

Abstract
The International Haplotype Map Project (HapMap) has provided an essential database for studies of human population genetics and genome-wide association. Phases I and II of the HapMap project generated genotype data across ∼3 million SNP loci in 270 individuals representing four populations. Phase III provides dense genotype data on ∼1.5 million SNPs, generated by Illumina and Affymetrix platforms in a larger set of individuals. Release 3 of phase III of the HapMap contains 1397 individuals from 11 populations, including 250 of the original 270 phase I and phase II individuals and 1147 additional individuals. Although some known relationships among the phase III individuals have been described in the data release, the genotype data that are currently available provide an opportunity to empirically ascertain previously unknown relationships. We performed a systematic analysis of genetic relatedness and were able not only to confirm the reported relationships, but also to detect numerous additional, previously unidentified pairs of close relatives in the HapMap sample. The inferred relative pairs make it possible to propose standardized subsets of unrelated individuals for use in future studies in which relatedness needs to be clearly defined.
Introduction
The International Haplotype Map Project (HapMap) database provides a catalog of common human genetic variants across several populations. Phases I and II of the HapMap project reported genome-wide SNP data at ∼3 million markers in 270 individuals from four populations: Yoruba in Ibadan, Nigeria (YRI); Japanese in Tokyo, Japan (JPT); Han Chinese in Beijing, China (CHB); and Utah residents with ancestry from Northern and Western Europe (CEU).1,2 Phase III of the HapMap project genotyped ∼1.5 million SNPs in an expanded set of samples that includes seven additional populations: Luhya in Webuye, Kenya (LWK); Maasai in Kinyawa, Kenya (MKK); Toscani in Italy (TSI); Gujarati Indians in Houston, Texas (GIH); Chinese in Metropolitan Denver, Colorado (CHD); Mexican Americans in Los Angeles, California (MXL); and African Americans from the Southwestern United States (ASW).3
Some of the individuals included in the HapMap sample are known relatives of each other,1,3 and these relationships have been described in the data release. With the inclusion of the seven new populations in phase III of the HapMap, it has been suggested (Dimitromanolakis et al., 2009, Am. Soc. Hum. Gen., abstract) that there exist additional, previously undocumented relative pairs in the data set. We have therefore used genome-wide genotype data for release 3 of HapMap phase III to comprehensively identify pairs of close relatives present among the 1397 sampled individuals. We have compared our results with sample descriptions in the data release, and we describe numerous newly identified relationships. Using the inferred relative pairs, we suggest standardized subsets of unrelated individuals from HapMap phase III for use in studies in which it is important for relatedness to be clearly defined. Our construction of these panels follows similar work in the Human Genome Diversity Panel (HGDP-CEPH).4
Material and Methods
Genotype Data
Release 3 of phase III of the HapMap contains 1397 individuals, each of which had been genotyped on both the Illumina Human 1M BeadChip and the Affymetrix Genome-Wide Human SNP Array 6.0 platforms. The genotype data set available on these 1397 individuals (downloaded from the Sanger Center FTP website on September 8, 2009) consists of 1,457,407 SNPs: 1,423,833 on the 22 autosomes and 33,574 on the X chromosome. After quality control, the final data set for our analyses consisted of 1,441,951 SNPs: 1,409,608 on the autosomes and 32,343 on the X chromosome. We first removed 58 SNPs (all on the X chromosome) that were monomorphic in the data set. An additional 15,398 SNPs were excluded after they failed a χ2 test of the null hypothesis of Hardy-Weinberg equilibrium (with the Yates continuity correction5) in at least one of the 11 HapMap populations. To be excluded on the basis of Hardy-Weinberg disequilibrium, a SNP had to have at least four copies of the minor allele in the population in which it was being assessed and possess at least one of the following properties: (1) a test statistic > 19.51142 (p < 10−5 for a χ2 distribution; 1 degree of freedom [df]) in at least one population or (2) a test statistic > 6.634897 (p < 10−2 for a χ2 distribution; 1 df) in at least two populations.
Multidimensional Scaling
To search for population-labeling errors, we performed classical metric multidimensional scaling (MDS). We constructed an allele-sharing distance matrix, examining all pairs of individuals and using in the calculation for a given pair only those SNPs (among the 1,409,608 autosomal SNPs considered) for which neither individual was missing genotypes. Following previously described methods,6,7 we applied MDS on the matrix using the cmdscale command in R version 2.8.1.8 Separate MDS analyses were performed on subsets of the distance matrix containing only those individuals with recent ancestry in Africa (ASW, LWK, MKK, YRI), Europe (CEU, TSI), and East Asia (CHB, CHD, JPT). An additional computation used only the MXL and GIH populations.
