

Abstract
By defining the chromosomal breakpoint of a balanced t(10;12) translocation from a subject with Kallmann syndrome and scanning genes in its vicinity in unrelated hypogonadal subjects, we have identified WDR11 as a gene involved in human puberty. We found six patients with a total of five different heterozygous WDR11 missense mutations, including three alterations (A435T, R448Q, and H690Q) in WD domains important for β propeller formation and protein-protein interaction. In addition, we discovered that WDR11 interacts with EMX1, a homeodomain transcription factor involved in the development of olfactory neurons, and that missense alterations reduce or abolish this interaction. Our findings suggest that impaired pubertal development in these patients results from a deficiency of productive WDR11 protein interaction.
Introduction
Human puberty is a dynamic process that initiates complex interactions of the hypothalamic-pituitary-gonadal axis, the purpose of which is to produce sex steroids for reproductive maturity and gametes for fertility. Any disruption of the development or regulation of this system, for which the hypothalamus serves as the master regulator through its pulsatile release of gonadotropin-releasing hormone (GnRH), can produce deleterious consequences for successful reproduction.1 Patients with idiopathic hypogonadotropic hypogonadism (IHH, MIM 146110) show clinical signs and symptoms of GnRH deficiency: delayed puberty due to low sex steroid production along with low levels of serum gonadotropins. Patients with Kallmann syndrome (KS, MIM 308700, 147950, 244200, 610628, 612370, 612702) have IHH but also display an impaired sense of smell, thought to be due to the developmental failure of the migration of GnRH neurons along the olfactory axonal projections.
IHH/KS is one of the most common causes of hypogonadism and is genetically heterogeneous. Researchers have used a variety of strategies to find IHH- and/or KS-causing mutations in a number of genes. Such strategies have included linkage analysis, deletion mapping, and candidate gene analysis. The discovery of a rare family with males displaying both X-linked KS and ichthyosis led to the identification of KAL1 (MIM 308700) by positional cloning,2,3 and characterization of deletions4 and a balanced translocation5 involving chromosome 8 facilitated the cloning of FGFR1 (KAL2, MIM 136350), associated with both IHH and KS.6 Positional cloning in consanguineous autosomal-recessive IHH families revealed KISS1R (MIM 604161) encoding GPR54,7,8 TAC39 (MIM 162330), and TACR39 (MIM 162332) and candidate-gene approaches identified mutations in GNRHR10,11 (MIM 138850), NELF12 (MIM 608137), CHD7(KAL5)13 (MIM 608892), FGF8 (KAL6)14 (MIM 600483), and GNRH115,16 (MIM 152760); analogous mouse phenotypes pointed to PROKR2 (KAL3)17 (MIM 607123), and PROK2 (KAL4)17 (MIM 607002), as well as KISS1R.8 Despite these significant advances in the past two decades, however, the genetic etiology remains unknown for about two-thirds of all IHH and KS patients.
To identify chromosomal rearrangements in IHH and KS as a basis for disease gene identification, we previously karyotyped 76 patients and found one male KS patient with chromosome translocation reported as: 46,XY, t(10;12)(q26.3;q13.1)dn (Figure 1A).18 The chromosome 10q26 region has been associated previously with abnormal male genital development resulting from interstitial or terminal deletions as well as a balanced translocation. Abnormal development has included cryptorchidism,19,20 small testes,21 sperm defects and infertility,22 micropenis and hypogonadism,23 hypogenitalism,24 and sparse sexual hair.25 All of these phenotypic features overlap with characteristics of IHH and KS. Importantly, a trisomy involving 10q26 has been reported in a KS patient with an unbalanced chromosome translocation,26 suggesting the presence of a new KS gene, which can involve dysregulation as a result of either reduced or increased dosage, in this critical region. Therefore, we postulated that a single disease gene disrupted or dysregulated by a position effect27 is located at or near the 10q26 breakpoint in our KS patient with a balanced translocation.
Figure 1.
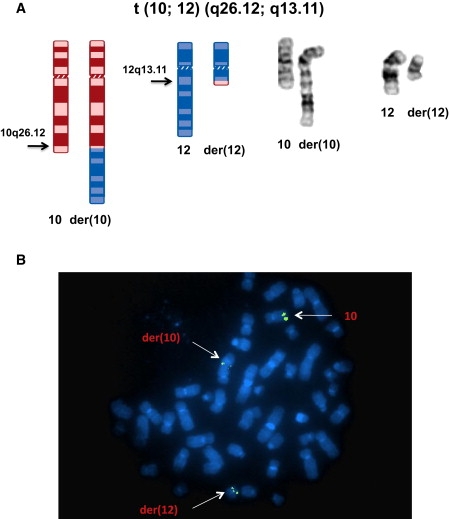
Balanced Chromosome Translocation and FISH Mapping of the Chromosome 10 Breakpoint
(A) Ideogram and composite chromosomes illustrating the balanced t(10;12)(q26.12;q13.11)dn (revised in this paper from the original karyotypic assessment, t(10;12)(q26.3;q13.1)dn) in the KS patient.
(B) FISH mapping with BAC clone RP11-254K03, labeled with SpectrumGreen to metaphase spreads of the KS patient, resulted in hybridization to the normal chromosome 10, as well as the der(10) and der(12) chromosomes, indicating that the translocation breakpoint of chromosome 10 is located within the sequence of this BAC clone.
Material and Methods
Patients
IHH was diagnosed in males who were 18 years old or younger and had delayed puberty, testosterone levels <100 ng/dl (normal is 300–1100 ng/dl), and low or normal serum gonadotropins. In females, IHH was defined as primary amenorrhea, nearly always with absent breast development at ≥17 years of age and low estradiol (<30 pg/ml).18,28 All patients had normal pituitary function, including normal thyroid-stimulating hormone, thyroxin, cortisol, and prolactin. No pituitary tumor was present by radiographic imaging. Complete IHH or KS is a more severe phenotype defined as the complete lack of puberty with absent breast development (Tanner 1) in females and testis size ≤3 ml bilaterally in males. Incomplete IHH or KS was defined as partial breast development in females and testis size ≥ 4 ml bilaterally in males.18 Olfaction was either tested with the University of Pennyslvania Smell Identification Test when available or documented by history. Lymphoblastoid cell lines were generated from patients, and DNA, RNA, and/or protein was extracted by standard methods as described previously.18 All patients signed an informed consent approved by the Human Assurance Committee of the Medical College of Georgia.
Tissue Culture and Lymphoblastoid Cell Lines
Epstein-Barr-virus-immortalized lymphoblastoid cell lines used in this study were established from peripheral blood lymphocytes of individuals. Once established, these cell lines were cultured in RPMI 1640 (Mediatech) supplemented with 10% FCS, 2 mM l-glutamine, and 0.017 mg/ml benzylpenicillin (CLS) and grown at 37°C with 5% CO2.
SNP Oligonucleotide Microarray Analysis
The Affymetrix Human Mapping 500K Array Set (Affymetrix, Santa Clara, CA) is comprised of two arrays, each capable of genotyping on average 250,000 SNPs (approximately 262,000 for the Nsp arrays and 238,000 for the Sty arrays). The mapping 500K array set has a mean spacing of 5.8 kb. Specimens were assayed with both arrays. The 500K assay was performed according to the manufacturer's protocol, beginning with 250 ng DNA. Ninety μg of PCR product were fragmented and labeled. Hybridization was carried out in the Affymetrix GeneChip hybridization oven 640. Posthybridization washing and processing were performed with the Affymetrix GeneChip Fluidics Station 450. The arrays were then scanned with the Affymetrix GeneChip Scanner 3000 7G. Image processing was performed with GCOS 1.4, and genotypes were called with GTYPE 4.1 software and default settings. Detection of copy-number changes was performed in the Chromosome Copy Number Analysis Tool (CNAT) version 4.0, with a reference set of 42 normal males. Data from the separate Mapping 250K arrays were virtually combined after normalization but before smoothing in CNAT 4.0.
Fluorescence In Situ Hybridization Analysis
The clones were labeled with either Spectrum Orange or Spectrum Green direct-labeled dUTP via the nick-translation Labeling Kit (Abbott Molecular). The procedure involved incubation of the DNA extracted from the clone with dATP, dCTP, dGTP, and fluorescent dUTP in the presence of DNA polymerase I and DNase I at 15°C for 8–16 hr. Heating the mixture in a 70°C water bath for 10 min stopped the reaction.
The slides from the harvested lymphoblastoid cell suspension were prepared according to standard procedures. The cells on the slides were denatured in 70% formamide and 2× SSC at 70°C for 3 min and dehydrated serially in cold ethanol at increasing concentrations up to absolute ethanol. The fluorescently labeled probe mix was then applied to the slides, which were covered with a coverslip and hybridized overnight in a moist chamber at 37°C. After overnight hybridization, the slides were washed in a solution of 4× SSC and 0.3% NP 40 at 72°C for 2 min and then washed with 2× SSC and 0.1% NP40 at room temperature for 2 min. The metaphases were counterstained with 0.4 μg/ml DAPI (4,6-diamidino-2-phenylindole) and analyzed under a fluorescence microscope. The images were photographed with a CCD camera with an Applied Imaging system.
Mouse In Situ Hybridization Analysis
Two primer pairs were used for amplification of two independent Wdr11 probes spanning nucleotides 641–1035 and 2892–3492 of the murine Wdr11 transcript (NM_172255.3). The primers used are listed in Table S4. Amplicons were ligated to a Topo-TA vector (Invitrogen) and subcloned into pBluescript via EcoRI and XbaI (restriction sites are underlined in Table S4). The probes were labeled with [α-35S]UTP for hybridization on sections or with Digoxigenin29 for whole-mount in situ hybridization as well as for cryosections. Murine embryos were frozen on solid CO2, and 10 μm sections were prepared on a cryostat. Adult mouse brains were sectioned at 15 μm. Sections hybridized with the [α-35S]UTP probe were exposed to Kodak Biomax MR film for 3 days. No specific signals were detected with the respective sense probes. Whole-mount in situ hybridization with Digoxigenin-UTP-labeled probes was performed on total mouse embryos with the in situ probe spanning nucleotides 2892–3492. The day of plug was not counted for specification of embryonic stages.
