

Abstract
Charcot-Marie-Tooth (CMT) disease comprises a genetically and clinically heterogeneous group of peripheral nerve disorders characterized by impaired distal motor and sensory function. Mutations in three genes encoding aminoacyl-tRNA synthetases (ARSs) have been implicated in CMT disease primarily associated with an axonal pathology. ARSs are ubiquitously expressed, essential enzymes responsible for charging tRNA molecules with their cognate amino acids. To further explore the role of ARSs in CMT disease, we performed a large-scale mutation screen of the 37 human ARS genes in a cohort of 355 patients with a phenotype consistent with CMT. Here we describe three variants (p.Leu133His, p.Tyr173SerfsX7, and p.Ile302Met) in the lysyl-tRNA synthetase (KARS) gene in two patients from this cohort. Functional analyses revealed that two of these mutations (p.Leu133His and p.Tyr173SerfsX7) severely affect enzyme activity. Interestingly, both functional variants were found in a single patient with CMT disease and additional neurological and non-neurological sequelae. Based on these data, KARS becomes the fourth ARS gene associated with CMT disease, indicating that this family of enzymes is specifically critical for axon function.
Main Text
Charcot-Marie-Tooth (CMT) disease represents a genetically and clinically heterogeneous group of peripheral neuropathies, with a prevalence of 1 in 2500 individuals.1 The major clinical features of CMT include distal muscular weakness and wasting, impaired sensation, steppage gait, pes cavus, and diminished deep-tendon reflexes.2,3 Broadly, CMT can be subdivided into two classes according to electrophysiological criteria.2 In CMT1, patients exhibit decreased motor nerve conduction velocities (MNCVs) and demyelination of peripheral nerve axons. In CMT2, patients do not show primary demyelination but do exhibit axonal loss accompanied by decreased amplitudes of evoked nerve responses.
Aminoacyl-tRNA synthetases (ARSs) are a ubiquitously expressed, essential family of enzymes responsible for charging tRNA molecules with their cognate amino acids in the cytoplasm and mitochondria.4 Interestingly, mutations in three genes encoding aminoacyl-tRNA synthetases have been implicated in CMT disease characterized by an axonal pathology: glycyl- (GARS [MIM 601472]), tyrosyl- (YARS [MIM 608323]), and alanyl- (AARS [MIM 613287]) tRNA synthetase.5–7 Although the molecular pathology of axonopathy associated with ARS mutations remains unclear, several mutant forms of GARS and YARS impair tRNA charging, cell viability in yeast assays, and cellular localization in mammalian cells, suggesting that impaired enzyme function may play a role in disease onset, with neurons harboring very long axons more susceptible to tRNA charging deficits.7,8 Combined, these findings strongly suggest that all genes encoding an ARS are excellent candidates for CMT disease. We therefore carried out a sequencing-based mutation screen of the 37 ARS genes in a cohort of 355 patients with a phenotype consistent with CMT and no known disease-causing mutation. The appropriate, institute-specific review boards approved all studies performed herein, and informed consent was obtained from all subjects. This study revealed four protein-coding variants (including one previously described polymorphism) in the lysyl-tRNA synthetase (KARS [MIM 601421]) gene. KARS resides on chromosome 16q23.1 and encodes the enzyme responsible for charging tRNALys molecules. Importantly, KARS is the only locus in the human genome encoding an enzyme responsible for tRNALys charging and is required in both the cytoplasm and mitochondria for protein translation.9
One KARS variant was identified in the heterozygous state in patient BAB663 (Figure 1A; BAB663): c.906C>G, which predicts p.Ile302Met. This patient's pedigree indicates an apparent autosomal-dominant mode of inheritance (see Figure S1 available online). Electrophysiological studies revealed that BAB663 exhibited normal MNCVs in all nerves tested, accompanied by normal amplitudes of evoked nerve response (6 mV, 7 mV, 8 mV, 11 mV, 7 mV, and 4 mV in the left median, left ulnar, right median, right ulnar, left peroneal, and left post-tibial nerves, respectively). Distal motor latencies were prolonged (7.2 ms in the right and left median nerves, 3.6 ms in the left ulnar nerve, 4.2 ms in the left ulnar nerve, 7.6 ms in the left peroneal nerve, and 5.8 ms in the left tibial nerve). Thus, this patient has a phenotype consistent with hereditary neuropathy, with liability to pressure palsies (HNPP [MIM 162500]).10
Figure 1.