Heterozygosity on the X Chromosome
To assess the accuracy of reported sex information, we determined the proportions of homozygous, heterozygous, and missing genotypes on the X chromosome (xhom, xhet, and xmiss, respectively) and on the autosomes (ahom, ahet, and amiss, respectively) for each of the 1397 HapMap individuals. This analysis used the 32,343 SNPs on the X chromosome and the 1,409,608 autosomal SNPs. We constructed two scatterplots to investigate sex assignment: (1) xmiss versus xhet/xhom and (2) ahet/ahom versus xhet/xhom. Male individuals are expected to have xhet/xhom ≈ 0 because xhet can differ from 0 only in the pseudoautosomal region, because of genotyping errors, or in heterozygous genomic duplications. They should therefore cluster near 0 on the x axis in both scatterplots. Female individuals are expected to have values of xhet/xhom substantially greater than 0 because xhet is expected to be a sizeable positive number in females.
RELPAIR Analysis
We identified relative pairs by using RELPAIR9,10 (version 2.0.1). In this analysis, only the 1,012,200 autosomal SNPs that had passed quality control and were separately polymorphic in each of the 11 HapMap populations were considered. To investigate the robustness of the inference of relative pairs to the choice of markers included in the analysis, we examined five different values for the number of SNPs: 1999, 3999, 5999, 7999, and 9999 (9999 being the maximum number of markers allowed by RELPAIR). For each choice d of the number of markers, five different SNP panels with even SNP spacing were created as follows: For a given value of d, a marker spacing s was calculated as ⌊1,012,200/d⌋ (for example, 1,012,200/1999 = 506.4, s = 506 for d = 1999). An offset g for number of SNPs d was calculated as ⌊s/5⌋ (for example, 506/5 = 101.2, g = 101 for d = 1999). For SNP panel n (where 1 ≤ n ≤ 5), markers were chosen every s SNPs along a vector of the considered 1,012,200 SNPs, starting at position g(n − 1). In this vector, SNPs were numbered starting at 0 and ordered from chromosome 1 to 22 and by increasing distance along each chromosome (with genomic positions provided by the HapMap, NCBI database build 36, dbSNP b126). For SNP panel n with number of markers d, SNP p (where 1 ≤ p ≤ d) was chosen as position s(p − 1) + g(n − 1). For example, for d = 1999, the first three SNPs chosen for the vector with n = 1 were 0 (506(1 − 1) + 101(1 − 1)), 506 (506(2 − 1) + 101(1 − 1)), and 1012 (506(3 − 1) + 101(1 − 1)), and the first three SNPs for n = 2 were 101 (506(1 − 1) + 101(2 − 1)), 607 (506(2 − 1) + 101(2 − 1)), and 1113 (506(3 − 1) + 101(2 − 1)). Thus, five nonoverlapping panels were chosen for each number of markers. For each pair of individuals, inference was performed with the putative relationship set to “unrelated.” For pairs of individuals in a given population, allele frequencies were set to the count estimates in their population. We used 0.001 for the genotyping error rate, a likely overestimate, and the critical value for the likelihood ratio computation in RELPAIR was set to 100. The genetic-map position of each marker was determined by interpolation on the Rutgers combined linkage-physical map.11
Allele-Sharing Analysis
The proportions of the SNPs at which a pair of individuals shared 0, 1, and 2 alleles identically by state—denoted p0, p1, and p2, respectively—were determined for each pair of individuals. Of the 1,409,608 autosomal SNPs considered, only those SNPs for which neither individual was missing genotypes were included in the calculation. We expect parent/offspring (PO) pairs to have low values of p0 because in such pairs, p0 can differ from 0 only as a result of genotyping errors or mutations. We expect full sibling (FS) pairs to have large values of p2 because such pairs share both alleles at a locus identically by descent for 25% of loci on average. Although second-degree relative pairs (half sibling, HS; avuncular, AV; grandparent-grandchild, GG) cannot be identified as confidently as PO and FS pairs by allele-sharing analysis, we expect HS, AV, and GG pairs to have values of p2 and p0 that are intermediate between those of PO and FS pairs and those of “unrelated” (UN) pairs.
Results
In five of the 11 HapMap populations (ASW, CEU, MKK, MXL, and YRI), many pairs of first-degree relatives have been well documented, because subject recruitment included parent/parent/offspring trios and parent/offspring duos.1,3 The genotype data provide an opportunity to systematically estimate relatedness solely on the basis of genetic inference, both to verify reported relationships and to search for unreported relationships. We first examined the population labels and sex assignment.
Population Affiliation
If the affiliation of an individual was mislabeled, on the basis of genetic similarity, we would expect the individual to not cluster with other individuals sharing the same population label. MDS analysis, both for the whole data set and for various subsets, can then reveal whether any mislabeling is likely.
Classical MDS analysis on the entire data set shows that, without exception, individuals from the same geographic region cluster together (Figure 1A). Individuals from populations with African, European, and East Asian ancestry form distinct clusters, in agreement with similar analyses performed on the HGDP-CEPH panel6,7,12,13 and release 2 of phase III of the HapMap.3 In the first two dimensions, individuals from the MXL and GIH populations form overlapping clusters between those of European and East Asian individuals.
Figure 1.