Isolation of Zebrafish wdr11 Gene and Whole-Mount In Situ Hybridization
The zebrafish wdr11 gene (XM_682139.3) was isolated from the 24 hpf zebrafish cDNA library by RT-PCR with a BamHI-linked forward primer and an NcoI-linked reverse primer (Table S4; restriction sites are underlined), subcloned into the pGEM T-easy vector, and then sequenced by automatic sequencer. To examine the spatiotemporal expression patterns of wdr11, whole-mount in situ hybridization was performed as previously described.30 Antisense digoxigenin-labeled RNA probes for wdr11 and emx131 were produced with a DIG-RNA labeling kit (Roche, Germany) according to the manufacturer's instructions.
Mutation Analysis
Genomic DNA extracted from 201 unrelated IHH and/or KS patients was amplified with WDR11 primer pairs flanking the 29 exons and splice junction sites by nested PCR. Oligo 6.0 or Primer 3 was used for primer design, and conditions were optimized for each PCR as described previously.11,13 Nested-PCR products were electrophoresed on agarose gels, precipitated via ethanol, quantified, and sequenced with the Big Dye Terminator Kit (Applied Biosystems; Foster City, CA).32 Sequencing reactions were purified with Centrosep columns, lyophilized, and placed on an ABI 3730 automated DNA sequencer (Applied Biosystems; Foster City, CA). All exons were sequenced at least twice; mutations were confirmed 3-4 times by independent PCR. Any putative mutation was sequenced in >420 healthy white controls and >400 healthy Turkish controls. The CodonCode Aligner Program (CodonCode Corporation; Dedham, MA) was used for analysis of the sequencing data.
Semiquantitative RT-PCR in Rat Tissue
Total RNA was extracted from lymphoblastoid cell lines and tissues with TRI Reagent (Molecular Research Center) and treated with DNase I (QIAGEN). We primed 100 ng of RNA with gene-specific primers by using Superscript III one-step RT-PCR kit (Invitrogen) and Mastercycler Gradient (Eppendorf). Typical conditions were as follows: 60°C for 30 min followed by 94°C for 2 min (one cycle), then 94°C for 15 s followed by 60°C for 30 s and 68°C for 50 s (30 cycles). Then 68°C for 5 min followed by cooling to 4°C. For semiquantitative RT-PCR analysis in rat tissues (Figure S1), we used two primers to amplify nucleotides 2651–3357 of the rat WDR11 transcript (XM_219377.5) (Table S4).
Yeast Two-hybrid Screening
For bait construction, a DNA fragment encoding amino acids 1–830 of WDR11 was subcloned into the BamHI and SalI sites of pGBKT7 (Clontech). Approximately 5 × 106 transformants from a mouse-brain cDNA library (Clontech) were screened in the AH109 yeast strain. The positive colonies were confirmed with β-galactosidase colony lift assays. The yeast plasmids were isolated and transformed into Escherichia coli DH5α cells. We cotransformed the isolated yeast DNA with a bait plasmid into AH109 to verify the interactions between WDR11 and the interacting proteins.
Preparation of WDR11 Mutant Constructs
A pFN21AA1351 clone that included WDR11 open-reading frame (ORF) sequence with the GenBank accession number of AB385454 was used for construction of the WDR11 mutant. DNA fragments containing a nucleotide substitution were amplified by two-step PCRs in which pFN21AA1351 served as a template; two separated DNA fragments were amplified by the first PCR with one of the mutant primers containing a nucleotide substitution (mR1 or mF1 series) and the corresponding opposite outside primer (F01, F06, R01 or R02), then two resultant amplified PCR products were combined by the second PCR with the outside primers and cloned into pUC118 vector. The cloned mutant DNA fragment was cut with EcoNI for m1, m2, m3, and m4 constructs, and the mutant EcoNI fragment was inserted between EcoNI sites of the WDR11 ORF. For m5 and m6, the cloned DNA fragment was cut with SwaI and PvuII, and the 1422 bp SwaI-PvuII fragment of WDR11 was replaced with the mutant fragment. The sequences of primers used for the construction are listed in Table S4.
Plasmid Construction
The full-length human WDR11 and EMX1 clones were purchased from Openbiosystems (clone ID 34306203 and 5260039, respectively). The full ORFs of WDR11 and EMX1 were amplified from purchased cDNA clones by PCR with specific primers, and the DNA fragment was inserted into BamHI/NotI and EcoRI/XhoI sites of pEntr3C, respectively. The Myc-WDR11, HA-EMX1, and GST-EMX1 plasmids were constructed by Gateway Technology (Invitrogen) with pEntr-WDR11 and pEntr-EMX1.
Coimmunoprecipitation and Immunoblotting
Coimmunoprecipitation was performed after the lysis of 2 × 107 cells with lysis buffer (50 mM HEPES, 150 mM NaCl, 10% glycerol, 1% Nonidet P-40, and 1 mM EDTA). After incubation on ice for 10 min and centrifugation for 10 min at 4°C, equal volumes of protein were incubated overnight with antibody and protein A G-Sepharose beads at 4°C on a rotating wheel. The beads were washed three times with lysis buffer. The whole-cell lysates and immunoprecipitates were separated by SDS-PAGE and then transferred onto polyvinylidene difluoride membranes. The membranes were immunoblotted with anti-Myc (Invitrogen) or anti-HA (Invitrogen) antibodies.
In Vitro Pull-Down Assays
The GST-fusion plasmids were expressed in E. coli. BL21(DE3) cells and were purified with glutathione-Sepharose beads according to the instructions of the manufacturer. The Myc-EMX1 proteins were synthesized via the coupled TNT in vitro translation system (Promega, Madison, WI). We performed pull-down assays by incubating equal amounts of GST or GST-WDR11 fusion proteins immobilized onto glutathione-Sepharose beads with in vitro translated Myc-Emx1 and washing the beads three times with phosphate-buffered saline and 0.5% Triton X-100. After washing, the bound proteins were resolved by SDS-PAGE and detected via immunoblotting with anti-Myc antibody.
Immunohistochemistry
U2OS cells were transfected with expression plasmids encoding HA-EMX1 and GFP-WDR11. Twenty-four hours after transfection, cells were treated with leptomycin B (10 ng/ml) for 12 hr before being fixed with 4% paraformaldehyde for 5 min and incubated with anti-HA antibody. Fluorescence microscopy was conducted with a Zeiss Axioskop 2 microscope; excitation wavelengths of 543 nm (rhodamine red) and 488 nm (GFP) were used. The acquired images were processed with Adobe Photoshop. For subcellular localization for FCNB4-hTERT cells, approximately 2 ml of cell suspension was plated into each well of a microscopy cover slide, incubated for 24 hr in a 37°C CO2 incubator, and then fixed with 4% paraformaldehyde (PFA) for 5 min at room temperature. Cells were washed three times with PBS and permeabilized with 0.1% Triton X-100 in PBS for 5 min. After two washes with PBS, nonspecific binding sites were blocked with 1% BSA (Sigma) in PBS for 5 min. Samples were then rinsed with PBS and incubated overnight at 4°C with primary antibody. After being washed with PBS, samples were incubated for 1 hr at room temperature with the appropriate Alexa Fluor dye (Molecular Probes)-coupled anti-rabbit 488 (green) or anti-mouse 589 (red) secondary antibodies. After three PBS washes, nuclei were counterstained with To-pro3 (1 nM). Cover slips were mounted with anti-fade vector shield (Molecular Probe) reagent and viewed with the Zeiss LSM 510 Meta Confocal Microscope.
Exon Trapping
We used the exon-trapping system (Invitrogen GIBCO) to investigate the effect of the heterozygous c.2932A>C nucleotide variant (p.K978Q) in the acceptor splice site of exon 24 of WDR11. A genomic fragment of 2411 bp encompassing exons 23–25 along with flanking intronic sequences and a 1286 bp fragment comprising exon 25 along with flanking intronic sequences were amplified by PCR with two primer pairs (Table S4). The resulting PCR products were cloned into exon-trapping vector pSPL3 via restriction sites EcoRI (5′ end) and BamHI (3′ end). We verified sequences for all constructs to exclude PCR-induced mutations and to select constructs with or without the splice site variant c.2932A>C (p.K978Q). COS-7 cells were grown in 100 mm dishes in DMEM (Invitrogen) supplemented with 10% fetal calf serum (Invitrogen) in a 10% CO2 atmosphere at 37°C. At approximately 60% confluency, cells were each transfected with 8 μg of exon-trapping construct DNA via Lipofectamine 2000 (Invitrogen). Cells were collected 30 hr after transfection, and cytoplasmic RNA was isolated with RNeasy (QIAGEN) according to the manufacturer's instructions. After transcription into cDNA via Omniscript Reverse Transcriptase (QIAGEN), two rounds of PCR with vector-specific oligonucleotides were performed. PCR products were subcloned into TA cloning vector pCR2.1-Topo (Invitrogen) and directly sequenced with the ABI BigDye Terminator Sequencing Kit (Applied Biosystems) and the automated capillary sequencer ABI 3130 (Applied Biosystems).
Anti-WDR11 Antibody Generation
A new WDR11 polyclonal antibody was generated against the N-terminal residues 32–48 of NP_060587.8 (QGLIAYGCHSLVVVIDS); an N-terminal cysteine was added for conjugation purposes. Rabbits were injected with Freund's adjuvant containing the peptide sequence. After the second bleed, affinity purification was performed. The peptide sequence used fop raising the antibody was conserved in ten species (human, chimpanzee, cow, horse, panda, pig, dog, rat, mouse, and rabbit).
Array Painting
Flow-sorting of metaphase chromosomes was performed as described previously.33 Both derivative chromosomes were amplified with the GenomiPhi V2 DNA Amplification kit (GE Healthcare, Piscataway, NJ, USA) according to the manufacturer's protocol. One microgram of each amplification product was labeled with Cy3 or Cy5 via Agilent's Genomic DNA Enzymatic Labeling Kit Plus (Agilent). To each labeling reaction, adding 100 ng of genomic control DNA ensured proper placement of the grid after image analysis. A customized oligonucleotide array that represented chromosomal regions chr10:121,855,186–122,255,228 and chr12:45,828,906–46,226,893 (hg18) was designed by means of eArray (Agilent, Santa Clara, CA); there was an average oligonucleotide spacing of 200–300 bases. Hybridization and washing steps were performed as previously described.34
Quantitative Real-Time RT-PCR (RT-qPCR) of WDR11 in the t(10;12) Patient
cDNA from lymphoblastoid cells was used for real-time RT-qPCR with the StepOnePlus Real-Time PCR system (Applied Biosystems) and Power SYBR Green PCR Master Mix (Applied Biosystems) and two primer pairs (WDR11-1:3117forward+3213reverse; WDR11-2: 3221forward+3370reverse; Table S4). RT-qPCR was performed in triplicate with same amount of cDNA, and WDR11 levels were normalized to GAPDH expression. WDR11 levels in t(10;12) were normalized to those in a male control with StepOne Software (Applied Biosystems).