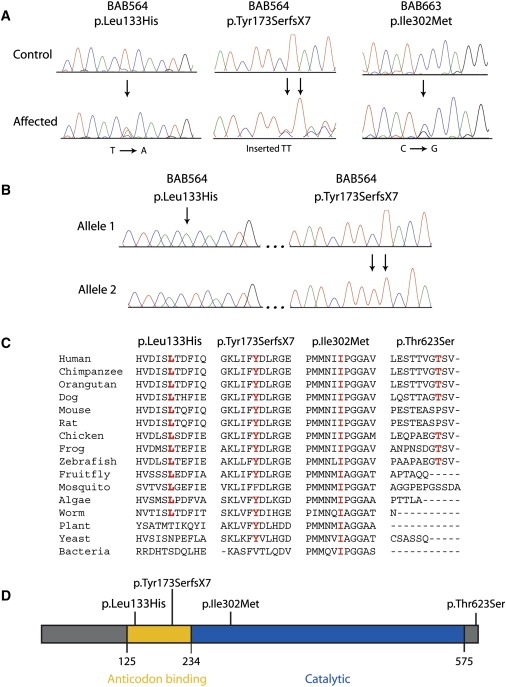
Characterization, Conservation, and Localization of KARS Variants
(A) Representative sections of sequence chromatograms are shown for the regions encompassing each identified KARS variant in the indicated individuals. Arrows denote the variant (present in the heterozygous state), with the predicted amino acid changes depicted above.
(B) Chromatograms from allele-specific sequencing of an ∼3.7 kb PCR-generated genomic segment spanning the two KARS variants (p.Leu133His and p.Tyr173SerfsX7) identified in patient BAB564. Arrows indicate each mutation. Note that each variant was identified on separate alleles, indicating that this patient is a compound heterozygote.
(C) For each of the four detected variants, the affected amino acid is shown along with the flanking KARS protein sequence in multiple, evolutionarily diverse species. Note that each specific amino acid change is given at the top, with the relevant position depicted in red for each protein sequence. Dashes indicate gaps in the sequence alignment.
(D) The known functional domains of the KARS protein are indicated in yellow (tRNALys-binding domain) and blue (core catalytic domain).
Two additional KARS variants were identified in a patient with intermediate CMT, developmental delay, self-abusive behavior, dysmorphic features, and vestibular Schwannoma (Figure 1A; BAB564): c.398T>A, which predicts p.Leu133His, and c.524_525insTT, which predicts a frameshift mutation p.Tyr173SerfsX7. BAB564 exhibited MNCVs of 39.5 m/s and 30.6 m/s in the median and ulnar nerves, consistent with an intermediate CMT phenotype.11 In addition, this patient displayed decreased amplitudes of evoked motor response in these nerves (0.5 mV). More detailed molecular analysis of BAB564 (involving PCR amplification, cloning, and sequencing the ∼3.7 kb segment encompassing the two detected variants) revealed that this individual is a compound heterozygote for p.Leu133His and p.Tyr173SerfsX7 (Figure 1B). Because this individual was adopted, these efforts were critical for distinguishing between a complex allele and compound heterozygosity (DNA samples from the biological parents are unavailable). Finally, p.Thr623Ser was identified in 31 out of 710 chromosomes studied (frequency = 0.044). This variant is present in dbSNP (rs6834), indicating that p.Thr623Ser represents a rare polymorphism.
Samples from patients BAB663 and BAB564 were screened for mutations in other genes previously implicated in CMT disease, including the CMT1A duplication and point mutations in AARS, EGR2 (MIM 129010), GARS, GDAP1 (MIM 606598), GJB1 (MIM 304040), MPZ (MIM 159440), NEFL (MIM 162280), PMP22 (MIM 601097), PRX (MIM 605725), SIMPLE (MIM 603795), SOX10 (MIM 602229), LMNA (MIM 150330), TDP1 (MIM 607198), MTMR2 (MIM 603557), and YARS. Patient BAB663 is heterozygous for p.Arg238His GJB1,12 p.Thr87Thr SIMPLE,13 and the 1.4 Mb PMP22 deletion.14 Interestingly, the PMP22 deletion and p.Arg238His GJB1 variant have previously been reported as pathogenic in HNPP and CMTX1 (MIM 302800), respectively.14,15 No mutations or copy number variations were detected in patient BAB564. Importantly, this includes MPZ and YARS, both of which have been associated with intermediate CMT.16 To further exclude known causes of intermediate CMT, we screened BAB564 for DNM2 (MIM 602378) mutations; these studies were also negative. Thus, BAB564 does not carry mutations in genes previously implicated in intermediate CMT.11,16 Finally, six variants in other ARS genes were identified in each individual, although none are likely to be pathogenic (Table 1).
Table 1.