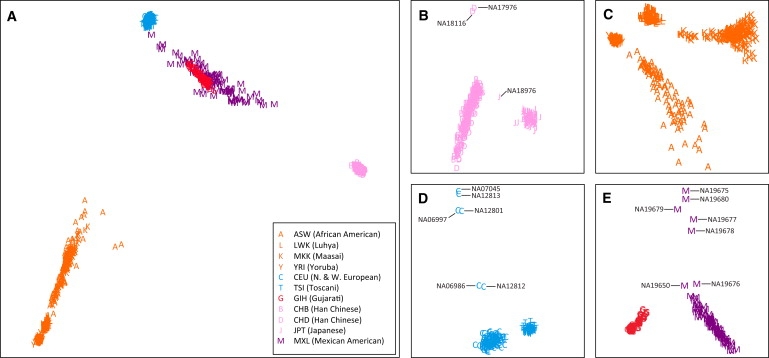
Classical Multidimensional Scaling
(A) The entire HapMap phase III release 3 data set of 1397 individuals.
(B) The 359 individuals from populations of East Asian descent (CHB, CHD, and JPT).
(C) The 584 individuals from populations of African descent (ASW, LWK, MKK, and YRI).
(D) The 267 individuals from populations of European descent (CEU and TSI).
(E) The 86 individuals from MXL and the 101 individuals from GIH.
Separate MDS analyses of individuals from each geographic region demonstrate that for each population, with the exception of the CHB and CHD populations, individuals from the population form a distinct cluster with respect to other populations from the same geographic region (Figures 1B–1E). The CHB and CHD populations, which are both composed of Han Chinese individuals (sampled at Beijing, China, and Denver, Colorado, respectively), form clusters that largely overlap. In contrast, the JPT individuals form a distinct cluster, with the exception that one individual (NA18976) is located between the JPT and Han Chinese clusters.
In the MDS analysis of the populations with African ancestry, including African Americans (LWK, MKK, YRI, and ASW; Figure 1C), individuals from the YRI and LWK populations form tight clusters. ASW individuals form a more dispersed cluster that reflects variability in the levels of African and European ancestry among these individuals. The MKK individuals are somewhat more dispersed than the YRI and LWK individuals, but they are more closely clustered than the ASW individuals.
In the MDS analysis of the two European populations (CEU and TSI; Figure 1D), the TSI are observed to form a single tight cluster, as are the CEU, with the exception of six individuals who are located outside of this cluster. These six individuals are members of two HapMap-reported trios (parents NA07045 and NA06986 and offspring NA06997 from trio 13291; parents NA12812 and NA12813 and offspring NA12801 from trio 1454). Similarly, the GIH and MXL individuals form separate clusters (Figure 1E), with the exception of seven MXL individuals who are located outside of this cluster. All seven of these individuals are members of HapMap-reported trios: parents NA19675 and NA19676 and offspring NA19677 from trio M004; parents NA19678 and NA19679 and offspring NA19680 from trio M009; offspring NA19650 from trio M001.
On the basis of the MDS analysis, because individuals clustered with other members of their same populations and no individuals clustered with members of other populations (other than CHB and CHD), we conclude that there is no evidence for population-labeling errors.
Sex Assignment
We used the proportions of homozygous, heterozygous, and missing genotypes calculated separately for the 22 autosomes and the X chromosome to examine the HapMap-reported sex assignment for each of the 1397 individuals. All individuals reported as male cluster with an xhet/xhom ratio near zero (0 < xhet/xhom < 0.0073; Figure 2A). All individuals reported as female cluster with an xhet/xhom ratio greater than 0.1857 (Figure 2A), with the exception of six individuals: NA10854 (CEU), NA19176 (YRI), NA19332 (LWK), NA20506 (TSI), NA20530 (TSI), and NA21424 (MKK). Other than NA19332, for which xhet/xhom was 0.0326, the remaining five exceptions had 0 < xhet/xhom < 0.0016, similar to the values for the individuals reported as male. All six of these exceptions had high proportions of missing genotypes (xmiss), between 0.6290 and 0.6718, suggesting that their low xhet/xhom ratios might have been due to the presence of a large amount of missing genotype data on the X chromosome. However, one other individual reported as female had a similarly high level of missing data as these six individuals (NA20821; xmiss = 0.6370) but a value of xhet/xhom similar to those of other putative females (xhet/xhom = 0.2512; Figure 2A). This result suggests that the low xhet/xhom observed in the six anomalous individuals is not caused by their high level of missing data and is instead evidence of misreported sex information. We also note that all six of the anomalous individuals had 0.3806 < ahet/ahom < 0.4146 and 0.0007 < amiss < 0.0044, consistent with the range of values observed across all individuals in the data set (0.3175 < ahet/ahom < 0.4623 and 0.0003 < amiss < 0.0283) and similar to the values of ahet/ahom and amiss observed for other individuals in their respective populations (Figure 2B). This observation argues against the possibility of sample mixing in the anomalous individuals, which would be expected to increase heterozygosity in a genome-wide fashion. Thus, the unusual X chromosomal values for the six anomalous individuals are not likely to be due to poor DNA quality or genome-wide systematic errors in genotyping. On the basis of our analysis of X chromosomal heterozygosity, although we have confirmed the reported sex information for most individuals, we have identified six individuals whose reported sex is likely to be erroneous.