Immunoblot Analysis of WDR11 in the t(10;12) Patient
Cells were washed with PBS twice and lysed in RIPA buffer (PIERCE) containing complete EDTA-free protease inhibitor (Roche Diagnostics). Total cellular lysates were separated by SDS-polyacrylamide gel electrophoresis and transferred to nitrocellulose membranes. Membranes were incubated with primary antibody overnight at 4°C. Membranes were washed with TBS-T buffer three times and incubated with a secondary antibody conjugated to horseradish peroxidase for 2 hr at room temperature. After three washes with TBS-T buffer, the blots were visualized by an enhanced chemiluminescence method (GE Healthcare).
Results
Delineation of the Breakpoint Region on 10q26
Affymetrix Human Mapping 500K Array SNP oligonucleotide microarray analysis (SOMA) of patient DNA was consistent with a balanced translocation and excluded copy-number variation (CNV) as the cause of the IHH or KS phenotype (data not shown). To identify the potentially disrupted genes, we first mapped the translocation breakpoints by using fluorescence in situ hybridization (FISH). Two BAC clones, RP11-592D19 in 10q25.3 and RP11-91E2 in 10q26.3, mapped as centromeric and telomeric, respectively, to the chromosome 10 breakpoint and thus flanked it within a 16.4 Mb region. Sequential rounds of FISH with BACs within this region narrowed the candidate region until a breakpoint-crossing BAC clone was identified. Of nineteen BACs examined, three were proximal and fifteen were distal to the breakpoint, whereas RP11-254K03 hybridized to the normal chromosome 10 and both der(10) and der(12) chromosomes, indicating that it spans the translocation breakpoint, as shown in Figures 1B and 2A.
Figure 2.
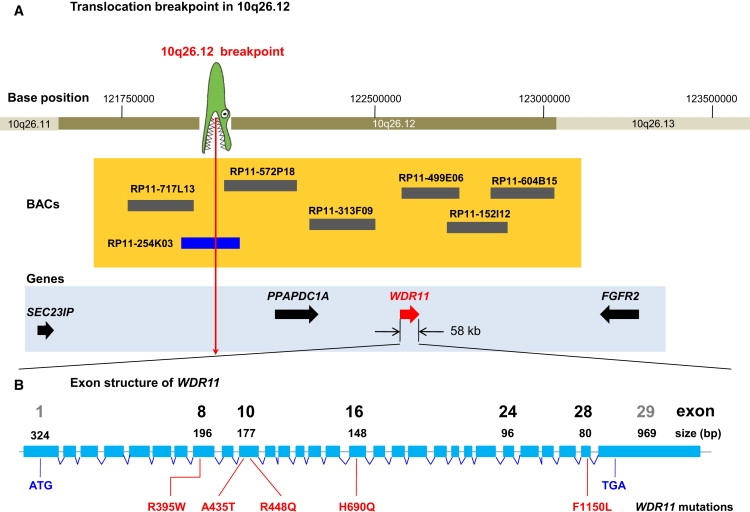
Genetic Mapping of the 10q26 Locus Involved in IHH and KS
(A) The translocation breakpoint, depicted as a crocodile head, is located between SEC23IP and PPAPDC1A. Horizontal bars at 10q26.12 show BACs used for FISH mapping. The size and location of BACs are to scale. The blue bar depicts a BAC clone spanning the breakpoint. Four positional candidate genes located proximal and distal to the breakpoint are shown as arrows in a 2 Mb region.
(B) Exon and intron structure of the 58 kb gene WDR11 (NM_018117.11). Locations of human missense mutations are identified in sporadic IHH and KS patients. Notable exons are shown to scale as blue rectangles and are numbered with exon size. The sizes of introns are not to scale. Mutation F1150L was identified in two independent sporadic IHH patients.
Delineation of the Breakpoint Region on 12q13
BAC clones RP11-88L2 from 12q12 and RP11-762I7 from 12q13.2, used as starting clones for FISH, mapped proximal and distal, respectively, to the breakpoint, confining the chromosome 12 breakpoint within an 11.7 Mb region. FISH experiments with twenty BACs within the region revealed twelve proximal and seven distal to the breakpoint, as well as RP11-464D5, which hybridized to the normal chromosome 12 and both der(10) and der(12) chromosomes, indicating that it spans the translocation breakpoint (data not shown). Sequencing of the breakpoint cloned as described below confirmed localization of the junction sequence within RP11-464D5, as shown in Figure 3A.
Figure 3.

Sequences of the Translocation Breakpoints and Protein Sequence Alignment of WDR11 Orthologs
(A) Genomic DNA sequence at the breakpoints from the normal and derivative chromosomes. The breakpoint on chromosome 10 is located between nucleotides 122,053,649 and 122,053,650, whereas on chromosome 12 it occurs between nucleotides 46,038,271 and 46,038,272 (UCSC Genome Browser NCBI hg18). The junction sequence reveals a 4 bp duplication at the breakpoint of the der(10) chromosome and a 1 bp deletion and a 57 bp insertion at the junction on the der(12) chromosome.
(B) ClustalW multiple alignment of partial protein sequences of WDR11 orthologs. The positions of three residues affected by missense mutations of WDR11 are marked by arrows and red letters in the corresponding segments of the multiple alignment. The amino acid residues that differ from the sequence of the human WDR11 protein are indicated in blue, and the ninth WD domain is indicated under the panel. All three mutated residues are evolutionarily fully conserved in all 13 available WDR11 orthologs.
Refining the Breakpoint Regions by Array Painting and Cloning of Junction Fragments
After narrowing down both breakpoint regions by FISH and before finding the breakpoint spanning clones, we performed array painting by using a customized oligonucleotide array that represented chromosomal regions chr10:121,855,186–122,255,228 and chr12:45,828,906–46,226,893 (hg18) with an average oligonucleotide spacing of 200–300 bases to determine the breakpoint regions more precisely and eventually clone the junction fragments. Based upon the array-painting results, the breakpoint regions were refined to 2.7 kb (between nucleotide positions 122,053,148 and 122,055,901) in 10q26.12 and 4.2 kb (between nucleotide positions 46,034,656 and 46,038,863 in 12q13.11) (UCSC Genome Browser, NCBI Build 36/hg18 assembly), resulting in restatement of the previously cytogenetically assigned karyotype to 46,XY, t(10;12)(q26.12;q13.11)dn. Five forward primers (12q13.2-10kb-4208f, -4562f, -5338f, -5961f, and -7351f) designed from 10 kb containing the 4.2 kb breakpoint region at 12q13.11 and three reverse primers (10q26-10kb-3497r, -5710r, and -5750r) designed from 10 kb encompassing the 2.7 kb breakpoint region at 10q26.12 were paired in a long-range PCR (QIAGEN LongRange PCR Kit 250). Annealing was performed at 60°C for 30 s with an extension for 7 min. All fifteen PCR reactions produced amplicons with sizes from 1.7–7.1 kb (consistent with the interprimer distances on each chromosome) from the subject's DNA but not from that of a normal control male. Therefore, the breakpoints at 10q26.12 and 12q13.11 were narrowed to 590 bp and 2.2 kb, respectively, confirming the array-painting result. Finally, the junction fragment of ∼1.8 kb from der(12) was amplified by normal PCR from one primer pair of 12q13.2-10kb-7351forward and 10q26-10kb-3497reverse and confirmed by sequencing and subsequent BLAST searching.
To amplify the junction fragment of der(10), we paired five forward primers (10q26-10kb -1851f, -2367f, -2801f, -2851f, and -2914f) designed from the 10 kb breakpoint region at 10q26 and five reverse primers (12q13.2-10kb-7630r, -8180r, -8836r, -9664r, and -9890r) from the 10 kb breakpoint region at 12q13.2 in a long-range PCR, which produced amplicons of 1–2.5 kb in combinations with two of the reverse primers (12q13.2-10kb-9664r and -9890r) but no amplicons with the other three (12q13.2-10kb-7630r, -8180r, and -8836r), further refining the breakpoint region. The 2 kb junction fragment of der(10) was amplified by primer set 10q26-10kb-2367f and 12q13.2-10kb-9890r. The partial sequences from chromosomes 10 and 12 of this junction fragment were confirmed by sequencing and BLAST searching. The breakpoint on chromosome 12 is located between nucleotides 46,038,271 and 46,038,272, whereas on chromosome 10 it occurs between nucleotides 122,053,649 and 122,053,650, 547 kb from the 5′end of WDR11. At the junction of the der(10) fragment, there was a CTTT duplication, and at the junction of der(12) fragment, there was a 1 bp G deletion and a 57 bp insertion (Figure 3A).
Candidate Gene Expression Pattern in Rat
The translocation did not directly disrupt a gene on either chromosome 10 or 12, but on the basis of the common chromosomal location associated with IHH and KS phenotypes, we hypothesized that 10q26 was more likely to harbor the causative gene; therefore, we did not study the 12q region further.
On 10q26.12, four candidate genes (FGFR2, PPAPDC1A, SEC23IP, and WDR11) were present in the vicinity of the breakpoint (Figure 2A). We examined their expression in the rat ovary, testis, olfactory bulb, hypothalamic preoptic area, medial basal hypothalamus, anterior pituitary, piriform cortex, temporal cortex, prefrontal cortex, hippocampus, and cerebellum using semiquantitative RT-PCR analysis. All four genes were expressed in all tissues tested; the highest Wdr11 (XM_219377.5) expression, in decreasing order, was in the ovary, olfactory bulb, and piriform cortex (Figure S1).
