

Abstract
Acrylamide (AA) is an industrial chemical, a by-product of fried starchy foods, and a mutagen and rodent carcinogen. It can also cause damage during spermatogenesis. In this study, we investigated whether AA and its metabolite glycidamide (GA) induce mutagenic effects in the germ cells of male mice. Male Big Blue transgenic mice were administered 1.4 or 7.0mM of AA or GA in the drinking water for up to 4 weeks. Testicular cII mutant frequency (MF) was determined 3 weeks after the last treatment, and the types of the mutations in the cII gene were analyzed by DNA sequencing. The testes cII MFs in mice treated with either the low or high exposure concentrations of AA and GA were increased significantly. There was no significant difference in the cII MFs between AA and GA at the low exposure concentration. The mutation spectra in mice treated with AA (1.4mM) or GA (both 1.4 and 7.0mM) differed significantly from those of controls, but there were no significant differences in mutation patterns between AA and GA treatments. Comparison of the mutation spectra between testes and livers showed that the spectra differed significantly between the two tissues following treatment with AA or GA, whereas the mutation spectra in the two tissues from control mice were similar. These results suggest that AA possesses mutagenic effects on testes by virtue of its metabolism to GA, possibly targeting spermatogonial stem cells, but possibly via different pathways when compared mutations in liver.
Keywords: acrylamide, glycidamide, Big Blue mice, testes, mutation frequency, mutation spectrum
Acrylamide (AA) is an industrial chemical and has been known as an occupational hazard for decades (Bull et al., 2005; Friedman, 2003). However, it was found in recent years that AA can be formed in fried and baked starchy foods during cooking (Rosen and Hellenas, 2002; Tareke et al., 2002). This latter finding has greatly raised public concerns over AA's potential risk to the health of people upon dietary exposure to the chemical.
AA is a neurotoxic chemical and can cause peripheral and central neuropathy in humans and laboratory animals (LoPachin, 2004). The interference of AA and its metabolite glycidamide (GA) with kinesin motor proteins in neurofilaments causes failure of the transport of nerve signals between axons, and this may be one of the mechanisms involved in its neurotoxicity (Sickles et al., 2007). AA is also a toxicant of the male reproductive system in rodents, but very little evidence is available about its toxic effects on the female reproductive system (Tyl and Friedman, 2003). The toxicities of AA in male animals include degeneration of the epithelial cells of the seminiferous tubules, decreased number of sperm, and abnormal sperm and result in decreased fertility rates and retarded development of pups (Hashimoto and Tanii, 1985; Tyl et al., 2000). These toxic effects may be attributed to the interfering effect of AA on the kinesin motor proteins, which also exist in the flagella of sperm, resulting in the reduction in sperm motility and fertilization events (Tyl and Friedman, 2003; Tyl et al., 2000).
AA has been shown to be genotoxic to sperm. Positive results were reported with AA in the dominant lethal test and heritable translocation assay and AA produced unscheduled DNA synthesis in the germ cells of male mice (Adler et al., 1994; Generoso et al., 1996). Exposure to AA increased the occurrence of micronuclei in sperm cells of mice and rats (Collins et al., 1992; Lahdetie et al., 1994). AA is therefore judged to be a classic clastogen, and its metabolite GA is required for this effect as there were no changes in fertility parameters with AA-treated CYP2E1-null male mice (Ghanayem et al., 2005). Biotransformation of AA to GA is exclusively mediated by CYP2E1 (Sumner et al., 1999). It has been postulated that the clastogenic effects of AA on sperm cells may not be by direct interaction with DNA; instead, these effects may be mediated through interference with the kinesin motor proteins that are involved in spindle fiber formation and chromosomal segregation during cell divisions or alkylation of protamines in sperm (Adler et al., 2000; Sega et al., 1989). However, this conclusion is incompatible with the requirement for metabolism to GA.
