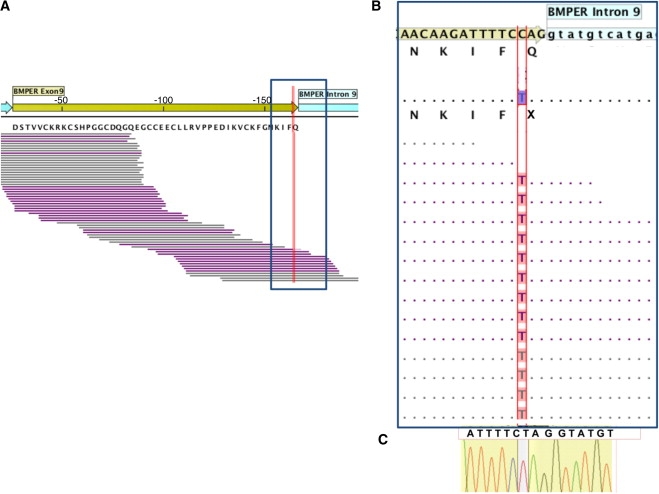
Molecular Basis of Diaphanospondylodysostosis
(A) The genomic structure and sequencing coverage for exon 9 of BMPER is shown, with the exon shown in yellow and the introns in blue (the specificity of capture for a larger region of BMPER is shown in Figure S1). Forward and reverse reads derived from the Solexa sequences are illustrated in purple and gray, respectively. The RefSeq BMPER amino acid sequence translation is directly under the reference nucleotide sequence, and the location of the variant identified is indicated by the red line.
(B) The zoomed panel of the region boxed in (A) shows the consensus of the Solexa sequence reads at each nucleotide position and the corresponding amino acid translation for individual II-2 below the reference sequence. Homozygosity for the pathological sequence change c.925C>T, which results in the p.Q309X premature termination codon, is shown.
(C) Confirmation of the mutation by Sanger sequence analysis of a PCR-amplified fragment containing exon 9 from II-2.
Four single-nucleotide variants that predicted homozygosity for nonsynonymous amino acid changes, all in genes in the interval on chromosome 7 and all of which were not found in dbSNP build 130, were identified (Table 1). Among the variants was a c.925C>T point mutation in exon 9 of BMPER (MIM 608699), which encodes the bone morphogenetic protein-binding endothelial cell precursor-derived regulator, predicting homozygosity for a p.Q309X null mutation (Figure 1D). Amplification and Sanger sequence analysis of exon 9 in the proband confirmed the mutation (Figure 1D), and both parents and the unaffected sibling were carriers of the change (data not shown).
Table 1.
SNPs Found in the Homozygous State by Targeted Resequencing that Were Not Identified in dbSNP130 and Predict Nonsynonymous Amino Acid Changes
| Chromosome | Gene | Exon | Coverage | DNA Sequence Change | Amino Acid Change |
|---|---|---|---|---|---|
| 7 | BMPER | 9 | 25 | C>T | p.Q309X |
| 7 | ELMO1 | 12 | 8 | A>C | p.I362S |
| 7 | ABCA13 | 15 | 17 | G>C | p.K886N |
| 7 | PKD1L1 | 17 | 24 | C>T | p.V906I |
To determine whether BMPER mutations were present in other DSD cases, we carried out mutation analysis of the 15 coding exons and flanking splice junctions in three genetically independent cases (Table 2). The 15 coding exons and flanking splice junctions of BMPER were amplified with the QIAGEN HotStart Hifidelity kit and primers that were designed with Primer3 (Table S3). Sequences of the PCR products were determined with the same amplification primers. Mutations are described relative to the BMPER cDNA (NM_133468.3), and protein sequence was numbered according to the first nucleotide of the translation start codon or the first amino acid, respectively.
Table 2.