Mutation Screening of Positional Candidate Genes
We screened 123 IHH and KS patients for FGFR2 (MIM 176943) mutations by direct sequencing of all coding exons and splice junctions (exons 2–18 of NM_022970.3 and the additional exon 8 of NM_000141.4) and found only known polymorphisms rs755793, rs1047100, rs3135774, rs41293763, and rs2278202. Exons 1–7 of PPAPDC1A (NM_001030059.1) were directly sequenced in 60 IHH or KS patients and were likewise negative for mutations other than reported polymorphisms rs10886691 and rs1047369 and additional synonymous variants F15F, L194L, and P270P. Exons 1–18 of SEC23IP (NM_007190.2) were screened in 120 IHH or KS patients. Only known polymorphisms rs34157476, rs3859163, rs3740569, rs3740570, rs17099368, rs34824340, rs2475298, rs2271123, rs12771873, rs58111481, and rs34826964 were seen as variants. Additionally, heterozygous c.760G>T of SEC23IP (p. V254F) was identified in one KS female and two IHH males but not in a second KS female within a family, indicating a lack of segregation with IHH or KS and suggesting that it is a rare sequence variant (absent from 182 normal white controls). Furthermore, heterozygous c.1191+38A>C identified in a male IHH patient was inherited from one of two healthy parents, both of whom are heterozygous for the same change, suggesting that it is also a polymorphism.
DNA sequencing of the protein-coding exons and splice junctions of the WDR11 gene with 29 exons (NM_018117.11, MIM 606417) was performed in 201 IHH or KS patients. Five different heterozygous missense variants that appear to be pathogenic mutations were identified in six unrelated probands (3%) (Table 1); none of these variants was present in ≥420 normal white controls or an additional 402 ethnically matched controls for the one proband who is of Turkish origin. Interestingly, the putative WDR11 mutations were found in both IHH (n = 5) and KS (n = 1) patients, as shown in Table 1, and are described below. We identified one additional heterozygous change, c.2932A>C (p.K978Q), in a Turkish male IHH patient. Residue K978 is completely conserved in all 13 higher vertebrates (Figure S2) and this same nucleotide change was not identified in an initial set of 180 white and Turkish controls. Although the K978Q alteration involves a base change in the first nucleotide of exon 24, exon-trapping experiments did not reveal any aberrant splicing in vitro (data not shown), suggesting that the variant does not affect splicing differences in vivo. Sequencing of additional Turkish control subjects identified the c.2932A>C variant in two Turkish controls (2/402). Thus, c.2932A>C (p.K978Q) is a sequence variant that is not sufficient to cause IHH or KS and has the characteristics of a rare polymorphism (Table S1). Whether it is associated with increased risk of IHH or KS will require investigation of additional IHH, KS, and ethnically matched control subjects.
Table 1.
WDR11 Mutations in IHH and KS Patients
| Patient | Gender and Phenotype | Geographic Origin | Location (Exon/Intron) | Nucleotide change (NM_018117.11) | Amino Acid Change (NP_060587.8) | Class of Mutation | Confirmatory Method |
|---|---|---|---|---|---|---|---|
| C37 | Male/KS; complete IHH; anosmia;testes, 1 ml bilaterally | United States | Translocation breakpoint is 547 kb away from the 5′ end of WDR11 | _ | _ | chromosomal structure mutation as a balanced translocation- t(10;12)(q26.12;q13.11)dn | FISH, array painting, and sequencing of junction fragment; de novo. |
| C71 | Male/complete IHH; testes, 2 ml; FSH = 3 mIU/ml; LH = 1.2 mIU/ml | United States | Exon 8 | c.1183C>T | R395W | missense | 0/420 white controls; detrimental in protein modeling; invariant in 13 available WDR11 orthologs. |
| T87 | Male/IHH; complete IHH; bilat cryptorchidism; FSH = 0.3 mIU/ml; LH < 0.3 mIU/ml | Turkey | Exon 10 | c.1303G>A | A435T | missense | 0/426 white controls; 0/402 Turkish controls; within sixth WD domain; detrimental in protein modeling; abolished EMX1 binding. |
| C99 | Female/IHH | United States | Exon 10 | c.1343G>A | R448Q | missense | 0/426 white controls; within sixth WD domain; detrimental in protein modeling; reduced EMX1 binding; predicted to destabilize the WDR11 dimer and impair actin binding of the complex. |
| C127 | Male/KS; anosmia | United States | Exon 16 | c.2070T>A | H690Q | missense | 0/420 white controls; within ninth WD domain; detrimental in protein modeling; abolished EMX1 binding; invariant in 13 available WDR11 orthologs. |
| C17 | Male/IHH; incomplete IHH; testes, 35, 25 ml; FSH = 6 mIU/ml | United States | Exon 28 | c.3450T>G | F1150L | missense | 0/420 white controls; invariant in 13 available WDR11 orthologs. |
| C100 | Female/IHH; complete IHH; breasts: Tanner 1; FSH < 1 mIU/ml; LH < 1 mIU/ml | United States | Exon 28 | c.3450T>G | F1150L | missense | 0/420 white controls; invariant in 13 available WDR11 orthologs. |
Complete IHH refers to the complete absence of sexual development, whereas incomplete IHH indicates partial pubertal development. Testis size is given (normal is 15–25 ml) when available. Breast development is given when available—Tanner 1 indicates no breast buds.
The five putative heterozygous missense variants that were observed in IHH and KS patients but absent from all controls are c.1183C>T (p.R395W) in an IHH male, c.1303G>A (p.A435T) in a Turkish IHH male, c.1343G>A (p.R448Q) in an IHH female, c.2070T>A (p.H690Q) in a KS male, and c.3450T>G (p.F1150L) in one male and one female IHH patient. Three of the missense changes altering amino acid residues R395, H690, and F1150 are completely conserved in all higher vertebrates (Figure 3B). Two unrelated patients had an identical heterozygous missense alteration, F1150L, so we performed haplotype analysis with seven informative SNP markers—rs7077126, rs10886800, rs41287988, rs1045154, rs12259815, rs10871, and rs1045170—from the WDR11 3′-UTR region. Genotyping of these two patients (C17 and C100) indicated that they share a haplotype around WDR11 and are therefore likely to be descended from a recent common ancestor (data not shown). All identified SNPs, including ten novel SNPs in WDR11, are listed in Table S1.
RT-qPCR and Immunoblot Analysis of WDR11 in the t(10;12) Patient
RT-qPCR and immunoblot analysis with antibody that we generated demonstrated reductions of approximately 20% for transcript and 10% for WDR11 in the lymphoblast cell line from our translocation patient compared to two gender-matched controls (Figure S3). Because this line has one normal allele and one translocated allele, if the entire effect were due to reduction of the mRNA from the translocated chromosome, as would be suspected, the 20% overall reduction in transcript would correspond to an approximate 40% reduction in expression from the translocated chromosome. Although this reduction is modest, it does demonstrate proof-of-principle for a potential position effect of the breakpoint, which is about 547 kb 5′ of WDR11. It is conceivable that, because of tissue-specific factors, the magnitude of the reduction might be significantly greater in some other tissues of the translocation subject, especially those tissues involved in the neuroendocrine control of reproduction, but it is not possible to measure this directly because the lymphoblasts represent the only tissue available from the subject. However, the small effect in lymphoblasts is consistent with an effect of the translocation on expression of this gene and therefore with its role in generating the phenotype in this patient.
Protein Structure Modeling of WDR11
To explore the potential structural features of WDR11, we generated a protein model by using multiple sequence alignment (ClustalW35) based on homology to the C. elegans Homolog of Yeast Actin Interacting Protein 1 (AIP1) (PDB code 1NR0),36 shown in Figure S4. The close structural similarity of the model of WDR11, which features two β propeller structures in each protein chain36,37 (Figure 4A), with the AIP1 dimer (PDB code 1PGU), indicates that WDR11 will also be an actin-binding protein38 (Figure S4). WDR11 contains twelve WD domains, nine (second to tenth repeats) that are confirmed on the basis of direct comparison with the template structure of AIP1 (Figures 4A and 4B) and three additional repeats (first, 11th, and 12th) detected by sequence comparison outside the region of the structural model (Figures 4A and 4B; Table S2). Like AIP1, WDR11 is predicted to exhibit two β propellers; WD domains 2–6 are predicted to constitute the first, and WD domains 7–10 are predicted to constitute the second (Figure 4A). The structural model for WDR11 overlaps well with the known AIP1 structure (Figure S4). This model predicts that WDR11 has twelve WD domains and that nine of them (second through tenth) participate in the genesis of two consecutive β propellers.
Figure 4.
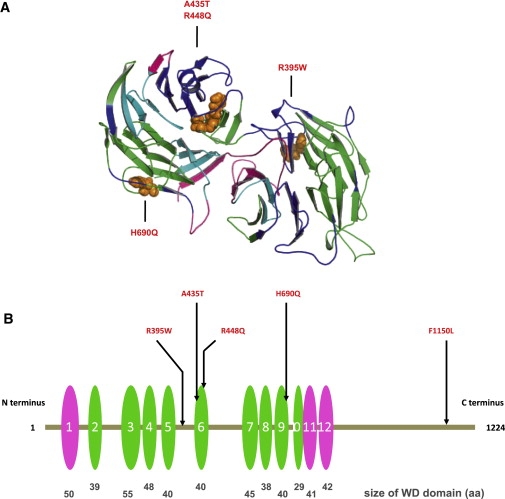
WDR11 Structural Model Indicating the Mutation Sites
(A) Model spanning amino acids 70–739 of WDR11. The model was obtained by alignment to 1NR0 via ClustalW and MOE (Molecular Operating Environment [MOE 2004.03], Chemical Computing Group, Montreal, Quebec, Canada H3B 3X3). WDR11 forms a double propeller structure, in which the WD domains indicated in (B) form the main structural constituent. The two propeller axes are tilted with respect to one another, so only the propeller structure on the left is clearly visible in this representation. Colors indicate side chains of the four mutations within the modeled sequence region, as follows: WD domains predicted on the basis of the model (green), on the basis of SMART (pink), or both (cyan). The sites of the mutations are indicated in orange.
(B) Positions of five missense mutations in WDR11; WD domains are depicted as ovals. The WD domains predicted on the basis of the model and by SMART are depicted in green and pink, respectively. The relative sizes and locations of WD domains are to scale. WDR11 contains twelve WD domains, nine (second to tenth repeats) that are confirmed on the basis of direct comparison with the template structure of AIP1 and three additional repeats (first, 11th, and 12th) detected by sequence comparison outside the region of the structural model. Note that three mutations directly affect WD domains 6 and 9.