ARS Missense Variants Identified in Patients BAB564 and BAB663
|
BAB564 |
BAB663 |
||||||
|---|---|---|---|---|---|---|---|
| Gene | MIM | Variant | Notes | Gene | MIM | Variant | Notes |
| EPRS | 138295 | p.Asp308Glu | rs22303011 | EPRS | 138295 | p.Asp308Glu | rs22303011 |
| FARS2 | 611592 | p.Asn280Ser | rs112430111 | FARSB | 609690 | p.Val585Ile | rs71851 |
| FARSB | 609690 | p.Val585Ile | rs71851 | IARS | 600709 | p.Lys1182Glu | rs5561551 |
| KARS | 601421 | p.Leu133His | This study | KARS | 601421 | p.Ile302Met | This study |
| RARS | 107820 | p.Val3Ile | rs2449031 | NARS | 108410 | p.Asn218Ser | 2/7102 |
| RARS | 107820 | p.Phe397Tyr | rs23057341 | RARS | 107820 | p.Val3Ile | rs2449031 |
| TARS | 187790 | p.Ala95Glu | 1/7102,3 | YARS2 | 610957 | p.Glu191Val | 10/7102 |
dbSNP accession number.
Number of chromosomes identified in total patient cohort.
Does not affect TARS enzyme function in aminoacylation assays (Jiqiang Ling and Dieter Söll, personal communication).
To determine whether the KARS variants are benign, we performed appropriate genotyping assays on DNA samples from neurologically normal controls of European decent (NINDS/Coriell). The p.Leu133His, p.Ile302Met, and p.Tyr173SerfsX7 variants were not detected in 1036, 1094, and 1098 chromosomes tested, respectively. We also screened all KARS protein-coding sequences for mutations in 95 individuals from the ClinSeq cohort.17 The only protein-coding variant identified was p.Thr623Ser, which occurred in 11 out of 190 chromosomes (frequency = 0.058).
The evolutionary conservation of each affected KARS residue was assessed by aligning protein sequences from KARS orthologs from multiple species (Figure 1C). Leucine 133 was conserved among all species analyzed, with the exception of plant, yeast, and bacteria. Isoleucine 302 was conserved among all species examined, including yeast and bacteria. Tyrosine 173 was conserved among all species analyzed, with the exception of mosquito and bacteria. In contrast, threonine 623 was not conserved between human and rodents and resides in a region that does not align with protein sequences from nonvertebrate species. Thus, the three rare KARS variants identified in our patient cohort reside at remarkably well-conserved amino acids, suggesting that they have a potential functional impact on the KARS protein.
We further computationally predicted the effect of each variant on protein function with the MuPro, PolyPhen, PolyPhen2, SIFT, Align GVGD, and CDPred algorithms (Table 2).18–23 It is notable that each of the known disease-associated GARS, YARS, and AARS mutations is predicted to be pathogenic by at least three of these six algorithms (Table 2). The p.Leu133His and p.Ile302Met KARS variants are predicted to interfere with protein function by four and three of the six algorithms, respectively. The p.Tyr173SerfsX7 KARS variant could not be analyzed, because these algorithms are unable to predict the effect of frameshift mutations; however, this variant is predicted to represent a null allele via nonsense-mediated decay. Importantly, the p.Thr623Ser polymorphism was predicted to be pathogenic by only one of the six algorithms.
Table 2.
Computational Predictions of KARS Variant Pathogenicity
| Gene | Variant | MUPro1 | PolyPhen2 | PolyPhen23 | SIFT4 | Align GVGD5 | CDPred6 |
|---|---|---|---|---|---|---|---|
| GARS | p.Glu71Gly | SVM = −1.007 | 2.217 | 0.997 | 0.017 | C657 | −77 |
| GARS | p.Leu129Pro | SVM = −1.007 | 2.217 | 1.007 | 0.007 | C657 | −87 |
| GARS | p.Gly240Arg | SVM = 0.30 | 2.317 | 0.997 | 0.007 | C657 | −87 |
| GARS | p.Gly526Arg | SVM = 0.00 | 2.797 | 1.007 | 0.007 | C657 | −117 |
| YARS | p.Gly41Arg | SVM = 0.84 | 2.497 | 1.007 | 0.007 | C657 | −127 |
| YARS | p.Glu196Lys | SVM = −1.007 | 0.63 | 0.997 | 0.17 | C557 | −2 |
| AARS | p.Arg329His | SVM = −0.777 | 3.047 | 1.007 | 0.007 | C25 | −107 |
| KARS | p.Leu133His | SVM = −0.617 | 0.11 | 0.997 | 0.06 | C657 | −47 |
| KARS | p.Ile302Met | SVM = −0.867 | 0.63 | 0.36 | 0.11 | C557 | −67 |
| KARS | p.Thr623Ser | SVM = −0.627 | 1.20 | 0.01 | 0.25 | C0 | +1 |
Support Vector Machine (SVM) scores < 0 indicate a decrease in protein stability.
PolyPhen scores ≥ 1.5 indicate a prediction of pathogenic.
PolyPhen2 scores of ∼1 indicate a prediction of pathogenic.
SIFT scores ≤ 0.05 indicate a prediction of pathogenic.