Mutations Identified in Four Patients with Diaphanospondylodysostosis
| Patient | Location | Mutation | Result |
|---|---|---|---|
| R05-062A | Exon 9 | c.925C>T | p.Q309X |
| Exon 9 | c.925C>T | p.Q309X | |
| R06-244A | Exon 1 | c.26_35delCTCTGGCTGAinsAGACCAGAGCGGCG | p.Ala9GlufsX44 |
| Intron 11 | c.1031+5G>A | Unknown | |
| R83-044A | Exon 6 | c.514C>T | p.Q172X |
| Unknown | Unknown | Unknown | |
| R94-127A | Exon 12 | c.1109C>T | p.P370L |
| Exon 13 | c.1638T>A | p.C546X |
In case R06-244A, compound heterozygosity for a paternally derived complex insertion-deletion mutation, c.26_35delCTCTGGCTGAinsAGACCAGAGCGGCG, predicted to result in a frameshift (p.Ala9GlufsX44), was found in exon 1. The second mutation was maternally derived (c.1031+5G>A) and altered the consensus splice donor at the +5 position of intron 11. Patient cells or tissues expressing BMPER were not available, but the skipping of exon 11 would be predicted to lead to an in-frame deletion of 35 codons and delete much of the fifth chordin-like cysteine-rich domain, which is important for binding to BMPER-regulated bone morphogenetic proteins (BMPs).11,12 Retention of intron 11 would be predicted to lead to a null allele. This allele was absent from dbSNP build 130 and was not identified in 210 alleles derived from individuals in the same ethnic group, indicating that it is not a frequent polymorphism in the population (data not shown). In case R83-044A, a point mutation (c.514C>T) in exon 6 predicting a p.Q172X change was found. The second mutation was not identified, suggesting that the other allele either results in a deletion or does not reside within the coding region. For case R94-127A, compound heterozygosity for a c.1638T>A mutation in exon 13, predicted to result in a p.C546X nonsense mutation, and a c.1109C>T point mutation, predicted to lead to a p.P370L missense substitution in exon 12, were found. The proline is evolutionarily conserved among 28 different vertebrates, from primates to fish, and is in the conserved von Willebrand factor type D domain of the protein. The mutation was not found among 216 alleles from an ethnically matched control panel, indicating that it is not a frequent polymorphism in the population (data not shown). Thus, all three patients carried mutations in BMPER, demonstrating that mutations in BMPER cause DSD. Furthermore, because four of the six mutations are predicted to lead to premature termination codons, we conclude that absent or markedly diminished BMPER activity produces DSD.
Recently, a number of genes associated with recessive disorders have been identified by high-throughput sequence analysis in regions of linkage identified by homozygosity/autozygosity mapping in large extended consanguineous families13 and by overlapping regions of autozygosity in sets of smaller consanguineous families.14 In some instances, whole-genome15 or exome16,17 sequence analysis has been used. Here we show that empirically testing for autozygosity that reflects unknown ancestral consanguinity combined with high-throughput sequence analysis of the genes in the autozygous region or regions is another way to localize and identify the loci in recessive disorders of unknown etiology.
For DSD, the parents of the proband in whom autozygosity was detected originated in the state of Sinaloa, Mexico. One parent was from the city of Mazatlan, and the other was from a village 195 km away. The parents were unable to identify a common relative, suggesting that there may be additional carriers for the DSD mutation in this region of Mexico. These data are similar to instances of unknown consanguinity in Mexican families with spondyloepimetaphyseal dysplasia aggrecan type (MIM 612813)7 and recessive osteogenesis imperfecta type III (MIM 259420).18 Furthermore, a recent study identified excess homozygosity among Latinos, including many individuals of Mexican descent, who recently moved to the United States.19 Also, an increased number of long regions of homozygosity has been observed in a set of samples derived from Guadalajara, Mexico, relative to European and Asian populations,20 further supporting the inference that ancestral consanguinity may be common in the Mexican population. Thus, although a higher frequency of recessive disorders is observed in regions of the world where known consanguineous matings are culturally favored, identifying autozygosity because of unknown ancestral consanguinity by genotyping cases derived from other parts of the world, including Mexico, may be advantageous. For DSD, detection of autozygosity in a single sporadic case significantly reduced the candidate genomic intervals likely to harbor the mutated gene, an enrichment that facilitated the targeted sequence analysis that led to identification of the causal mutation.
DSD phenocopies many of the abnormalities found in the Bmper (also known as Cv2) knockout mouse,21–23 further supporting the BMPER sequence changes as the disease-causing mutations. The characteristic features of the human phenotype, perinatal death due to respiratory insufficiency, craniofacial abnormalities, lack of vertebral ossification, rib gaps, and renal abnormalities were recapitulated in the mouse model. Many of the phenotypic abnormalities seen in both species appear to result from an early developmental abnormality in mesenchymal differentiation that is necessary for proper maturation of the vertebrae, kidney, and other tissues,21–23 confirming the importance of BMP signaling in these processes.