AA is a proven rodent carcinogen. In 2-year carcinogenicity studies in rats (Friedman et al., 1995; Johnson et al., 1986), AA induced tumors at multiple organ sites, including thyroid follicular cell tumors (males and females), testicular mesotheliomas (males), and mammary tumors (females). AA also initiated skin tumorigenesis in two different strains of mice, A/J and Swiss-ICR mice, with the greatest initiating effect where AA was administered orally and with significant formation of alveolar bronchiolar adenomas and carcinomas observed even in the absence of a promoting agent (Bull et al., 1984a,b). The carcinogenicity of AA is strongly linked with a genotoxic mode of action, such as DNA adduct–targeted mutagenesis, although non-genotoxic modes such as depletion of cellular glutathione stores and disruption in hormone secretion have been postulated with limited evidence (Besaratinia and Pfeifer, 2007; Dearfield et al., 1988,1995; Rice, 2005).
The genotoxicity of AA has been studied extensively in vitro and in vivo. In our previous study, groups of male and female Big Blue mice were treated with 100 or 500 mg/l of AA or equimolar doses of GA in drinking water for 4 weeks, and the average daily dose calculated from the amount of consumed water varied from 19 to 35 mg/kg and 88 to 111 mg/kg per body weight (bw) for the low and high exposure concentrations of AA and GA, respectively (Manjanatha et al., 2006). We observed that the high exposure concentrations of both AA and GA were mutagenic in the liver cII gene and lymphocyte Hprt gene (Manjanatha et al., 2006). Using Big Blue rats, AA and GA produced significant increases in lymphocyte Hprt mutant frequency (MF) and in bone marrow and thyroid cII MFs (Mei et al., 2010).
Despite the fact that AA by virtue of its metabolism to GA can induce base mutations in somatic cells and cause genetic damage to sperm cells through clastogenic effects, it is still not clear whether AA has direct mutagenic potential, i.e., producing base mutations in sperm DNA. In order to estimate the genetic risk associated with AA and GA exposure, it is necessary to obtain additional experimental data for treatment of spermatogonia as well as female germ cells (Favor and Shelby, 2005). To evaluate AA-related genotoxicity in germ cells, we analyzed the mutation frequency and mutation spectra in the testes of Big Blue mice after treatment with equimolar concentrations of AA and GA and compared the differences between the two chemicals as well as between these germinal cells and previously reported somatic cells.
MATERIALS AND METHODS
Chemicals and reagents.
AA (purity > 99.9%) was purchased from Sigma (St Louis, MO), and GA (purity > 98.5%, with AA present at ∼1%) was purchased from Toronto Research Chemicals (North York, ON). The RecoverEase DNA Isolation Kit, Transpack packaging extract, and the Escherichia coli G1250 strain were obtained from Stratagene (La Jolla, CA). PCR Master Mix was purchased from Promega (Madison, WI), and CEQ Dye Terminator Cycle Sequencing Kits were obtained from Beckman Coulter (Fullerton, CA).
Animals and treatments.
During the course of this experiment, we followed the recommendations set forth by our Institutional Animal Care and Use Committee for the handling, maintenance, treatment, and killing of the animals. Detailed information about animals and treatments has been reported previously (Manjanatha et al., 2006). Briefly, male Big Blue transgenic mice were obtained from Taconic Farms (Germantown, NY) as weanlings and housed two per cage. Mice were fed NIH-31IR diet (Purina Mills, Brentwood, MO), a diet selected because of its low AA content. At 7 weeks of age, the mice were exposed to 0, 1.4, or 7.0mM AA or GA, dissolved in the drinking water (100 or 120 mg/l and 500 or 600 mg/l, respectively), for up to 28 days (Fig. 1). The two concentrations of AA and GA in drinking water were about 2 and 10 times higher than the highest dose used in 2-year carcinogenesis studies of AA and GA in B6C3F1 mice. Chemical analysis indicated that AA and GA were stable in water for at least 1 week, and dosing solutions were prepared and changed weekly. Weekly individual bw gains and water consumption in each cage were monitored throughout the course of the experiment to estimate the amount of the chemicals consumed per mouse per kilogram bw. On the 21st day after the last treatment, the mice were killed, and the testes were isolated, frozen quickly in liquid nitrogen, and stored at −80°C. Because the entire spermatogenic process takes about 6 weeks in mice, proceeding from spermatogonial stem cells to mature sperm in the ejaculate (Ashby et al., 1997; Ghanayem et al., 2005; Shelby and Tindall, 1997; Singer et al., 2006), the total duration of spermatogenesis in mouse testes is about 35 days with additional 7 days in the epididymides (Fig. 1). Therefore, our experiment was conducted over 7 weeks including treatment and sampling time to excise the testes for the mutation assay. If any mutations were recovered from testes, they would have originated in spermatogonial stem cells because those from later stages of maturation at the time of treatment would have exited the testes.