Grantham variation (GV) and Grantham difference (GD) classes ≥ C55 indicate a prediction of pathogenic.
CDPred delta scores ≤ −3 indicate a prediction of pathogenic.
Denotes a pathogenic prediction.
The KARS holoenzyme exists in dimeric and tetrameric forms.24 We mapped each affected KARS residue onto the crystal structure of the human enzyme to examine the amino acid position and structural relationship to functional domains that could potentially alter enzyme function. Leucine 133 is located within an N-terminal anticodon-binding domain (Figure 1D) and is adjacent to the dimer-dimer interface (Figures 2A–2C). This interface may be involved in interactions between KARS and various binding partners, including p38 (MIM 600859) in the mammalian multisynthetase complex, the HIV-1 Gap protein, and mutant forms of SOD1 (MIM 147450) found in patients with amyotrophic lateral sclerosis (MIM 105400).24–26 The p.Leu133His mutation is likely to impact some of these interactions. The p.Tyr173SerfsX7 variant resides in the anticodon-binding domain (Figure 1D) and predicts a complete loss of the catalytic domain. Isoleucine 302 resides in the catalytic domain (Figure 1D) and is also adjacent to the dimer-dimer interface (Figures 2A–2C). Thus, p.Ile302Met may also affect the association between KARS and binding partners. Threonine 623 is unresolved in the crystal structure. Importantly, these analyses reveal similarities between p.Leu133His and p.Ile302Met KARS and disease-associated GARS mutations; most GARS mutations affect residues that reside on the dimer interface of the holoenzyme.27
Figure 2.
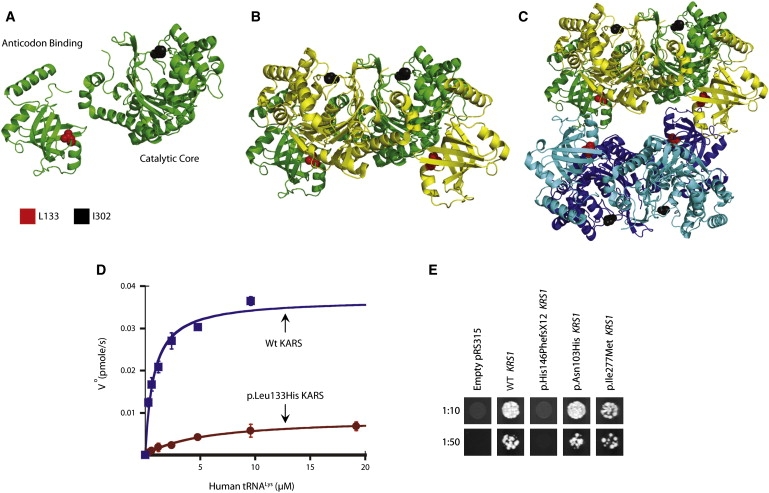
Functional Consequences of KARS Variants
(A–C) An illustration of the KARS protein crystal structure is shown for the monomer (A), dimer (B), and tetramer (C). The anti-codon binding and catalytic domains are indicated in (A). The position of residues L133 and I302 are indicated in red and black, respectively. Note that L133 resides at the tetramer interface in (C).
(D) Initial aminoacylation rates, V0 (pmol/s), of wild-type KARS (blue squares) and p.Leu133His KARS (red circles) were plotted against tRNA concentration and fit to the Michaelis-Menten equation. Error bars indicate standard deviation.
(E) Representative cultures of the indicated yeast strains were inoculated and grown on solid growth medium containing 5-FOA. Each strain was previously transfected with a vector containing no insert (pRS315), wild-type KRS1 (WT KRS1), or the indicated mutant form of KRS1 that modeled a human KARS mutation (see Table 4). Before inoculating on 5-FOA-containing medium, each strain was diluted 1:10 or 1:50 in water.
KARS catalyzes the aminoacylation of tRNALys in the cytoplasm and mitochondria via a two-step aminoacylation reaction.28 Importantly, 7 out of 10 disease-associated GARS and YARS mutations tested to date impair aminoacylation activity.4 We investigated the ability of each KARS variant to catalyze the aminoacylation reaction in vitro. Human cytoplasmic tRNALys was synthesized by in vitro transcription and was used as the substrate for aminoacylation. Analysis of the catalytic efficiency (kcat/Km) of aminoacylation showed that the p.Thr623Ser and p.Ile302Met variants maintain normal catalytic activity, indicating that these variants do not negatively affect aminoacylation. In contrast, p.Leu133His severely impairs enzyme activity, resulting in an ∼94% loss of catalytic efficiency of aminoacylation relative to wild-type KARS (Table 3; Figure 2D).
Table 3.