WDR11 Interacts and Colocalizes with EMX1 In Vivo and In Vitro
WDR11 is a 1,224 amino acid protein that contains multiple WD domains likely to mediate interaction with protein-binding partners (NCBI Conserved Domain Database cl02567). Consequently, we performed a yeast two-hybrid screen to identify potential cellular proteins whose interaction with WDR11 might be disrupted by the apparent missense mutations in IHH and KS patients. We identified EMX1 (MIM 600034), a human ortholog of Drosophila ems (empty spiracles), as a WDR11-interacting protein. Although four other proteins, including Hey1 (hairy/enhancer-of-split related with YRPW motif 1, MIM 602953), Tagln2 (Transgelin 2, MIM 604634), Ndrg4 (N-Myc downstream-regulated gene 4), and Nrxn3 (neurexin 3, MIM 600567) were also identified as potential WDR11 binding partners, we focused on EMX1 for further studies because of its functional relevance to the developing nervous system.39–43
Specific interaction between WDR11 and EMX1 was revealed by cotransformation of yeast with an expression plasmid encoding WDR11 fused to the GAL4 DNA-binding domain and a plasmid encoding EMX1 fused to the GAL4 activation domain (Figure 5A). The interaction was confirmed in mammalian cells by coimmunoprecipitation assays, which demonstrated that HA-tagged EMX1 was recovered from immunoprecipitates of coexpressed Myc-WDR11 (Figure 5B), and further confirmed by GST pull-down analysis (Figure 5C).
Figure 5.
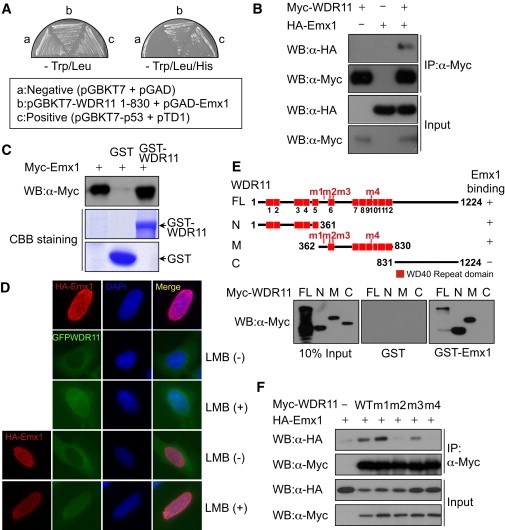
The WDR11 Interacts and Colocalizes with EMX1 In Vivo and In Vitro
(A) EMX1 was identified in the yeast two-hybrid screen as a WDR11-interacting protein. The specific interactions between these two proteins were confirmed by streaking of transformed yeast cells onto synthetic drop-out plates lacking TL (Trp/Leu) or TLH (Trp/Leu/His). Yeast AH109 cells were transformed with empty vectors (pGBKT7 and pGAD), and plasmids encoding SV40 T antigen (pTD1) and p53 (pGBKT7-p53) were utilized as a negative and positive control, respectively.
(B) Myc-WDR11 expression plasmids were transfected alone or along with HA-EMX1 into HeLa cells, the cell lysates were immunoprecipitated with anti-Myc antibody, and coprecipitated HA-EMX1 was detected via immunoblotting with anti-HA antibody.
(C) In-vitro-translated Myc-EMX1 was subjected to GST pull-down analysis with GST (lane 2) or GST-WDR11 (lane 3). The bound proteins were detected via immunoblotting with anti-Myc antibody.
(D) HA-EMX1 and GFP-WDR11 expression plasmids were transfected into U2OS cells, and then cells were treated with leptomycin B (LMB), an inhibitor of nuclear export. Fluorescence microscopy analysis helped determine localizations of HA-EMX1 and GFP-WDR11. Nuclei were stained with DAPI.
(E) Wild-type Myc-WDR11 and its deletion mutants were synthesized in vitro and subjected to a GST pull-down assay with GST (center panel) or GST-WDR11 (right panel). The N, M, and C denote the WDR11 N terminus (amino acids 1–361), middle portion (amino acids 362–830), and C terminus (amino acids 831–1224), respectively. The positions of missense mutations found within the N terminus and central region of WDR11 in IHH patients are marked as m1 through m4 on the schematic diagrams of WDR11.
(F) The wild-type and missense mutant WDR11 expression plasmids were transfected into HEK293 cells along with HA-EMX1 expression plasmids. The cell lysates were immunoprecipitated with anti-Myc antibody, and the association of EMX1 with wild-type WDR11 or missense mutants was determined via immunoblot analysis with anti-HA antibody.
To define the particular WDR11 domains necessary for interaction with EMX1, we generated WDR11 deletion mutants and performed GST pull-down assays to assess EMX1 binding. Both the N terminus and the central portion of WDR11 bound to EMX1, whereas the C terminus did not (Figure 5E). We next tested whether any of the four missense mutations in the N terminus and the central region of WDR11 interfered with EMX1 binding in vitro. Coimmunoprecipitation of EMX1 with the translated proteins from each of the four WDR11 mutant constructs demonstrated that mutants 2 (A435T) and 4 (H690Q) did not associate with EMX1 in mammalian cells, whereas mutant 3 (R448Q) reduced, but did not eliminate, the EMX1 binding. Mutant 1 (R395W) had no effect upon EMX1 binding (Figure 5F).
Subcellular Localization of WDR11 and EMX1
Because EMX1 is a homeodomain transcription factor that participates in the development of olfactory neurons,40 we examined the subcellular localization of both EMX1 and WDR11 in a physiologically relevant human cell system. Immunostaining with a polyclonal anti-WDR11 antibody (directed to N-terminal residues 32–48 of NP_060587.8: QGLIAYGCHSLVVVIDS) localized WDR11 to the cytoplasm of FCNB4-hTERT cells, immortalized human embryonic olfactory GnRH neuroblasts isolated from olfactory epithelium of an 8- to 12-week-old human embryo (data not shown).44 Similarly, fluorescence microscopy of GFP-WDR11 transfected into U2OS cells also yielded a cytoplasmic localization, whereas HA-EMX1 localized to the nucleus. However, when these cells were treated with leptomycin B, an inhibitor of nuclear export, both HA-EMX1 and GFP-WDR11 colocalized in the nucleus (Figure 5D), suggesting that WDR11 might be shuttling between the nucleus and cytoplasm. When each of the five WDR11 missense mutants was transfected into FCNB4-hTERT cells, none altered subcellular localization of the protein (data not shown).
Mouse and Zebrafish Whole-Mount In Situ Hybridization
To investigate the developmental expression of Wdr11, we performed whole-mount in situ hybridization analysis in mouse embryos from days E10.5–E14.5. The pattern of expression observed was consistent with a role for WDR11 in IHH and KS. As early as E10.5, the entire developing central nervous system, except for the spinal cord, revealed Wdr11 expression (Figures 6A–6C). The neuroepithelium, including the diencephalic region that gives rise to hypothalamic neurons where GnRH neurons reside, stained strongly for Wdr11 at E11.5 and E12.5. Mouse neuroendocrine GnRH neurons migrate from the olfactory placode region alongside olfactory neurons to cross the cribriform plate and finally reach the hypothalamus, a process that is active from E10.5–E14.5 and is usually completed by E18.5.45 At E14.5 high levels of Wdr11 expression were particularly noteworthy in the developing cortex and the olfactory bulb (Figures 6D and 6E). In the adult brain, intense Wdr11 expression was restricted to the olfactory bulb, the olfaction-related piriform cortex (Figure 6F), the granule cell layer of the cerebellum, and neurons of the hippocampal formation (Figure 6G). Increased signal intensities within the hypothalamus were observed with 35S-UTP labeled Wdr11 antisense probes (Figure 6G). For higher-resolution images of this region, digoxigenin labeling of sagittal sections of adult brains demonstrated signals scattered throughout the hypothalamus, sometimes in clusters of neurons (Figure 6H). Notably, in mouse, Emx1 expression is also detectable from day 9.5 of gestation in the presumptive cerebral cortex and olfactory bulbs.46
Figure 6.
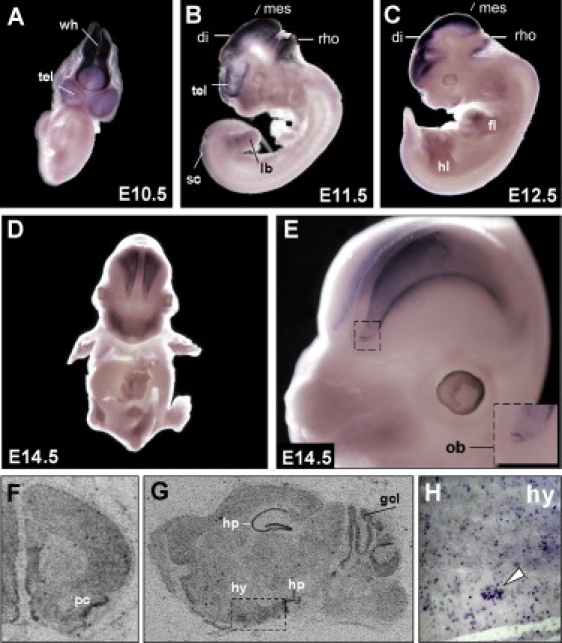
Wdr11 Expression during Murine Development
(A–E) DIG-labeled whole-mount in situ hybridization with a Wdr11 antisense probe at different embryonic stages. High expression levels are found in all structures of the developing brain as early as E10.5. Expression in the limbs is prominent at E12.5 and E13.5. Staining was also observed in both the hind and forelimb buds, but as limbs developed, it shifted toward the terminal phalanges. At E14.5 the olfactory bulb and the developing cortex show the highest expression levels. A magnification of the developing cortex and olfactory bulb is shown in (E).
(F–H) Expression of Wdr11 in the adult brain. [35S]-UTP-labeled in situ hybridizations show prominent Wdr11 signals in the piriform cortex (F) as well as in the hippocampus and cerebellum (G). Note the higher signal intensity in the hypothalamic region within the dotted rectangle in (G). Single cells as well as clusters of neurons within the hypothalamic nuclei also showed Wdr11 expression in DIG-labeled cryosections (H). Signals were absent with the sense control. Abbreviations are as follows: tel, telencephalic vesicle; wh, wall of hindbrain; di, diencephalon; mes, mesencephalon; rho, rhombencephalon; sc, spinal cord; lb, limb bud; hl, hind limb; fl, fore limb; ob, olfactory bulb; pc, piriform cortex; gcl, granule cell layer of the cerebellum; hp, hippocampus; and hy, hypothalamus.
In the zebrafish, wdr11 was expressed ubiquitously at 24 hpf; whereas emx1 was highly expressed in the forebrain, and a small group of cells was clustered around the same area of the diencephalic GnRH3 neurons at 30–36 hpf (Figure 7), again consistent with a potential role for a WDR11-EMX1 protein interaction.