FIG. 1.

Scheme for the treatment schedule and different stages of spermatogenesis in mouse testes. Big Blue mice were treated with AA or GA in drinking water for 4 weeks and killed 3 weeks after last treatment (above the arrow). Sampling time is the time between the last treatment and the time of sampling, which is also referred as manifestation time, fixation time, or expression time. Total duration of spermatogenesis in mouse testes is about 35 days and additional 1 week in the epididymis (below the arrow), which is indicated in retrograde chronological order. After 7 weeks of treatment and sampling time, all mutations recovered from testes would have originated in spermatogonial stem cells because those from later stages of maturation at the time of treatment would have exited the testes.
cII mutant assay.
Testes were decapsulated and the high–molecular weight genomic DNA was extracted using the RecoverEase DNA Isolation Kit. The packaging of the phage, plating the packaged DNA samples, and determination of mutants were carried out following the manufacturer’s instructions for the Select-cII Mutation Detection System for Big Blue Rodents (Stratagene). Briefly, the shuttle vector containing the cII target gene was rescued from total genomic DNA with phage packaging extract, and the resulting phage plated on E. coli host strain G1250. To determine the total titer of packaged phages, G1250 bacteria were mixed with 1:3000 dilutions of phage, plated on TB1 plates, and incubated overnight at 37°C (nonselective conditions). For mutant selection, the packaged phages were mixed with G1250, plated on TB1 plates, and incubated at 24°C for about 42 h (conditions for cII selection). Assays were repeated until a minimum of 35 mutant plaques were obtained from each group. The cII MF is defined as the total number of mutant plaques (determined at 24°C) divided by the total number of plaques screened (determined at 37°C) and expressed as mutants per million plaque-forming units (pfus). After sequencing the mutants (see below) for correcting MF, the cII mutation frequency is defined as the number of independent mutations divided by the total number of plaques screened.
Sequence analysis of cII mutants.
The cII mutant plaques from control and treated mice were isolated and replated at low density to verify the mutant phenotype. Single well-isolated plaques were transferred from these plates to a microcentrifuge tube containing 100 μl of sterile distilled water. The tube was heated at 100°C for 5 min and centrifuged at 12,000 × g for 3 min. cII target DNA released by this procedure was amplified by PCR using primers 5′-AAAAAGGGCATCAAATTAACC-3′ (upstream) and 5′-CCGAAGTTGAGTATTTTTGCTG-3′ (downstream) using procedures as previously reported (Mei et al., 2006). The PCR products were isolated using a QIAQuick PCR Product Purification Kit (Qiagen, Chatsworth, CA). The cII mutant DNA was sequenced with a CEQ Dye Terminator Cycle Sequencing Kit and a Beckman Coulter CEQ 8000 Genetic Analysis System. The primer for cII mutation sequencing was the upstream primer used for the PCR.
Statistical analyses.
Analyses were performed using SigmaStat 3.1 (SPSS, Chicago, IL). Data are expressed as the mean ± SD from six or seven mice per group. Statistical significance was determined by one-way ANOVA followed by the Holm-Sidak test for comparison of multiple treatment groups. Because the variance increased with magnitude of the mutation frequencies, the data were log-transformed before conducting the analysis. Mutation spectra were compared using the computer program written by Cariello et al. (1994) for the Monte Carlo analysis developed by Adams and Skopek (1987).