Enzyme Kinetics of Variant KARS Proteins in Aminoacylation Assays
| KARS Enzyme | Km (μM) | kcat (s−1) | kcat/Km (s−1μM−1) | Relative Activity |
|---|---|---|---|---|
| Wild-type | 1.8 ± 0.9 | 1.4 ± 0.1 | 0.8 | 1 |
| p.Leu133His | 5.8 ± 2.4 | 0.3 ± 0.3 | 0.05 | 0.06 |
| p.Ile302Met | 2.1 ± 0.8 | 2.7 ± 1.6 | 1.3 | 1.6 |
| p.Thr623Ser | 3.5 ± 0.1 | 4.0 ± 0.7 | 1.1 | 1.4 |
Many GARS and YARS mutations do not complement deletion of the corresponding yeast orthologs.7,8 To further assess for defects in KARS enzyme function, we modeled each KARS variant in the yeast ortholog (KRS1; Table 4) and determined the effect on yeast cell viability via complementation assays. A haploid yeast strain with the endogenous KRS1 deleted (krs1Δ) was maintained via transformation with a wild-type copy of KRS1 on a URA3-bearing vector (pRS316). Experimental alleles were generated on a LEU2-bearing vector (pRS315) and transformed into the above strain, and their viability was assessed by analysis of growth on 5-fluoroorotic acid (5-FOA). Wild-type KRS1 vector supported significant growth, whereas an insert-free pRS315 construct did not (Figure 2E), consistent with our experimental vector harboring functional KRS1 and with KRS1 being an essential gene, respectively. The p.Asn103His and p.Ile277Met KRS1 variants allowed growth in a manner consistent with wild-type KRS1. In contrast, p.His146PhefsX12 KRS1 could not complement the krs1Δ allele (Figure 2E), consistent with p.Tyr173SerfsX7 KARS representing a null allele.
Table 4.
Human KARS Variants Modeled in the Yeast Ortholog KRS1
| Human KARS1 | Yeast KRS12 |
|---|---|
| p.Leu133His | p.Asn103His |
| p.Tyr173SerfsX7 | p.His146PhefsX12 |
| p.Ile302Met | p.Ile277Met |
Amino acid coordinates correspond to GenBank accession number NP_001123561.1.
Amino acid coordinates correspond to GenBank accession number NP_010322.1.
In summary, we report three rare KARS variants in two patients with peripheral neuropathy. The p.Ile302Met variant was discovered in the heterozygous state in an individual with clinical electrophysiological evidence for HNPP and was molecularly found to harbor the common 1.4 Mb deletion, including PMP22. Although p.Ile302Met resides at a residue within the catalytic core of the enzyme that is conserved between human and bacteria, we were unable to show an effect on enzyme function via aminoacylation and yeast growth assays. Thus, the PMP22 deletion should be considered the primary pathogenic mutation in BAB663. However, it will be important to determine whether or not the PMP22 deletion, p.Arg238His GJB1, and p.Ile302Met KARS interact to modify the phenotype in this patient; several recent studies suggest the potential for a more severe neuropathy phenotype associated with variants at more than one CMT locus.29–31
The p.Leu133His and p.Tyr173SerfsX7 variants were identified in the compound heterozygous state in a patient with intermediate CMT, developmental delay, self-abusive behavior, dysmorphic features, and vestibular Schwannoma. The p.Tyr173SerfsX7 variant represents a loss-of-function allele, and p.Leu133His represents a severely hypomorphic allele in yeast growth and aminoacylation assays, respectively. Combined, these data indicate that the patient has a severe depletion of charged tRNALys in both the cytoplasm and mitochondria. It is important to consider these loss-of-function mutations in the context of the CMT and non-CMT phenotypes observed in this patient. To date, three ARS genes have been implicated in CMT.5–7 The encoded enzymes either are bifunctional (GARS charges tRNA in both the cytoplasm and mitochondria) or charge tRNA in the cytoplasm (YARS and AARS). In each case, the phenotypes are dominant and the mutations are missense or in-frame deletions. Furthermore, each mutation has been associated with a loss of function, as observed by impaired tRNA charging, inability to complement the deletion of the yeast ortholog, and/or reduced localization of the ARS enzyme to axons.5,7,8,27 Because haploinsufficiency for Gars does not cause a CMT-like phenotype in mouse,32 a dominant-negative effect has been proposed.4 Such an effect would reduce tRNA charging levels to ∼25%, a level that may breach a threshold required by neurons with particularly long axons.6 Our functional analyses suggest that KARS charging activity is reduced to well below 25% (∼6%) in patient BAB564. As such, compound heterozygosity for a null and severely hypomorphic KARS allele may be expected to cause a more severe or complex phenotype than heterozygosity for a dominant-negative ARS mutation. It is also possible that additional genetic complexities could be associated with the non-CMT sequelae—in particular, the vestibular Schwannoma often associated with NF2 mutations (MIM 607379).33 Therefore, detailed analysis in a vertebrate model system will be required to tease out the contribution of each KARS allele to the neuronal and non-neuronal phenotypes observed in patient BAB564.