Figure 2.
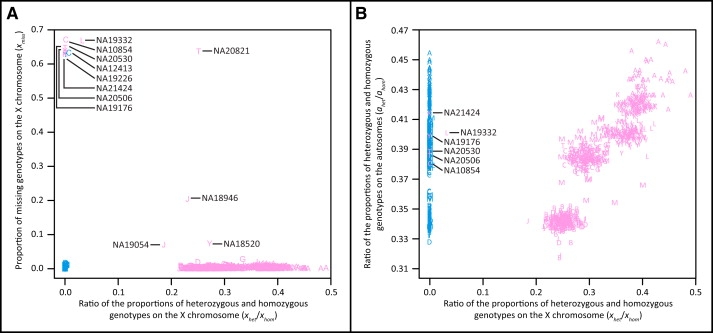
Missing Data and Heterozygosity on the X Chromosome
(A) The proportion of missing genotypes on the X chromosome versus the ratio of the proportions of heterozygous and homozygous genotypes on the X chromosome.
(B) The ratio of the proportions of heterozygous and homozygous genotypes on the autosomes versus the ratio of the proportions of heterozygous and homozygous genotypes on the X chromosome.
Pink letters represent HapMap-reported female individuals, and blue letters represent HapMap-reported male individuals. Letters used to signify population affiliations are as follows: A, ASW; C, CEU; B, CHB; D, CHD; G, GIH; J, JPT; L, LWK; K, MKK; M, MXL; T, TSI; Y, YRI.
Analysis of Relatives
To search for relative pairs, each pair of individuals in each population was evaluated both by the software package RELPAIR9,10 and by allele sharing. Because classical MDS analysis (Figure 1) shows that, except in the case of CHB and CHD, no individual clusters with a different population, it is sensible to carry out the inference of relatedness within populations, considering the possibility of interpopulation relationships only for CHB and CHD.
Eight different relationships are examined by RELPAIR: monozygotic twins (MZ), full siblings (FS), parent/offspring (PO), half siblings (HS), grandparent/grandchild (GG), avuncular (AV), first cousins (CO), and “unrelated” (UN). The following principles were used for identifying pairs of related individuals:
-
1.
If a relative pair inferred by RELPAIR was compatible with other RELPAIR-inferred relationships, it was accepted as “accurate.”
-
2.
If two or more relative pairs inferred by RELPAIR were incompatible (for example, if a parent in an inferred trio was also inferred to be FS with a second individual, but the offspring in that trio was inferred to be HS instead of AV with that second individual), the RELPAIR-inferred first-degree relationships were treated as “accurate” because these relationships are more confidently inferred by RELPAIR than are second-degree relationships.9,10 If one or more inferred second-degree relative pairs were incompatible with a first-degree relative pair and no further information on first-degree relative pairs was available to support the accuracy of the second-degree inferences, the second-degree pairs were treated as second-degree relative pairs of unknown relationship.
-
3.
In populations for which the number of relationships was particularly large, namely MKK, RELPAIR inference was particularly difficult. In such populations, the allele-sharing analysis was used to assist in decisions about the type of relationship.
RELPAIR Analysis
Pairs for which the inferred relationship differed from “unrelated” were identified for each of the 25 SNP panels (see Material and Methods). Some pairs yielded two or more distinct relationships (other than “unrelated”) across the different SNP panels. Such discrepancies typically occur when the likelihood-ratio statistics calculated by RELPAIR for different relationships are similar, resulting in different SNP panels reporting two, or in a few cases three or more, relationships. To arrive at a consensus inference for such pairs, the relationship supported by the largest number of SNP panels was used.
The relationships closer than first cousins inferred via RELPAIR are summarized in Tables S1–S5 (available online). We do not report first cousins, because inferences of cousin relationships are less reliable than those for closer relationships.9,10 All previously documented relative pairs were confirmed by our analysis (Table S1), except for four discrepancies that are not included in Table S1:
-
1.
NA20281 was identified in the HapMap genotype data files as the father of NA20284 in trio 2469 (ASW). However, their relationship was inferred as UN by RELPAIR. The Coriell Institute for Medical Research lists these individuals as unrelated. We conclude that this pair is UN.
-
2.
NA21410 and NA21434 had the same family ID (2600; MKK) in the HapMap genotype data files, but neither was identified as the parent of the other. However, RELPAIR analysis identified this pair as PO. This inferred relationship is compatible with the Coriell Institute for Medical Research listing, in which NA21410 is the father of NA21434. We conclude that this pair is PO.
-
3.
NA19984 was given as the father of NA19714 in trio 2437 (ASW) by the Coriell Institute for Medical Research. However, RELPAIR analysis identified these individuals as UN, in agreement with their assignment in the HapMap genotype data files. We conclude that this pair is UN.