BMPs are members of the TGF-β superfamily, play a pivotal role in the signaling networks in many organs and tissues, and are required for normal skeletal morphogenesis.24 BMP pathways have been linked to regulating cell proliferation, apoptosis, differentiation, and morphogenesis.25 Bmper is a paralog of chordin and kielin forming a family of molecules, which has been thought to function as BMP antagonists by binding BMPs to limit their activities.26 However, recent data suggest that BMPER has both antagonistic and agonistic mechanisms of action on BMP signaling,27 with the relative concentrations of BMPER and its ligands determining whether Bmper has a pro- or anti-BMP signaling role.21–23,27 Abnormalities in vertebral development in the Bmper knockout mice share similarities with Bmp428 and Bmp729 knockout mice, models of deficient BMP signaling, thus supporting a predominantly pro-BMP role in vivo for BMPER in skeletal development. In the Bmper knockout mice, the defects in the vertebrae are enhanced by a Bmp4 null allele21 and the kidney abnormalities are enhanced by absence of Bmp7,22 further suggesting that BMPER plays a pro-BMP role in these contexts. Similarly, absence of BMPER leads to reduced SMAD1 phosphorylation during vertebral and nephrogenic development,22,23 again suggesting that decreased BMP signaling results in the developmental defects observed in the mouse. Although the precise effects on BMP signaling that produce DSD in humans have not yet been characterized, it is likely that absence of BMPER disrupts the tight regulation of BMP signaling that governs development of the skeleton and other organs.30
Defects in vertebral segmentation, although morphologically distinct from those observed in DSD, have been associated with defects in components of the Notch signaling pathway, including DLL3 (MIM 602768), LNFG (MIM 602576), MESP2 (MIM 605195), and HES7 (MIM 608059).31–33 In the Bmper null mice, Pax1, an early marker of vertebral development, was normally expressed,21 indicating that Bmper functions downstream of the early stages of vertebral segmentation. The temporospatial relationship between the defect in Bmper and the Notch pathway is uncharacterized.
In summary, this study used empiric identification of autozygosity due to ancestral consanguinity in an autosomal-recessive disease to significantly refine candidate intervals containing the mutated gene and thus the genomic regions for targeted resequencing with a next-generation sequencing platform. Using this approach, we identified mutations in BMPER as the cause for diaphanospondylodysostosis and thus a requirement for BMPER-mediated signaling in human vertebral development.
Acknowledgments
We thank the families for their active participation in this work. We thank Stephen Moore and Keith Brown for providing resources that contributed greatly to the project. Microarray data were generated and analyses were performed within the UCLA DNA Microarray Facility. This work was supported in part by grants from the National Institutes of Health (DE019567, HD22657, and M01-RR00425).
Contributor Information
Vincent A. Funari, Email: vincent.funari@cshs.org.
Daniel H. Cohn, Email: dan.cohn@cshs.org.
Supplemental Data
Web Resources
The URL for data presented herein is as follows:
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/
Accession Numbers
The human BMPER NCBI reference sequence has accession number NM_133468.3.
References
- 1.Lander E.S., Botstein D. Homozygosity mapping: A way to map human recessive traits with the DNA of inbred children. Science. 1987;236:1567–1570. doi: 10.1126/science.2884728. [DOI] [PubMed] [Google Scholar]
- 2.Woods C.G., Cox J., Springell K., Hampshire D.J., Mohamed M.D., McKibbin M., Stern R., Raymond F.L., Sandford R., Malik Sharif S. Quantification of homozygosity in consanguineous individuals with autosomal recessive disease. Am. J. Hum. Genet. 2006;78:889–896. doi: 10.1086/503875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gonzales M., Verloes A., Saint Frison M.H., Perrotez C., Bourdet O., Encha-Razavi F., Joyé N., Taillemite J.L., Walbaum R., Pfeiffer R., Maroteaux P. Diaphanospondylodysostosis (DSD): Confirmation of a recessive disorder with abnormal vertebral ossification and nephroblastomatosis. Am. J. Med. Genet. A. 2005;136A:373–376. doi: 10.1002/ajmg.a.30537. [DOI] [PubMed] [Google Scholar]