The studies presented here describe the fourth association between ARS gene mutations and CMT and outline the importance of using informative and relevant functional assays as a follow-up to large-scale mutation screens. Indeed, our efforts illustrate some of the issues common to contemporary human genetics and genomics; for example, although we have the capacity to sequence large cohorts of patients, the analysis of small families and sporadic cases is often key for assessing the role of specific genes in human disease. In these and other “gene discovery” cases, relevant and informative functional assays provide critical evidence for the pathogenicity of uncharacterized variants.
KARS protein orthologs from multiple species were derived from the following GenBank accession numbers: human (Homo sapiens, NP_00112356), chimpanzee (Pan troglodytes, XP_511115.2), orangutan (Pongo abelii, NP_001123561), dog (Canis familiaris, XP_536777.2), mouse (Mus musculus, NP_444322), rat (Rattus norvegicus, NP_001006968), chicken (Gallus gallus, NP_001025754), frog (Xenopus laevis, NP_001080633), zebrafish (Danio rerio, NP_001002386), fruitfly (Drosophila melanogaster, NP_572573), mosquito (Anopheles gambiae, XP_310792), algae (Chlamydomonas reinhardtii, XP_001697493), worm (Caenorhabditis elegans, NP_495454), plant (Arabidopsis thaliana, NP_187777), yeast (Saccharomyces cerevisiae, NP_010322), bacteria (Escherichia sp. 1_1_43, ZP_04871218).
Acknowledgments
We are indebted to the patients and their families for their participation in this study. We thank Ellen Pederson, Bob Lyons, and the University of Michigan DNA Sequencing Core for sequencing and genotyping assistance, Jeffrey Innis and the Michigan Medical Genetics Laboratory for anonymized DNA samples, Giovanni Manfredi and Kiyotaka Shiba for KARS cDNA constructs, and Jiqiang Ling and Dieter Söll for sharing unpublished data. We are also very grateful to the two anonymous reviewers for their helpful comments and suggestions, which dramatically improved the study. This work was supported in part by grant R00NS060983 from the National Institute of Neurological Diseases and Stroke (AA) and by the Intramural Research Program of the National Human Genome Research Institute (NIH). The ClinSeq cohort and sequencing were also supported by Intramural Funding from the National Human Genome Research Institute. H.M.M. was supported by the Rackham Merit Fellowship and the NIH Genetics Training Grant T32 GM007544-32. Y.-M.H. and R.S. were supported by a grant from the Muscular Dystrophy Association (157681 to Y.-M.H.).
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Align GVGD, http://agvgd.iarc.fr/agvgd_input.php
Conserved Domain-Based Prediction (CDPred), http://research.nhgri.nih.gov/software/CDPred/
MUpro: Prediction of Protein Stability Changes for Single-Site Mutations from Sequences, http://www.ics.uci.edu/∼baldig/mutation.html
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/
PolyPhen-2, http://genetics.bwh.harvard.edu/pph2/
SIFT Sequence, http://sift.jcvi.org/www/SIFT_seq_submit2.html
References
- 1.Skre H. Genetic and clinical aspects of Charcot-Marie-Tooth's disease. Clin. Genet. 1974;6:98–118. doi: 10.1111/j.1399-0004.1974.tb00638.x. [DOI] [PubMed] [Google Scholar]
- 2.Dyck P.J., Lambert E.H. Lower motor and primary sensory neuron diseases with peroneal muscular atrophy. II. Neurologic, genetic, and electrophysiologic findings in various neuronal degenerations. Arch. Neurol. 1968;18:619–625. doi: 10.1001/archneur.1968.00470360041003. [DOI] [PubMed] [Google Scholar]
- 3.Murakami T., Garcia C.A., Reiter L.T., Lupski J.R. Charcot-Marie-Tooth disease and related inherited neuropathies. Medicine (Baltimore) 1996;75:233–250. doi: 10.1097/00005792-199609000-00001. [DOI] [PubMed] [Google Scholar]
- 4.Antonellis A., Green E.D. The role of aminoacyl-tRNA synthetases in genetic diseases. Annu. Rev. Genomics Hum. Genet. 2008;9:87–107. doi: 10.1146/annurev.genom.9.081307.164204. [DOI] [PubMed] [Google Scholar]