-
4.
NA19195 and NA19196 were identified as parents in trio Y108 (YRI) by the Coriell Institute for Medical Research and were listed as unrelated in the HapMap genotype data files. However, RELPAIR analysis identified this pair as PO. In this case, all three sources of information were discordant. We conclude that this pair is PO.
In CHB and JPT, no relationships were inferred. Similarly, in a combined analysis of CHB and CHD, we did not identify any relationships in which one individual was from CHB and the other was from CHD. In TSI, no relationships closer than CO were inferred. In the remaining populations, varying numbers of both first- and second-degree relationships were inferred, the largest number of cases being observed in MKK (Table 1). One pair of individuals in MKK, NA21344 and NA21737, was inferred by RELPAIR to have an MZ relationship in all 25 SNP panels analyzed. Although it is possible that these two individuals are indeed MZ, a perhaps more likely explanation is that they are duplicate samples.
Table 1.
Summary of Previously Reported and RELPAIR-Inferred Relatives Present in Release 3 of Phase III of the HapMap
|
Population |
Reported Relatives |
Previously Unreported RELPAIR-Inferred Relative Pairs |
||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ID | Name | Sample Size | Trios | Duos | FS | AV | Trios | Duos | MZ | FS | HS | AV | GG | CP |
| ASW | African American | 87 | 10 | 21 | 2 | 0 | 0 | 0 | 0 | 3 | 2 | 11 | 1 | 0 |
| CEU | Northern and Western European | 165 | 44 | 8 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 2 | 0 | 0 |
| CHB | Han Chinese | 137 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| CHD | Han Chinese | 109 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 1 | 0 | 1 | 0 | 0 |
| GIH | Gujarati Indians | 101 | 0 | 0 | 0 | 0 | 1 | 1 | 0 | 1 | 0 | 1 | 0 | 0 |
| JPT | Japanese | 113 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| LWK | Luhya | 110 | 0 | 0 | 0 | 0 | 0 | 4 | 0 | 6 | 1 | 3 | 0 | 0 |
| MKK | Maasai | 184 | 28 | 1 | 0 | 0 | 2 | 7 | 1 | 16 | 19 | 35 | 22 | 4a |
| MXL | Mexican | 86 | 24 | 4 | 0 | 0 | 2 | 0 | 0 | 3 | 0 | 6 | 4 | 0 |
| TSI | Toscani | 102 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| YRI | Yoruba | 203 | 51 | 6 | 0 | 1 | 0 | 2 | 0 | 3 | 1 | 3 | 2 | 0 |
| Total | 1397 | 157 | 40 | 3 | 1 | 5 | 15 | 1 | 33 | 23 | 62 | 29 | 4 | |
MZ, monozygotic twins; FS, full siblings; HS, half siblings; AV, avuncular; GG, grandparent/grandchild; CP, complex; Trios, parent/parent/offspring; Duos, parent/offspring.
For three pairs of individuals inferred by RELPAIR as HS (NA21453 and NA21378; NA21617 and NA21520; NA21617 and NA21613) and one pair inferred as GG (NA21453 and NA21493), the relationships were incompatible in the constructed pedigrees. These four relationships were treated as uncertain second-degree relationships when constructing set HAP1117.
Inferences regarding first-degree relatives were highly reproducible. Nearly all inferences of first-degree relative pairs (PO and FS) were supported in all 25 SNP panels analyzed, with the exception of two previously documented PO pairs and one previously unreported pair:
-
1.
Reported PO pair NA12874 and NA12865 (CEU) was inferred to have a GG relationship in one of the five SNP panels with d = 9999. We conclude that this pair is PO.
-
2.
Reported PO pair NA12889 and NA12877 (CEU) was inferred to have a GG relationship in three SNP panels, one with d = 7999 and two with d = 9999. We conclude that this pair is PO.
-
3.
NA21362 and NA21438 (MKK) were inferred as FS by RELPAIR in all five SNP panels with d = 9999, four SNP panels with d = 7999, and one SNP panel with d = 5999. However, they were inferred as HS (nine SNP panels), AV (five SNP panels), and CO (one SNP panel with d = 1999) in the remaining SNP panels. Because true FS relationships tend to be inferred much more definitively, we tentatively conclude that this pair has a second-degree relationship. However, we are unable to determine the exact nature of the relationship.
While the first two cases are likely due to chance fluctuations in signal for genuine PO pairs, the third case likely reflects more complex background relatedness in the MKK population. Because no additional relative pairs that could support inferences about this latter pair were identified, we do not make a specific claim for the relationship of this pair. In all cases other than these three, because inferences of first-degree relative pairs were seen in all 25 SNP panels, we conclude that the inference of first-degree relatives is likely to be accurate.