RESULTS
The Change in the Testes and bw
Previously, we reported that the average daily dose calculated from the amount of consumed water varied from 19 to 25 mg/kg and 88–98 mg/kg bw for the male mice treated with the low and high exposure concentrations of AA and GA, respectively (Manjanatha et al., 2006). The treatment of mice administered with the high exposure concentration of AA was halted after 3 weeks of treatment because of hind-leg paralysis and sluggish movement, probably associated with the neurotoxicity of AA. These mice appeared to become normal after cessation of the treatment and were killed along with the mice in other groups, i.e., 4 weeks after the last AA treatment (Manjanatha et al., 2006). There were no significant differences in bw between the control and any of the treated groups. When compared with the controls, the ratio of testes to bw did not change in the low–exposure concentration groups but significantly decreased in the high–AA and –GA exposure groups by 33 and 53%, respectively (Fig. 2).
FIG. 2.

Ratios of mouse testes to bw in control and AA- or GA-treated Big Blue mice. Each bar represents the mean ± SD from six or seven mice in each group. *Significantly different from the control group (p < 0.05).
cII Mutant Assay
Table 1 shows the results of the cII mutant assays. To obtain a reasonable number of mutants for sequencing, more than 9 million pfus were screened for the control group and more than 3 million pfus for the treatment groups. The MF of control mouse testes was 3.8 ± 1.6 × 10−6, which was much lower than that in the liver (28.4 ± 3.1 × 10−6; Manjanatha et al., 2006). The MFs were significantly increased in all treatment groups (Table 1). After the confirmation of the mutations via DNA sequencing (see below), the number of independent cII mutations was obtained and the cII mutation frequencies (note that corrected for siblings and clonal expansion) in the testes were determined (Table 1). The mutation frequency was 3.3 × 10−6 in the controls, and it was elevated by 170 and 255% in the low- and high-AA exposure concentrations, respectively, and by 179 and 458% in the low– and high–GA treatment groups, respectively. There was no significant difference in the mutation frequencies between the two AA dose groups, probably because of a 3-week treatment period for the high–exposure concentration group compared with a 4-week treatment period for the low–exposure concentration group. On the other hand, the difference in mutation frequencies between the two GA groups was significant (Table 1). In addition, there was no difference in mutation frequencies between low exposure concentration of AA and GA.
TABLE 1.
Testis cII Mutation Frequencies in AA- or GA-Treated Male Big Blue Mice
| Group | Total plaques screened (×103) | Mutant plaquesa | MFb (×10−6) | Independent mutations | Mutation frequencyc (×10−6) |
| Control | 1206 | 7 | 5.8 | 7 | 5.8 |
| 1133 | 3 | 2.6 | 3 | 2.6 | |
| 1245 | 4 | 3.2 | 4 | 3.2 | |
| 1387 | 6 | 4.3 | 5 | 3.6 | |
| 1168 | 2 | 1.7 | 2 | 1.7 | |
| 1357 | 8 | 5.9 | 4 | 2.9 | |
| 1577 | 5 | 3.2 | 5 | 3.2 | |
| Mean ± SD | 3.8 ± 1.6 | 3.3 ± 1.3 | |||
| AA—1.4mM | 659 | 10 | 15.2 | 9 | 13.7 |
| 672 | 46 | 68.5 | 9 | 13.4 | |
| 753 | 8 | 10.6 | 6 | 8.0 | |
| 1042 | 29 | 27.8 | 4 | 3.8 | |
| 571 | 6 | 10.5 | 4 | 7.0 | |
| 786 | 6 | 7.6 | 6 | 7.6 | |
| Mean ± SD | 23.4 ± 25.6 | 8.9 ± 3.9d | |||
| AA—7.0mM | 795 | 15 | 18.9 | 13 | 16.4 |
| 660 | 12 | 18.2 | 7 | 10.6 | |
| 1086 | 13 | 12.0 | 10 | 9.2 | |
| 577 | 11 | 19.1 | 7 | 12.1 | |
| 989 | 14 | 14.2 | 10 | 10.1 | |
| 766 | 11 | 14.4 | 9 | 11.7 | |
| Mean ± SD | 16.1 ± 3.0 | 11.7 ± 2.5d | |||
| GA—1.4mM | 892 | 8 | 9.0 | 6 | 6.7 |
| 683 | 14 | 20.5 | 9 | 13.2 | |
| 660 | 8 | 12.1 | 6 | 9.1 | |
| 735 | 8 | 10.9 | 7 | 9.5 | |
| 908 | 11 | 12.1 | 9 | 9.9 | |
| 1376 | 9 | 6.5 | 9 | 6.5 | |
| Mean ± SD | 11.9 ± 4.7 | 9.2 ± 2.4d | |||
| GA—7.0mM | 405 | 9 | 22.2 | 8 | 19.8 |
| 428 | 20 | 46.7 | 16 | 37.4 | |
| 688 | 12 | 17.4 | 11 | 16.0 | |
| 635 | 15 | 23.6 | 10 | 15.7 | |
| 1238 | 13 | 10.5 | 13 | 10.5 | |
| 1009 | 11 | 10.9 | 11 | 10.9 | |
| Mean ± SD | 21.9 ± 13.3 | 18.4 ± 9.9d |
Sibling mutants or clonal expansions isolated from a single mouse with same type of mutations were included.