- 5.Latour P., Thauvin-Robinet C., Baudelet-Méry C., Soichot P., Cusin V., Faivre L., Locatelli M.C., Mayençon M., Sarcey A., Broussolle E. A major determinant for binding and aminoacylation of tRNA(Ala) in cytoplasmic Alanyl-tRNA synthetase is mutated in dominant axonal Charcot-Marie-Tooth disease. Am. J. Hum. Genet. 2010;86:77–82. doi: 10.1016/j.ajhg.2009.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Antonellis A., Ellsworth R.E., Sambuughin N., Puls I., Abel A., Lee-Lin S.Q., Jordanova A., Kremensky I., Christodoulou K., Middleton L.T. Glycyl tRNA synthetase mutations in Charcot-Marie-Tooth disease type 2D and distal spinal muscular atrophy type V. Am. J. Hum. Genet. 2003;72:1293–1299. doi: 10.1086/375039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jordanova A., Irobi J., Thomas F.P., Van Dijck P., Meerschaert K., Dewil M., Dierick I., Jacobs A., De Vriendt E., Guergueltcheva V. Disrupted function and axonal distribution of mutant tyrosyl-tRNA synthetase in dominant intermediate Charcot-Marie-Tooth neuropathy. Nat. Genet. 2006;38:197–202. doi: 10.1038/ng1727. [DOI] [PubMed] [Google Scholar]
- 8.Antonellis A., Lee-Lin S.Q., Wasterlain A., Leo P., Quezado M., Goldfarb L.G., Myung K., Burgess S., Fischbeck K.H., Green E.D. Functional analyses of glycyl-tRNA synthetase mutations suggest a key role for tRNA-charging enzymes in peripheral axons. J. Neurosci. 2006;26:10397–10406. doi: 10.1523/JNEUROSCI.1671-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tolkunova E., Park H., Xia J., King M.P., Davidson E. The human lysyl-tRNA synthetase gene encodes both the cytoplasmic and mitochondrial enzymes by means of an unusual alternative splicing of the primary transcript. J. Biol. Chem. 2000;275:35063–35069. doi: 10.1074/jbc.M006265200. [DOI] [PubMed] [Google Scholar]
- 10.Li J., Krajewski K., Shy M.E., Lewis R.A. Hereditary neuropathy with liability to pressure palsy: The electrophysiology fits the name. Neurology. 2002;58:1769–1773. doi: 10.1212/wnl.58.12.1769. [DOI] [PubMed] [Google Scholar]
- 11.Nicholson G., Myers S. Intermediate forms of Charcot-Marie-Tooth neuropathy: A review. Neuromolecular Med. 2006;8:123–130. doi: 10.1385/nmm:8:1-2:123. [DOI] [PubMed] [Google Scholar]
- 12.Bergoffen J., Scherer S.S., Wang S., Scott M.O., Bone L.J., Paul D.L., Chen K., Lensch M.W., Chance P.F., Fischbeck K.H. Connexin mutations in X-linked Charcot-Marie-Tooth disease. Science. 1993;262:2039–2042. doi: 10.1126/science.8266101. [DOI] [PubMed] [Google Scholar]
- 13.Street V.A., Bennett C.L., Goldy J.D., Shirk A.J., Kleopa K.A., Tempel B.L., Lipe H.P., Scherer S.S., Bird T.D., Chance P.F. Mutation of a putative protein degradation gene LITAF/SIMPLE in Charcot-Marie-Tooth disease 1C. Neurology. 2003;60:22–26. doi: 10.1212/wnl.60.1.22. [DOI] [PubMed] [Google Scholar]
- 14.Chance P.F., Alderson M.K., Leppig K.A., Lensch M.W., Matsunami N., Smith B., Swanson P.D., Odelberg S.J., Disteche C.M., Bird T.D. DNA deletion associated with hereditary neuropathy with liability to pressure palsies. Cell. 1993;72:143–151. doi: 10.1016/0092-8674(93)90058-x. [DOI] [PubMed] [Google Scholar]
- 15.Nelis E., Simokovic S., Timmerman V., Löfgren A., Backhovens H., De Jonghe P., Martin J.J., Van Broeckhoven C. Mutation analysis of the connexin 32 (Cx32) gene in Charcot-Marie-Tooth neuropathy type 1: Identification of five new mutations. Hum. Mutat. 1997;9:47–52. doi: 10.1002/(SICI)1098-1004(1997)9:1<47::AID-HUMU8>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 16.Reilly M.M. Sorting out the inherited neuropathies. Pract. Neurol. 2007;7:93–105. [PubMed] [Google Scholar]
- 17.Biesecker L.G., Mullikin J.C., Facio F.M., Turner C., Cherukuri P.F., Blakesley R.W., Bouffard G.G., Chines P.S., Cruz P., Hansen N.F., NISC Comparative Sequencing Program The ClinSeq Project: Piloting large-scale genome sequencing for research in genomic medicine. Genome Res. 2009;19:1665–1674. doi: 10.1101/gr.092841.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Adzhubei I.A., Schmidt S., Peshkin L., Ramensky V.E., Gerasimova A., Bork P., Kondrashov A.S., Sunyaev S.R. A method and server for predicting damaging missense mutations. Nat. Methods. 2010;7:248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cheng J., Randall A., Baldi P. Prediction of protein stability changes for single-site mutations using support vector machines. Proteins. 2006;62:1125–1132. doi: 10.1002/prot.20810. [DOI] [PubMed] [Google Scholar]