The inference of second-degree relationships (HS, GG, and AV) did not exhibit the same level of consistency over different marker densities and SNP panels, and for second-degree pairs, it was generally harder to assign a consensus relationship. Of the 118 inferred second-degree relative pairs, only 13 AV relationships and 12 GG relationships were inferred unanimously by all 25 SNP panels analyzed. For each of the 93 remaining second-degree relative pairs, we tentatively assigned the most frequently inferred relationship across the 25 SNP panels. In order to identify discrepancies and resolve conflicting results, we used the inferred pairwise relationships to construct pedigrees. We then used the combined information from multiple pairs to corroborate or clarify many of these second-degree relationships and to revise the initial assignments. An example is shown in Figure 3. Individual NA19685, who is the father in a HapMap-reported trio (M011), was inferred to have an unreported PO relationship with both NA19660 and NA19661, who were themselves reported as parents of NA19662 in another HapMap-reported trio (M008). These inferred PO relationships are supported by an inferred FS relationship between NA19685 and NA19662. Furthermore, individual NA19686, the reported offspring of NA19685 in trio M011, was inferred to have a GG relationship with both NA19660 and NA19661, as well as an AV relationship with NA19662. All of these inferences are compatible.
Figure 3.
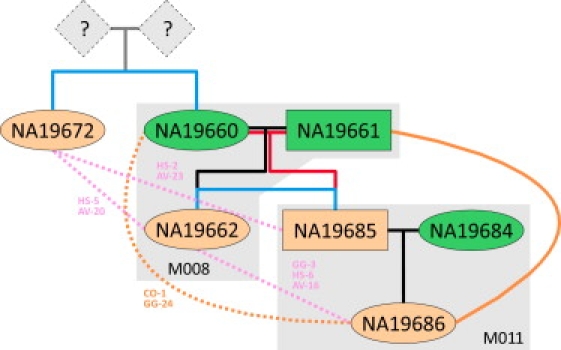
An Example Pedigree Created from RELPAIR-Inferred Relative Pairs in the MXL Population
Individuals shaded in green were retained in both sets, HAP1161 and HAP1117, and individuals shaded lightly in orange were removed in sets HAP1161 and HAP1117 because of first-degree RELPAIR-inferred relationships. A black line represents a previously reported relationship; red line, a RELPAIR-inferred PO relationship; blue line, a RELPAIR-inferred FS relationship; pink line, a RELPAIR-inferred AV relationship; orange line, a RELPAIR-inferred GG relationship. Solid lines indicate a relationship inferred in all 25 SNP panels. Dashed lines indicate a relationship inferred in a majority of SNP panels; the number of times a relationship was inferred is shown adjacent to the line. Pedigree numbers appear along the bottom of the shaded gray regions, each of which represents a HapMap-reported pedigree.
In some instances, the RELPAIR-inferred second-degree relationship may differ from the final relationship reported in Table S4. These differences arise from incompatibilities between two or more RELPAIR-inferred second-degree relationships. In each case, the relationship that is compatible with other relationships is reported in Table S4. Overall, 27 of the 118 inferred second-degree relative pairs had their relationship changed after pedigree-assisted revision. This rate is comparable to the previously reported accuracy of RELPAIR in inferring second-degree relationships.10
Allele-Sharing Analysis
Figure 4 displays the levels of allele sharing for pairs of individuals from the MXL population. Similar plots appear for the other ten HapMap populations in Figures S1–S4. In all populations with HapMap-reported PO pairs (ASW, CEU, MKK, MXL, and YRI), these pairs form a cluster with p0 near 0 (p0 < 0.00095), as expected from the fact that parents and offspring share at least one allele at a locus identically by descent. Similarly, previously unreported PO pairs that were inferred by RELPAIR all had p0 close to 0 (p0 < 0.00041) and were observed to cluster with HapMap-reported PO pairs. In each population, RELPAIR-inferred FS pairs were observed to form a distinct cluster with high values of p2 (0.711 < p2 < 0.786) and low values of p0 (0.01019 < p0 < 0.01975), with the exception of one MKK pair with p0 and p2 outside of these ranges, as discussed below. In all populations, with the exception of MKK, RELPAIR-inferred second-degree relative pairs were observed to cluster separately from the “unrelated” individuals. In MKK, the RELPAIR-inferred second-degree pairs with the highest values of p0 were situated close to pairs whose RELPAIR-inferred relationship was CO or more distant.
Figure 4.
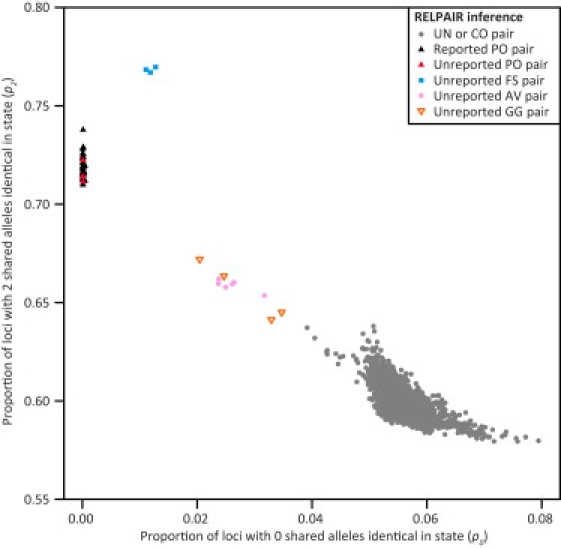
Allele Sharing for Pairs of Individuals in MXL
The plot contains 52 previously reported PO pairs together with 17 RELPAIR-inferred pairs: four PO pairs, three FS pairs, six AV pairs, and four GG pairs.