Figure 7.
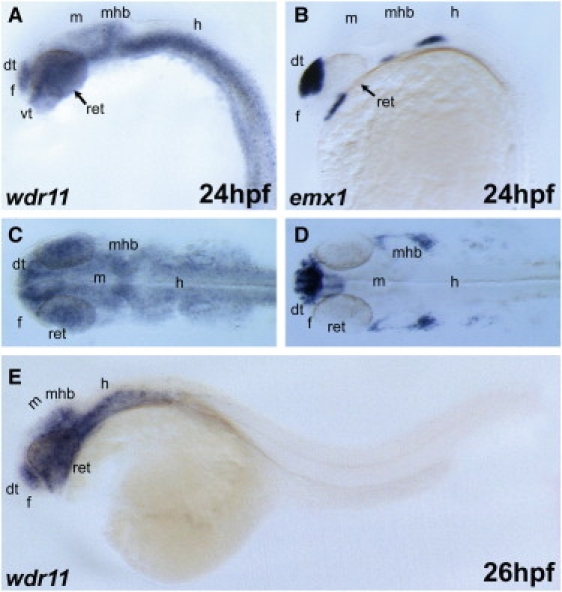
Expression of wdr11 in Developing Zebrafish Embryos
At 24 hpf, wdr11 transcripts were broadly detected in forebrain, midbrain, and hindbrain.
(A, B, and E) Lateral view; anterior is to the left. (C and D) Dorsal view. The expression domain of wdr11 in the brain partially overlapped with that of emx1 (B and D), a dorsal telencephalon marker. Abbreviations are as follows: dt, dorsal telencephalon; f, forebrain; m, midbrain; h, hindbrain; mhb, midbrain-hindbrain boundary; vt, ventral telencephalon; and ret, retina.
Discussion
Our mapping, cloning, and sequencing of the breakpoints of the de novo balanced chromosome translocation from a KS male with karyotype 46,XY,t(10;12)(q26.12;q13.11)dn and the consequent detection of multiple independent missense variants in WDR11 in KS and IHH patients argues strongly for a causative role for WDR11 in this disorder. Regrettably, DNA samples of the patients’ parents are not available, and all efforts to track the parents have been unsuccessful. However, because all WDR11 mutations were heterozygous, autosomal-dominant inheritance of the IHH phenotype is likely. Consistent with this interpretation, no WDR11 mutation was found in any of 29 exons or splice junctions on the untranslocated second allele from our translocation patient, suggesting that a reduction in functional WDR11 as a result of dysregulation of the gene via a position effect of the translocation is the cause of KS in this subject (see Results and Figure S3). The absence of truncating nonsense and frameshift mutations indicates that these might produce a more severe phenotype or embryonic lethality.
WDR11 was originally identified as a potential tumor suppressor by positional cloning of a somatically acquired t(10;19) chromosome translocation that generated an intragenic deletion in human glioblastoma cells.47 This deletion disrupted WDR11 and fused it to the ZNF320 gene (MIM 606427). Interestingly, other genes associated with mammalian puberty are also known for their involvement in tumorigenesis.48 For example, hypothalamic expression of certain tumor suppressor genes is increased at puberty in monkeys or decreased in mice with delayed puberty.49 Similarly, KISS1 (MIM 603286), a tumor metastasis suppressor gene50 in melanomas51 and breast carcinomas52 encodes the peptide ligand of G-protein-coupled receptor 54 (GPR54),53 which plays a critical role in the initiation of puberty. KISS1R (encoding GPR54) mutations cause autosomal-recessive IHH in consanguineous families and mice.7,8
WDR11's function is unknown, but it contains twelve WD domains and is highly conserved throughout vertebrate evolution. The gene was previously annotated as BRWD2 (Bromodomain and WD repeat domain containing 2) but has recently been more appropriately designated WDR11 because it has WD domains but no Bromo domains.54 Mutations of other WD proteins have been associated previously with mammalian reproduction. Both genders of Repro5 mice, which have ENU-induced Brwd1 (Wdr9) mutations, demonstrate infertility. In addition, patients with mutations of BRWD3 (MIM 300553) have undescended testes and minimal facial or axillary hair.55 However, this is the first report of a specific role for a WD protein in IHH or KS, and it implicates protein-protein interaction mediated by WD repeats of WDR11 as a critical requirement for normal puberty.
Proteins with repeated WD domains, each of which consists of four antiparallel β strands, form β propeller structures to support interactions with protein-binding partners and to organize and stabilize multiprotein complexes.56 A β propeller is characterized by 4–8 blade-shaped β sheets arranged around a central axis; each sheet of four antiparallel β strands is twisted so that the first and fourth sheets are close to perpendicular. The last β strand of one WD repeat, and the first three β strands of the WD repeat form a blade of the β propeller (Figure 4A).
Three of the WDR11 missense mutations leading to R395W, H690Q, and F1150L alter amino acid residues that are completely conserved in all 13 available mammalian and avian orthologs (human, chimpanzee, cow, horse, panda, pig, dog, rat, mouse, rabbit, opossum, chicken, and finch), and a fourth change, A435T, is shared in 11 out of 13 species, suggesting that these substitutions in six independent sporadic patients are very to be detrimental. Three of the missense alterations are located directly in the predicted propeller regions of WDR11: A435T and R448Q are in the sixth WD domain, and H690Q is in the ninth WD domain (Figure 4B). These three mutations are predicted by SPPIDER to alter protein-protein binding domains defined by protein modeling and therefore are likely to disrupt normal protein function (Figure S5).
The similarity in structural modeling between WDR11 and the known structure of AIP1 suggests that WDR11, like AIP1, may form a dimer stabilized by interaction with two zinc ions (Figure S6). Because the two protein structures are not identical, deviations arising from their alignment make the position of the Zn ion uncertain to a few angstroms (Figure S4). However, WDR11 has the required residues (Asp377, Glu384, His501, His508, and Glu510) for zinc binding in the vicinity of the putative zinc position (Figure S7). The R448Q mutation is less than 5 Å from the predicted zinc binding site. Arg residues near Zn coordination sites do not directly interact with the Zn, but they stabilize their environment because Arg is highly positively charged. Replacing Arg with the much smaller Gln residue could influence the zinc-binding propensity of WDR11 and affect its dimer formation and interactions, including a potential actin interaction predicted by analogy with AIP1.57
In view of the importance of protein-protein interactions for the function of WD proteins, we sought binding partners for WDR11 and identified the transcription factor EMX1 as a novel interactor. EMX1 is a homeobox transcription factor involved in specifying cell fates in the developing central nervous system,39 and it participates in the development of olfactory neurons.40 This putative transcription factor has been shown to be one of the downstream target genes for Gli-Kruppel family member 3 (Gli3, MIM 165240) transcription factor,41,42 which is a part of the Sonic hedgehog-Patched-Gli (Shh-Ptch-Gli) signaling pathway43 important in endocrine signaling.
Analysis of the expression patterns of WDR11 in human embryonic olfactory GnRH neuroblasts as well as in mouse and zebrafish development revealed overlapping patterns of expression with EMX1 in regions critical for formation of the hypothalamus,45 supporting the opportunity for the two proteins to interact in vivo and to act together during development. Deletion analysis revealed that WDR11 interacts with EMX1 via both its N terminus and its central region, where four (R395W, A435T, R448Q, and H690Q) of the five WDR11 missense alterations were found. R448Q reduced and both A435T and H690Q abolished binding to EMX1, physically decreasing the opportunity for productive interaction. Interestingly, the mutant R395W did not appear to affect EMX1 binding, but this represents alteration of an amino acid that is invariant in higher vertebrates, suggesting that this alteration permits binding but impairs the as-yet-undefined functional consequences of the WDR11-EMX1 interaction.
Taken together, our genetic and functional data provide strong evidence for missense sequence variants of WDR11 as a cause of IHH and KS in a proportion of cases of this genetically heterogeneous condition. This adds to the growing list of genes known to be mutated in IHH and KS and will open new investigative routes for understanding the development of normal human puberty and reproduction. Importantly, the identification of EMX1 as a binding partner of WDR11 whose interaction can be disrupted by IHH- and KS-associated mutations is significant for two principal reasons. First, it places WDR11, whose biological function is not well understood, as a potential player in the Sonic hedgehog-Patched-Gli-Emx signaling pathway via its interaction with EMX1. Second, the demonstration of a developmental role for WDR11 in IHH and KS suggests a possible connection between Shh signaling and pubertal development. The potential for defects in this pathway in IHH and KS is worthy of further exploration.
Acknowledgments
This paper is dedicated to the 80th birthday of Paul G. McDonough. We thank all families for their kind participation in this study. We are indebted to the following colleagues for this study: Stefanie Meien, Milton David Stuart, Karen Norris, Ihn-Sik Seong, Jae Ho Lee, Gabriela Alexandru, Wei Chen, Ines Müller, Fei Lan, Hans-Jürgen Kreienkamp, Enno Gößling, Duygu Duman, Lynn Chorich, Sandra Tho, Jill Mokry, Lisa Halvorson, and Temple Smith. We acknowledge support by the Landesstiftung Baden-Würtemberg, the Deutsche Akademische Austauschdienst (German Academic Exchange Service) and the volunteers of POEM@HOME. This study was supported in part by a Korea Science and Engineering Foundation (KOSEF) grant funded by the government of the Republic of Korea (The Ubiquitome Research Program, 2009-00983 to C.Y.C.) J.F.G. was supported by National Institutes of Health grant GM061354 for the Developmental Genome Anatomy Project. We also acknowledge support to L.C.L. from National Institutes of Health grants HD33004 and HD040287.
Contributor Information
Hyung-Goo Kim, Email: hkim@chgr.mgh.harvard.edu.
Lawrence C. Layman, Email: llayman@mcg.edu.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
NCBI Single Nucleotide Polymorphism Database, http://www.ncbi.nlm.nih.gov.ezp-prod1.hul.harvard.edu/projects/SNP/
NCBI BLAST, http://blast.ncbi.nlm.nih.gov/
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/
Primer design, http://www.es.embnet.org/cgi-bin/primer3_www.cgi
UCSC Genome Browser, http://genome.ucsc.edu/
Accession Numbers
Human WDR11 mRNA, NM_018117.11; human WDR11 protein, NP_060587.8; mouse WDR11 mRNA, NM_172255.3; mouse WDR11 protein, NP_758459.2; rat WDR11 mRNA, XM_219377.5; rat WDR11 protein, XP_219377.5; chimpanzee WDR11 protein, XP_508077.2; cow WDR11 protein, NP_001094592.1; horse WDR11 protein, XP_001496034.1; panda WDR11 protein, EFB13673.1; pig WDR11 protein, XP_001924391.1; dog WDR11 protein, XP_535039.2; rabbit WDR11 protein, XP_002718739.1; opossum WDR11 protein, XP_001376637.1; chicken WDR11 protein, XP_421795.2; zebra finch WDR11 protein, XP_002188976.1; zebrafish wdr11 mRNA, XM_682139.3; zebrafish wdr11 protein, XP_687231.2; human PPAPDC1A mRNA, NM_001030059.1; human SEC23IP mRNA, NM_007190.2.