- 20.Johnston J.J., Teer J.K., Cherukuri P.F., Hansen N.F., Loftus S.K., Chong K., Mullikin J.C., Biesecker L.G., NIH Intramural Sequencing Center Massively parallel sequencing of exons on the X chromosome identifies RBM10 as the gene that causes a syndromic form of cleft palate. Am. J. Hum. Genet. 2010;86:743–748. doi: 10.1016/j.ajhg.2010.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mathe E., Olivier M., Kato S., Ishioka C., Hainaut P., Tavtigian S.V. Computational approaches for predicting the biological effect of p53 missense mutations: A comparison of three sequence analysis based methods. Nucleic Acids Res. 2006;34:1317–1325. doi: 10.1093/nar/gkj518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ng P.C., Henikoff S. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003;31:3812–3814. doi: 10.1093/nar/gkg509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ramensky V., Bork P., Sunyaev S. Human non-synonymous SNPs: Server and survey. Nucleic Acids Res. 2002;30:3894–3900. doi: 10.1093/nar/gkf493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Guo M., Ignatov M., Musier-Forsyth K., Schimmel P., Yang X.L. Crystal structure of tetrameric form of human lysyl-tRNA synthetase: Implications for multisynthetase complex formation. Proc. Natl. Acad. Sci. USA. 2008;105:2331–2336. doi: 10.1073/pnas.0712072105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kovaleski B.J., Kennedy R., Hong M.K., Datta S.A., Kleiman L., Rein A., Musier-Forsyth K. In vitro characterization of the interaction between HIV-1 Gag and human lysyl-tRNA synthetase. J. Biol. Chem. 2006;281:19449–19456. doi: 10.1074/jbc.M601189200. [DOI] [PubMed] [Google Scholar]
- 26.Kunst C.B., Mezey E., Brownstein M.J., Patterson D. Mutations in SOD1 associated with amyotrophic lateral sclerosis cause novel protein interactions. Nat. Genet. 1997;15:91–94. doi: 10.1038/ng0197-91. [DOI] [PubMed] [Google Scholar]
- 27.Nangle L.A., Zhang W., Xie W., Yang X.L., Schimmel P. Charcot-Marie-Tooth disease-associated mutant tRNA synthetases linked to altered dimer interface and neurite distribution defect. Proc. Natl. Acad. Sci. USA. 2007;104:11239–11244. doi: 10.1073/pnas.0705055104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Delarue M. Aminoacyl-tRNA synthetases. Curr. Opin. Struct. Biol. 1995;5:48–55. doi: 10.1016/0959-440x(95)80008-o. [DOI] [PubMed] [Google Scholar]
- 29.Hodapp J.A., Carter G.T., Lipe H.P., Michelson S.J., Kraft G.H., Bird T.D. Double trouble in hereditary neuropathy: Concomitant mutations in the PMP-22 gene and another gene produce novel phenotypes. Arch. Neurol. 2006;63:112–117. doi: 10.1001/archneur.63.1.112. [DOI] [PubMed] [Google Scholar]
- 30.Meggouh F., de Visser M., Arts W.F., De Coo R.I., van Schaik I.N., Baas F. Early onset neuropathy in a compound form of Charcot-Marie-Tooth disease. Ann. Neurol. 2005;57:589–591. doi: 10.1002/ana.20434. [DOI] [PubMed] [Google Scholar]
- 31.Chung K.W., Sunwoo I.N., Kim S.M., Park K.D., Kim W.K., Kim T.S., Koo H., Cho M., Lee J., Choi B.O. Two missense mutations of EGR2 R359W and GJB1 V136A in a Charcot-Marie-Tooth disease family. Neurogenetics. 2005;6:159–163. doi: 10.1007/s10048-005-0217-4. [DOI] [PubMed] [Google Scholar]
- 32.Seburn K.L., Nangle L.A., Cox G.A., Schimmel P., Burgess R.W. An active dominant mutation of glycyl-tRNA synthetase causes neuropathy in a Charcot-Marie-Tooth 2D mouse model. Neuron. 2006;51:715–726. doi: 10.1016/j.neuron.2006.08.027. [DOI] [PubMed] [Google Scholar]
- 33.Rouleau G.A., Merel P., Lutchman M., Sanson M., Zucman J., Marineau C., Hoang-Xuan K., Demczuk S., Desmaze C., Plougastel B. Alteration in a new gene encoding a putative membrane-organizing protein causes neuro-fibromatosis type 2. Nature. 1993;363:515–521. doi: 10.1038/363515a0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.