Several discrepancies appeared in MKK between RELPAIR-inferred relative pairs and the allele-sharing analysis. As noted above, NA21362 and NA21438 were inferred as FS by RELPAIR in ten SNP panels but as AV, HS, and CO in the remaining 15 SNP panels. In the allele-sharing analysis (Figure S1C), this pair is found to cluster with pairs with inferred HS, AV, and GG relationships. It is therefore unlikely that this pair is FS, and we find that allele-sharing analysis supports a second-degree relationship whose exact nature we are unable to determine. Additionally, five pairs inferred by RELPAIR to be GG (NA21357 and NA21576, NA21420 and NA21524, NA21509 and NA21576, NA21519 and NA21635, NA21519 and NA21678) were found to cluster among pairs with inferred CO and UN relationships and away from other pairs with inferred HS, AV, and GG relationships. On the basis of the allele-sharing patterns, it is therefore possible that these five pairs are more distantly related than GG. However, to be conservative in identifying relative pairs for potential exclusion from standardized sets of individuals, we treat these pairs as GG.
Proposed Subsets of Unrelated Individuals
In order to facilitate future studies that require well-defined relatedness, we used the inferred relationships to assemble maximal sets of unrelated individuals. Because different studies may require different levels of stringency, we propose two standard sets, following a strategy similar to that previously used for the HGDP-CEPH panel.4 First, we propose HAP1161, a set of 1161 individuals that we selected by removing a member of every first-degree relative pair (PO and FS). Second, we propose HAP1117, consisting of 1117 individuals selected by the additional removal from HAP1161 of a member of every second-degree relative pair.
We do not exclude CO relationships, because inferences of CO pairs by RELPAIR often do not have sufficient support to warrant their exclusion. Among the 16 pairs that were inferred by RELPAIR as CO in 18 or more SNP panels and for which a conclusive determination of the relationship could be made solely on the basis of first-degree relationships in the full sample, seven inferred CO relationships conflicted with a first-degree relative pair and are therefore very likely to be erroneous. CO relationships are the most distant relationship investigated by RELPAIR, and more distant relationships not tested by RELPAIR, such as great-aunt/great-nephew and second cousins, can potentially give rise to an incorrect inference of CO for a pair of individuals.
For each pair, to minimize the number of exclusions required, we adopted procedures similar to those of Rosenberg4 to decide which individuals to exclude. Individuals with multiple inferred relationships were preferentially removed before individuals with a single inferred relationship. If either individual could be removed, the individual with the higher amount of missing data was excluded. A full list of the 236 individuals removed when constructing HAP1161 can be found in Table S6, and the 44 individuals removed from HAP1161 when constructing HAP1117 are given in Table S7.
Discussion
Our study provides a comprehensive evaluation of genetic relatedness between individuals in release 3 of phase III of the HapMap. Although most previously reported relationships described in the data release have been confirmed by our analysis, four such relationships were found not to be empirically supported. Furthermore, we have identified an additional 177 relationships that included five trios, 16 duos, 33 FS pairs, and 118 second-degree relative pairs (Table 1 and Tables S1–S5).
Generally, marker density did not greatly affect RELPAIR inferences. All inferred PO and FS pairs, with a few exceptions described above, were obtained in all SNP panels at all marker densities. The inference of second-degree relative pairs was less consistent, and only 13 AV relative pairs and 12 GG relative pairs were inferred in all SNP panels at all marker densities. Of the 27 second-degree relative pairs whose relationships were revised after pedigree construction (Table S4), in all but one case (NA19677 and NA19679), the relationship to which a pair was changed was the second most frequent relationship inferred for that pair. However, no general trend was observed between marker density and the inference of the relationship supported by the constructed pedigrees. Although fewer markers can often be used to infer second-degree relatives from genome-wide SNP data, with 5999 or more markers, SNP panels generally produced greater concordance in the type of relationship inferred (data not shown).
On the basis of the relative pairs we have identified, we have proposed standardized subsets of unrelated individuals for use in future studies in which relatedness needs to be clearly defined. Set HAP1161 has been constructed such that it contains no known pairs of individuals with a first-degree relationship (PO and FS), and set HAP1117 has been constructed such that it contains no known pairs of individuals with a relationship closer than first cousins. Our construction of these panels follows similar work for identifying related individuals in the HGDP-CEPH panel4 to ensure compatibility of sample sets when jointly analyzing these two widely used resources.