References
- 1.Kim H.G., Bhagavath B., Layman L.C. Clinical manifestations of impaired GnRH neuron development and function. Neurosignals. 2008;16:165–182. doi: 10.1159/000111561. [DOI] [PubMed] [Google Scholar]
- 2.Franco B., Guioli S., Pragliola A., Incerti B., Bardoni B., Tonlorenzi R., Carrozzo R., Maestrini E., Pieretti M., Taillon-Miller P. A gene deleted in Kallmann's syndrome shares homology with neural cell adhesion and axonal path-finding molecules. Nature. 1991;353:529–536. doi: 10.1038/353529a0. [DOI] [PubMed] [Google Scholar]
- 3.Legouis R., Hardelin J.P., Levilliers J., Claverie J.M., Compain S., Wunderle V., Millasseau P., Le Paslier D., Cohen D., Caterina D. The candidate gene for the X-linked Kallmann syndrome encodes a protein related to adhesion molecules. Cell. 1991;67:423–435. doi: 10.1016/0092-8674(91)90193-3. [DOI] [PubMed] [Google Scholar]
- 4.Dodé C., Levilliers J., Dupont J.M., De Paepe A., Le Dû N., Soussi-Yanicostas N., Coimbra R.S., Delmaghani S., Compain-Nouaille S., Baverel F. Loss-of-function mutations in FGFR1 cause autosomal dominant Kallmann syndrome. Nat. Genet. 2003;33:463–465. doi: 10.1038/ng1122. [DOI] [PubMed] [Google Scholar]
- 5.Kim H.G., Herrick S.R., Lemyre E., Kishikawa S., Salisz J.A., Seminara S., MacDonald M.E., Bruns G.A., Morton C.C., Quade B.J., Gusella J.F. Hypogonadotropic hypogonadism and cleft lip and palate caused by a balanced translocation producing haploinsufficiency for FGFR1. J. Med. Genet. 2005;42:666–672. doi: 10.1136/jmg.2004.026989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pitteloud N., Acierno J.S., Jr., Meysing A., Eliseenkova A.V., Ma J., Ibrahimi O.A., Metzger D.L., Hayes F.J., Dwyer A.A., Hughes V.A. Mutations in fibroblast growth factor receptor 1 cause both Kallmann syndrome and normosmic idiopathic hypogonadotropic hypogonadism. Proc. Natl. Acad. Sci. USA. 2006;103:6281–6286. doi: 10.1073/pnas.0600962103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.de Roux N., Genin E., Carel J.C., Matsuda F., Chaussain J.L., Milgrom E. Hypogonadotropic hypogonadism due to loss of function of the KiSS1-derived peptide receptor GPR54. Proc. Natl. Acad. Sci. USA. 2003;100:10972–10976. doi: 10.1073/pnas.1834399100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Seminara S.B., Messager S., Chatzidaki E.E., Thresher R.R., Acierno J.S., Jr., Shagoury J.K., Bo-Abbas Y., Kuohung W., Schwinof K.M., Hendrick A.G. The GPR54 gene as a regulator of puberty. N. Engl. J. Med. 2003;349:1614–1627. doi: 10.1056/NEJMoa035322. [DOI] [PubMed] [Google Scholar]
- 9.Topaloglu A.K., Reimann F., Guclu M., Yalin A.S., Kotan L.D., Porter K.M., Serin A., Mungan N.O., Cook J.R., Ozbek M.N. TAC3 and TACR3 mutations in familial hypogonadotropic hypogonadism reveal a key role for Neurokinin B in the central control of reproduction. Nat. Genet. 2009;41:354–358. doi: 10.1038/ng.306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.de Roux N., Young J., Misrahi M., Genet R., Chanson P., Schaison G., Milgrom E. A family with hypogonadotropic hypogonadism and mutations in the gonadotropin-releasing hormone receptor. N. Engl. J. Med. 1997;337:1597–1602. doi: 10.1056/NEJM199711273372205. [DOI] [PubMed] [Google Scholar]
- 11.Layman L.C., Cohen D.P., Jin M., Xie J., Li Z., Reindollar R.H., Bolbolan S., Bick D.P., Sherins R.R., Duck L.W. Mutations in gonadotropin-releasing hormone receptor gene cause hypogonadotropic hypogonadism. Nat. Genet. 1998;18:14–15. doi: 10.1038/ng0198-14. [DOI] [PubMed] [Google Scholar]
- 12.Miura K., Acierno J.S., Jr., Seminara S.B. Characterization of the human nasal embryonic LHRH factor gene, NELF, and a mutation screening among 65 patients with idiopathic hypogonadotropic hypogonadism (IHH) J. Hum. Genet. 2004;49:265–268. doi: 10.1007/s10038-004-0137-4. [DOI] [PubMed] [Google Scholar]
- 13.Kim H.G., Kurth I., Lan F., Meliciani I., Wenzel W., Eom S.H., Kang G.B., Rosenberger G., Tekin M., Ozata M. Mutations in CHD7, encoding a chromatin-remodeling protein, cause idiopathic hypogonadotropic hypogonadism and Kallmann syndrome. Am. J. Hum. Genet. 2008;83:511–519. doi: 10.1016/j.ajhg.2008.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Falardeau J., Chung W.C., Beenken A., Raivio T., Plummer L., Sidis Y., Jacobson-Dickman E.E., Eliseenkova A.V., Ma J., Dwyer A. Decreased FGF8 signaling causes deficiency of gonadotropin-releasing hormone in humans and mice. J. Clin. Invest. 2008;118:2822–2831. doi: 10.1172/JCI34538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bouligand J., Ghervan C., Tello J.A., Brailly-Tabard S., Salenave S., Chanson P., Lombès M., Millar R.P., Guiochon-Mantel A., Young J. Isolated familial hypogonadotropic hypogonadism and a GNRH1 mutation. N. Engl. J. Med. 2009;360:2742–2748. doi: 10.1056/NEJMoa0900136. [DOI] [PubMed] [Google Scholar]
- 16.Chan Y.M., de Guillebon A., Lang-Muritano M., Plummer L., Cerrato F., Tsiaras S., Gaspert A., Lavoie H.B., Wu C.H., Crowley W.F., Jr. GNRH1 mutations in patients with idiopathic hypogonadotropic hypogonadism. Proc. Natl. Acad. Sci. USA. 2009;106:11703–11708. doi: 10.1073/pnas.0903449106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dodé C., Teixeira L., Levilliers J., Fouveaut C., Bouchard P., Kottler M.L., Lespinasse J., Lienhardt-Roussie A., Mathieu M., Moerman A. Kallmann syndrome: Mutations in the genes encoding prokineticin-2 and prokineticin receptor-2. PLoS Genet. 2006;2:e175. doi: 10.1371/journal.pgen.0020175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bhagavath B., Podolsky R.H., Ozata M., Bolu E., Bick D.P., Kulharya A., Sherins R.J., Layman L.C. Clinical and molecular characterization of a large sample of patients with hypogonadotropic hypogonadism. Fertil. Steril. 2006;85:706–713. doi: 10.1016/j.fertnstert.2005.08.044. [DOI] [PubMed] [Google Scholar]
- 19.Suzuki Y., Sasagawa I., Nakada T., Onmura Y. Bilateral cryptorchidism associated with terminal deletion of 10q. Urol. Int. 1998;61:186–187. doi: 10.1159/000030321. [DOI] [PubMed] [Google Scholar]
- 20.Mutoh A., Sasagawa I., Tateno T., Sawamura T., Nakada T. Long arm deletion of chromosome 10 in a boy with monorchidism. Scand. J. Urol. Nephrol. 1999;33:77–78. doi: 10.1080/003655999750016357. [DOI] [PubMed] [Google Scholar]
- 21.Leonard N.J., Harley F.L., Lin C.C. Terminal deletion of chromosome 10q at band 26.1: Follow-up in an adolescent male with high-output renal failure from congenital obstructive uropathy. Am. J. Med. Genet. 1999;86:115–117. [PubMed] [Google Scholar]
- 22.Baccetti B., Bruni E., Collodel G., Gambera L., Moretti E., Marzella R., Piomboni P. 10, 15 reciprocal translocation in an infertile man: Ultrastructural and fluorescence in-situ hybridization sperm study: case report. Hum. Reprod. 2003;18:2302–2308. doi: 10.1093/humrep/deg460. [DOI] [PubMed] [Google Scholar]
- 23.Ogata T., Muroya K., Sasagawa I., Kosho T., Wakui K., Sakazume S., Ito K., Matsuo N., Ohashi H., Nagai T. Genetic evidence for a novel gene(s) involved in urogenital development on 10q26. Kidney Int. 2000;58:2281–2290. doi: 10.1046/j.1523-1755.2000.00412.x. [DOI] [PubMed] [Google Scholar]
- 24.Lukusa T., Fryns J.P. Pure distal monosomy 10q26 in a patient displaying clinical features of Prader-Willi syndrome during infancy and distinct behavioural phenotype in adolescence. Genet. Couns. 2000;11:119–126. [PubMed] [Google Scholar]
- 25.Bofinger M.K., Opitz J.M., Soukup S.W., Ekblom L.S., Phillips S., Daniel A., Greene E.W. A familial MCA/MR syndrome due to translocation t(10;16) (q26;p13.1): report of six cases. Am. J. Med. Genet. 1991;38:1–8. doi: 10.1002/ajmg.1320380102. [DOI] [PubMed] [Google Scholar]
- 26.Schinzel A., Lorda-Sanchez I., Binkert F., Carter N.P., Bebb C.E., Ferguson-Smith M.A., Eiholzer U., Zachmann M., Robinson W.P. Kallmann syndrome in a boy with a t(1;10) translocation detected by reverse chromosome painting. J. Med. Genet. 1995;32:957–961. doi: 10.1136/jmg.32.12.957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Higgins A.W., Alkuraya F.S., Bosco A.F., Brown K.K., Bruns G.A., Donovan D.J., Eisenman R., Fan Y., Farra C.G., Ferguson H.L. Characterization of apparently balanced chromosomal rearrangements from the developmental genome anatomy project. Am. J. Hum. Genet. 2008;82:712–722. doi: 10.1016/j.ajhg.2008.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Crowley W.F., Jr., Filicori M., Spratt D.I., Santoro N.F. The physiology of gonadotropin-releasing hormone (GnRH) secretion in men and women. Recent Prog. Horm. Res. 1985;41:473–531. doi: 10.1016/b978-0-12-571141-8.50015-9. [DOI] [PubMed] [Google Scholar]