- 4.Nisbet D.L., Griffin D.R., Chitty L.S. Prenatal features of Noonan syndrome. Prenat. Diagn. 1999;19:642–647. doi: 10.1002/(sici)1097-0223(199907)19:7<642::aid-pd610>3.0.co;2-1. [DOI] [PubMed] [Google Scholar]
- 5.Prefumo F., Homfray T., Jeffrey I., Moore I., Thilaganathan B. A newly recognized autosomal recessive syndrome with abnormal vertebral ossification, rib abnormalities, and nephrogenic rests. Am. J. Med. Genet. A. 2003;120A:386–388. doi: 10.1002/ajmg.a.20097. [DOI] [PubMed] [Google Scholar]
- 6.Vatanavicharn N., Graham J.M., Jr., Curry C.J., Pepkowitz S., Lachman R.S., Rimoin D.L., Wilcox W.R. Diaphanospondylodysostosis: Six new cases and exclusion of the candidate genes, PAX1 and MEOX1. Am. J. Med. Genet. A. 2007;143A:2292–2302. doi: 10.1002/ajmg.a.31934. [DOI] [PubMed] [Google Scholar]
- 7.Tompson S.W., Merriman B., Funari V.A., Fresquet M., Lachman R.S., Rimoin D.L., Nelson S.F., Briggs M.D., Cohn D.H., Krakow D. A recessive skeletal dysplasia, SEMD aggrecan type, results from a missense mutation affecting the C-type lectin domain of aggrecan. Am. J. Hum. Genet. 2009;84:72–79. doi: 10.1016/j.ajhg.2008.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lee H., O'Connor B.D., Merriman B., Funari V.A., Homer N., Chen Z., Cohn D.H., Nelson S.F. Improving the efficiency of genomic loci capture using oligonucleotide arrays for high throughput resequencing. BMC Genomics. 2009;10:646. doi: 10.1186/1471-2164-10-646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., 1000 Genome Project Data Processing Subgroup The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kent W.J., Sugnet C.W., Furey T.S., Roskin K.M., Pringle T.H., Zahler A.M., Haussler D. The human genome browser at UCSC. Genome Res. 2002;12:996–1006. doi: 10.1101/gr.229102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Conley C.A., Silburn R., Singer M.A., Ralston A., Rohwer-Nutter D., Olson D.J., Gelbart W., Blair S.S. Crossveinless 2 contains cysteine-rich domains and is required for high levels of BMP-like activity during the formation of the cross veins in Drosophila. Development. 2000;127:3947–3959. doi: 10.1242/dev.127.18.3947. [DOI] [PubMed] [Google Scholar]
- 12.Zhang J.L., Huang Y., Qiu L.Y., Nickel J., Sebald W. von Willebrand factor type C domain-containing proteins regulate bone morphogenetic protein signaling through different recognition mechanisms. J. Biol. Chem. 2007;282:20002–20014. doi: 10.1074/jbc.M700456200. [DOI] [PubMed] [Google Scholar]
- 13.Rehman A.U., Morell R.J., Belyantseva I.A., Khan S.Y., Boger E.T., Shahzad M., Ahmed Z.M., Riazuddin S., Khan S.N., Riazuddin S., Friedman T.B. Targeted capture and next-generation sequencing identifies C9orf75, encoding taperin, as the mutated gene in nonsyndromic deafness DFNB79. Am. J. Hum. Genet. 2010;86:378–388. doi: 10.1016/j.ajhg.2010.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Reversade B., Escande-Beillard N., Dimopoulou A., Fischer B., Chng S.C., Li Y., Shboul M., Tham P.Y., Kayserili H., Al-Gazali L. Mutations in PYCR1 cause cutis laxa with progeroid features. Nat. Genet. 2009;41:1016–1021. doi: 10.1038/ng.413. [DOI] [PubMed] [Google Scholar]
- 15.Roach J.C., Glusman G., Smit A.F., Huff C.D., Hubley R., Shannon P.T., Rowen L., Pant K.P., Goodman N., Bamshad M. Analysis of genetic inheritance in a family quartet by whole-genome sequencing. Science. 2010;328:636–639. doi: 10.1126/science.1186802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ng S.B., Turner E.H., Robertson P.D., Flygare S.D., Bigham A.W., Lee C., Shaffer T., Wong M., Bhattacharjee A., Eichler E.E. Targeted capture and massively parallel sequencing of 12 human exomes. Nature. 2009;461:272–276. doi: 10.1038/nature08250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Choi M., Scholl U.I., Ji W., Liu T., Tikhonova I.R., Zumbo P., Nayir A., Bakkaloğlu A., Ozen S., Sanjad S. Genetic diagnosis by whole exome capture and massively parallel DNA sequencing. Proc. Natl. Acad. Sci. USA. 2009;106:19096–19101. doi: 10.1073/pnas.0910672106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Alanay Y., Avaygan H., Camacho N., Utine G.E., Boduroglu K., Aktas D., Alikasifoglu M., Tuncbilek E., Orhan D., Bakar F.T. Mutations in the gene encoding the RER protein FKBP65 cause autosomal-recessive osteogenesis imperfecta. Am. J. Hum. Genet. 