Although we have taken care to ensure that HAP1117 contains no relatives closer than first cousins, in MKK it is difficult to be certain that after exclusions in Tables S6 and S7 are made, no relative pairs closer than first cousins are present. The large number of relative pairs identified in MKK suggests that there exists considerable background relatedness in this sample. Although this result could be a consequence of sampling procedures employed during recruitment, it could also be due to sociogenetic outcomes of cultural practices with regard to marriage and reproduction14,15 or to recent demographic events in the history of the Maasai population.16 We recommend that particular caution should be exercised when interpreting patterns of genetic variation in the MKK sample.
Acknowledgments
The authors would like to thank William Duren and Michael Boehnke for their assistance with RELPAIR and Zachary Szpiech for providing the C program used to calculate allele-sharing distance. This investigation was supported by NIH grant GM081441 (N.A.R.), and by grants from the Burroughs Wellcome Fund (N.A.R.) and the Alfred P. Sloan Foundation (N.A.R.).
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Coriell Institute for Medical Research, http://www.coriell.org/
The International HapMap Project, http://hapmap.ncbi.nlm.nih.gov/
Sanger Center FTP website, ftp://ftp.sanger.ac.uk/pub/hapmap3/r3/
References
- 1.International HapMap Consortium A haplotype map of the human genome. Nature. 2005;437:1299–1320. doi: 10.1038/nature04226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.International HapMap Consortium A second generation human haplotype map of over 3.1 million SNPs. Nature. 2007;449:851–861. doi: 10.1038/nature06258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.International HapMap 3 Consortium Integrating common and rare genetic variation in diverse human populations. Nature. 2010;467:52–58. doi: 10.1038/nature09298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rosenberg N.A. Standardized subsets of the HGDP-CEPH Human Genome Diversity Cell Line Panel, accounting for atypical and duplicated samples and pairs of close relatives. Ann. Hum. Genet. 2006;70:841–847. doi: 10.1111/j.1469-1809.2006.00285.x. [DOI] [PubMed] [Google Scholar]
- 5.Weir B.S. Sinauer; Sunderland, MA: 1996. Genetic data analysis II. [Google Scholar]
- 6.Jakobsson M., Scholz S.W., Scheet P., Gibbs J.R., VanLiere J.M., Fung H.C., Szpiech Z.A., Degnan J.H., Wang K., Guerreiro R. Genotype, haplotype and copy-number variation in worldwide human populations. Nature. 2008;451:998–1003. doi: 10.1038/nature06742. [DOI] [PubMed] [Google Scholar]
- 7.Wang C., Szpiech Z.A., Degnan J.H., Jakobsson M., Pemberton T.J., Hardy J.A., Singleton A.B., Rosenberg N.A. Comparing spatial maps of human population-genetic variation using Procrustes analysis. Stat. Appl. Genet. Mol. Biol. 2010;9 doi: 10.2202/1544-6115.1493. Article 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.R Development Core Team . R Foundation for Statistical Computing; Vienna, Austria: 2009. R: A Language and Environment for Statistical Computing. [Google Scholar]
- 9.Boehnke M., Cox N.J. Accurate inference of relationships in sib-pair linkage studies. Am. J. Hum. Genet. 1997;61:423–429. doi: 10.1086/514862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Epstein M.P., Duren W.L., Boehnke M. Improved inference of relationship for pairs of individuals. Am. J. Hum. Genet. 2000;67:1219–1231. doi: 10.1016/s0002-9297(07)62952-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Matise T.C., Chen F., Chen W., De La Vega F.M., Hansen M., He C., Hyland F.C.L., Kennedy G.C., Kong X., Murray S.S. A second-generation combined linkage physical map of the human genome. Genome Res. 2007;17:1783–1786. doi: 10.1101/gr.7156307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li J.Z., Absher D.M., Tang H., Southwick A.M., Casto A.M., Ramachandran S., Cann H.M., Barsh G.S., Feldman M., Cavalli-Sforza L.L., Myers R.M. Worldwide human relationships inferred from genome-wide patterns of variation. Science. 2008;319:1100–1104. doi: 10.1126/science.1153717. [DOI] [PubMed] [Google Scholar]
- 13.Biswas S., Scheinfeldt L.B., Akey J.M. Genome-wide insights into the patterns and determinants of fine-scale population structure in humans. Am. J. Hum. Genet. 2009;84:641–650. doi: 10.1016/j.ajhg.2009.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Coast E. Maasai marriage: a comparative study of Kenya and Tanzania. J. Comp. Fam. Stud. 2006;37:399–420. [Google Scholar]
- 15.Talle A. ‘Serious games’: licences and prohibitions in Maasai sexual life. Africa. 2007;77:351–370. [Google Scholar]
- 16.Waller R. Acceptees and Aliens: Kikuyu settlement in Maasailand. In: Spear T., Waller R., editors. Being Maasai: Ethnicity And Identity In East Africa. James Currey Ltd; London, United Kingdom: 1993. pp. 226–257. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.