- 29.Hermans-Borgmeyer I., Hampe W., Schinke B., Methner A., Nykjaer A., Süsens U., Fenger U., Herbarth B., Schaller H.C. Unique expression pattern of a novel mosaic receptor in the developing cerebral cortex. Mech. Dev. 1998;70:65–76. doi: 10.1016/s0925-4773(97)00177-9. [DOI] [PubMed] [Google Scholar]
- 30.Kim H.T., Kim E.H., Yoo K.W., Lee M.S., Choi J.H., Park H.C., Yeo S.Y., Lee D.S., Kim C.H. Isolation and expression analysis of Alzheimer's disease-related gene xb51 in zebrafish. Dev. Dyn. 2008;237:3921–3926. doi: 10.1002/dvdy.21806. [DOI] [PubMed] [Google Scholar]
- 31.Morita T., Nitta H., Kiyama Y., Mori H., Mishina M. Differential expression of two zebrafish emx homeoprotein mRNAs in the developing brain. Neurosci. Lett. 1995;198:131–134. doi: 10.1016/0304-3940(95)11988-9. [DOI] [PubMed] [Google Scholar]
- 32.Bhagavath B., Ozata M., Ozdemir I.C., Bolu E., Bick D.P., Sherins R.J., Layman L.C. The prevalence of gonadotropin-releasing hormone receptor mutations in a large cohort of patients with hypogonadotropic hypogonadism. Fertil. Steril. 2005;84:951–957. doi: 10.1016/j.fertnstert.2005.04.029. [DOI] [PubMed] [Google Scholar]
- 33.Kalscheuer V.M., FitzPatrick D., Tommerup N., Bugge M., Niebuhr E., Neumann L.M., Tzschach A., Shoichet S.A., Menzel C., Erdogan F. Mutations in autism susceptibility candidate 2 (AUTS2) in patients with mental retardation. Hum. Genet. 2007;121:501–509. doi: 10.1007/s00439-006-0284-0. [DOI] [PubMed] [Google Scholar]
- 34.Tzschach A., Bisgaard A.M., Kirchhoff M., Graul-Neumann L.M., Neitzel H., Page S., Ahmed A., Müller I., Erdogan F., Ropers H.H. Chromosome aberrations involving 10q22: Report of three overlapping interstitial deletions and a balanced translocation disrupting C10orf11. Eur. J. Hum. Genet. 2010;18:291–295. doi: 10.1038/ejhg.2009.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li K.B. ClustalW-MPI: ClustalW analysis using distributed and parallel computing. Bioinformatics. 2003;19:1585–1586. doi: 10.1093/bioinformatics/btg192. [DOI] [PubMed] [Google Scholar]
- 36.Mohri K., Vorobiev S., Fedorov A.A., Almo S.C., Ono S. Identification of functional residues on Caenorhabditis elegans actin-interacting protein 1 (UNC-78) for disassembly of actin depolymerizing factor/cofilin-bound actin filaments. J. Biol. Chem. 2004;279:31697–31707. doi: 10.1074/jbc.M403351200. [DOI] [PubMed] [Google Scholar]
- 37.Voegtli W.C., Madrona A.Y., Wilson D.K. The structure of Aip1p, a WD repeat protein that regulates Cofilin-mediated actin depolymerization. J. Biol. Chem. 2003;278:34373–34379. doi: 10.1074/jbc.M302773200. [DOI] [PubMed] [Google Scholar]
- 38.Franke W.W. Actin's many actions start at the genes. Nat. Cell Biol. 2004;6:1013–1014. doi: 10.1038/ncb1104-1013. [DOI] [PubMed] [Google Scholar]
- 39.Bishop K.M., Garel S., Nakagawa Y., Rubenstein J.L., O'Leary D.D. Emx1 and Emx2 cooperate to regulate cortical size, lamination, neuronal differentiation, development of cortical efferents, and thalamocortical pathfinding. J. Comp. Neurol. 2003;457:345–360. doi: 10.1002/cne.10549. [DOI] [PubMed] [Google Scholar]
- 40.Lichtneckert R., Nobs L., Reichert H. Empty spiracles is required for the development of olfactory projection neuron circuitry in Drosophila. Development. 2008;135:2415–2424. doi: 10.1242/dev.022210. [DOI] [PubMed] [Google Scholar]
- 41.Theil T., Alvarez-Bolado G., Walter A., Rüther U. Gli3 is required for Emx gene expression during dorsal telencephalon development. Development. 1999;126:3561–3571. doi: 10.1242/dev.126.16.3561. [DOI] [PubMed] [Google Scholar]
- 42.Tole S., Ragsdale C.W., Grove E.A. Dorsoventral patterning of the telencephalon is disrupted in the mouse mutant extra-toes(J) Dev. Biol. 2000;217:254–265. doi: 10.1006/dbio.1999.9509. [DOI] [PubMed] [Google Scholar]
- 43.Villavicencio E.H., Walterhouse D.O., Iannaccone P.M. The sonic hedgehog-patched-gli pathway in human development and disease. Am. J. Hum. Genet. 2000;67:1047–1054. doi: 10.1016/s0002-9297(07)62934-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hu Y., Guimond S.E., Travers P., Cadman S., Hohenester E., Turnbull J.E., Kim S.H., Bouloux P.M. Novel mechanisms of fibroblast growth factor receptor 1 regulation by extracellular matrix protein anosmin-1. J. Biol. Chem. 2009;284:29905–29920. doi: 10.1074/jbc.M109.049155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schwarting G.A., Wierman M.E., Tobet S.A. Gonadotropin-releasing hormone neuronal migration. Semin. Reprod. Med. 2007;25:305–312. doi: 10.1055/s-2007-984736. [DOI] [PubMed] [Google Scholar]
- 46.Simeone A., Gulisano M., Acampora D., Stornaiuolo A., Rambaldi M., Boncinelli E. Two vertebrate homeobox genes related to the Drosophila empty spiracles gene are expressed in the embryonic cerebral cortex. EMBO J. 1992;11:2541–2550. doi: 10.1002/j.1460-2075.1992.tb05319.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chernova O.B., Hunyadi A., Malaj E., Pan H., Crooks C., Roe B., Cowell J.K. A novel member of the WD-repeat gene family, WDR11, maps to the 10q26 region and is disrupted by a chromosome translocation in human glioblastoma cells. Oncogene. 2001;20:5378–5392. doi: 10.1038/sj.onc.1204694. [DOI] [PubMed] [Google Scholar]
- 48.Ojeda S.R., Dubay C., Lomniczi A., Kaidar G., Matagne V., Sandau U.S., Dissen G.A. Gene networks and the neuroendocrine regulation of puberty. Mol. Cell. Endocrinol. 2010;324:3–11. doi: 10.1016/j.mce.2009.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Parent A.S., Matagne V., Westphal M., Heger S., Ojeda S., Jung H. Gene expression profiling of hypothalamic hamartomas: A search for genes associated with central precocious puberty. Horm. Res. 2008;69:114–123. doi: 10.1159/000111815. [DOI] [PubMed] [Google Scholar]
- 50.Lee J.H., Miele M.E., Hicks D.J., Phillips K.K., Trent J.M., Weissman B.E., Welch D.R. KiSS-1, a novel human malignant melanoma metastasis-suppressor gene. J. Natl. Cancer Inst. 1996;88:1731–1737. doi: 10.1093/jnci/88.23.1731. [DOI] [PubMed] [Google Scholar]
- 51.Lee J.H., Welch D.R. Identification of highly expressed genes in metastasis-suppressed chromosome 6/human malignant melanoma hybrid cells using subtractive hybridization and differential display. Int. J. Cancer. 1997;71:1035–1044. doi: 10.1002/(sici)1097-0215(19970611)71:6<1035::aid-ijc20>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- 52.Lee J.H., Welch D.R. Suppression of metastasis in human breast carcinoma MDA-MB-435 cells after transfection with the metastasis suppressor gene, KiSS-1. Cancer Res. 1997;57:2384–2387. [PubMed] [Google Scholar]
- 53.Ohtaki T., Shintani Y., Honda S., Matsumoto H., Hori A., Kanehashi K., Terao Y., Kumano S., Takatsu Y., Masuda Y. Metastasis suppressor gene KiSS-1 encodes peptide ligand of a G-protein-coupled receptor. Nature. 2001;411:613–617. doi: 10.1038/35079135. [DOI] [PubMed] [Google Scholar]
- 54.Philipps D.L., Wigglesworth K., Hartford S.A., Sun F., Pattabiraman S., Schimenti K., Handel M., Eppig J.J., Schimenti J.C. The dual bromodomain and WD repeat-containing mouse protein BRWD1 is required for normal spermiogenesis and the oocyte-embryo transition. Dev. Biol. 2008;317:72–82. doi: 10.1016/j.ydbio.2008.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Field M., Tarpey P.S., Smith R., Edkins S., O'Meara S., Stevens C., Tofts C., Teague J., Butler A., Dicks E. Mutations in the BRWD3 gene cause X-linked mental retardation associated with macrocephaly. Am. J. Hum. Genet. 2007;81:367–374. doi: 10.1086/520677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Higa L.A., Zhang H. Stealing the spotlight: CUL4-DDB1 ubiquitin ligase docks WD40-repeat proteins to destroy. Cell Div. 2007;2:5. doi: 10.1186/1747-1028-2-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kudryashov D.S., Sawaya M.R., Adisetiyo H., Norcross T., Hegyi G., Reisler E., Yeates T.O. The crystal structure of a cross-linked actin dimer suggests a detailed molecular interface in F-actin. Proc. Natl. Acad. Sci. USA. 2005;102:13105–13110. doi: 10.1073/pnas.0506429102. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.