2010;86:551–559. doi: 10.1016/j.ajhg.2010.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shtir C.J., Marjoram P., Azen S., Conti D.V., Le Marchand L., Haiman C.A., Varma R. Variation in genetic admixture and population structure among Latinos: The Los Angeles Latino eye study (LALES) BMC Genet. 2009;10:71. doi: 10.1186/1471-2156-10-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Auton A., Bryc K., Boyko A.R., Lohmueller K.E., Novembre J., Reynolds A., Indap A., Wright M.H., Degenhardt J.D., Gutenkunst R.N. Global distribution of genomic diversity underscores rich complex history of continental human populations. Genome Res. 2009;19:795–803. doi: 10.1101/gr.088898.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ikeya M., Kawada M., Kiyonari H., Sasai N., Nakao K., Furuta Y., Sasai Y. Essential pro-Bmp roles of crossveinless 2 in mouse organogenesis. Development. 2006;133:4463–4473. doi: 10.1242/dev.02647. [DOI] [PubMed] [Google Scholar]
- 22.Ikeya M., Fukushima K., Kawada M., Onishi S., Furuta Y., Yonemura S., Kitamura T., Nosaka T., Sasai Y. Cv2, functioning as a pro-BMP factor via twisted gastrulation, is required for early development of nephron precursors. Dev. Biol. 2010;337:405–414. doi: 10.1016/j.ydbio.2009.11.013. [DOI] [PubMed] [Google Scholar]
- 23.Zakin L., Metzinger C.A., Chang E.Y., Coffinier C., De Robertis E.M. Development of the vertebral morphogenetic field in the mouse: Interactions between Crossveinless-2 and Twisted Gastrulation. Dev. Biol. 2008;323:6–18. doi: 10.1016/j.ydbio.2008.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mizutani C.M., Nie Q., Wan F.Y., Zhang Y.T., Vilmos P., Sousa-Neves R., Bier E., Marsh J.L., Lander A.D. Formation of the BMP activity gradient in the Drosophila embryo. Dev. Cell. 2005;8:915–924. doi: 10.1016/j.devcel.2005.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hogan B.L. Bone morphogenetic proteins: Multifunctional regulators of vertebrate development. Genes Dev. 1996;10:1580–1594. doi: 10.1101/gad.10.13.1580. [DOI] [PubMed] [Google Scholar]
- 26.Walsh D.W., Godson C., Brazil D.P., Martin F. Extracellular BMP-antagonist regulation in development and disease: Tied up in knots. Trends Cell Biol. 2010;20:244–256. doi: 10.1016/j.tcb.2010.01.008. [DOI] [PubMed] [Google Scholar]
- 27.Kelley R., Ren R., Pi X., Wu Y., Moreno I., Willis M., Moser M., Ross M., Podkowa M., Attisano L., Patterson C. A concentration-dependent endocytic trap and sink mechanism converts Bmper from an activator to an inhibitor of Bmp signaling. J. Cell Biol. 2009;184:597–609. doi: 10.1083/jcb.200808064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lawson K.A., Dunn N.R., Roelen B.A., Zeinstra L.M., Davis A.M., Wright C.V., Korving J.P., Hogan B.L. Bmp4 is required for the generation of primordial germ cells in the mouse embryo. Genes Dev. 1999;13:424–436. doi: 10.1101/gad.13.4.424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jena N., Martín-Seisdedos C., McCue P., Croce C.M. BMP7 null mutation in mice: Developmental defects in skeleton, kidney, and eye. Exp. Cell Res. 1997;230:28–37. doi: 10.1006/excr.1996.3411. [DOI] [PubMed] [Google Scholar]
- 30.Wan M., Cao X. BMP signaling in skeletal development. Biochem. Biophys. Res. Commun. 2005;328:651–657. doi: 10.1016/j.bbrc.2004.11.067. [DOI] [PubMed] [Google Scholar]
- 31.Turnpenny P.D., Alman B., Cornier A.S., Giampietro P.F., Offiah A., Tassy O., Pourquié O., Kusumi K., Dunwoodie S. Abnormal vertebral segmentation and the notch signaling pathway in man. Dev. Dyn. 2007;236:1456–1474. doi: 10.1002/dvdy.21182. [DOI] [PubMed] [Google Scholar]
- 32.Sparrow D.B., Guillén-Navarro E., Fatkin D., Dunwoodie S.L. Mutation of Hairy-and-Enhancer-of-Split-7 in humans causes spondylocostal dysostosis. Hum. Mol. Genet. 2008;17:3761–3766. doi: 10.1093/hmg/ddn272. [DOI] [PubMed] [Google Scholar]
- 33.Dunwoodie S.L. The role of Notch in patterning the human vertebral column. Curr. Opin. Genet. Dev. 2009;19:329–337. doi: 10.1016/j.gde.2009.06.005. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.