The MF is defined as the total number of mutant plaques divided by the total number of plaques screened.
The mutation frequency is defined as the number of independent mutations divided by the total number of plaques screened.
Significantly different from control at p < 0.01 (ANOVA, followed by the Holm-Sidak test).
Sequence Analysis of cII Mutants
All mutants obtained from control and AA/GA-treated testes were analyzed for sequence alterations in the cII gene. At least one base pair substitution, insertion, or deletion was detected in each mutant. Mutations that were found more than once among the mutants isolated from a single mouse were assumed to be siblings (two to three times) resulting from the amplification of a single independent mutation. A clonal expansion was found in two mice treated with 1.4mM AA, with 25 and 37 repeated copies of a point mutation in each animal (Table 1). As a result, we identified 30 independent cII mutations for control mice, 38 and 56 for low and high doses of AA-treated mice, and 46 and 69 for the low and high doses of GA-treated mice (Table 2).
TABLE 2.
Summary of Independent Mutations in the Testis cII Gene of Male Big Blue Mice Treated with AA or GA
| AAa |
GAa |
||||
| Control | 1.4mMb | 7.0mM | 1.4mMb | 7.0mMb | |
| Type of mutation | n (%) | n (%) | n (%) | n (%) | n (%) |
| G:C → A:T | 16 (53.3) | 15 (39.5) | 24 (42.9) | 17 (37.0) | 21 (30.4) |
| G:C → T:A | 6 (20.0) | 4 (10.5) | 8 (14.3) | 3 (6.5) | 8 (11.6) |
| G:C → C:G | 0 (0) | 6 (15.8) | 2 (3.6) | 4 (8.7) | 11 (15.9) |
| A:T → G:C | 0 (0) | 9 (23.7) | 5 (8.9) | 7 (15.2) | 7 (10.1) |
| A:T → C:G | 1 (3.3) | 0 (0) | 4 (7.1) | 4 (8.7) | 7 (10.1) |
| A:T → T:A | 1 (3.3) | 2 (5.3) | 6 (10.7) | 3 (6.5) | 7 (10.1) |
| Frameshift | 6 (20.0) | 2 (5.3) | 7 (12.5) | 8 (17.4) | 8 (11.6) |
| Total mutations | 30 | 38 | 56 | 46 | 69 |
There were no significant differences between low and high exposure concentrations of AA- or GA-treated groups.
The mutation spectra from AA-treated (1.4mM) and GA-treated (1.4 and 7.0mM) groups differed significantly from those of control group (p < 0.05).
Frameshift (insertion and deletion) mutation was 20% or less in control and AA/GA-treated groups, and base pair substitutions comprised the majority of the mutations. Substitution at G:C accounted for 73% of the mutations in the controls, and the most frequent type was G:C → A:T transition (53%). The mutations at A:T were less than 10% in the control group (Table 2). With AA and GA treatments, mutations at A:T were increased to more than 25% in both treatment groups, especially the A:T → G:C transition and A:T → T:A transversion. The mutations at G:C and frameshifts in treated groups were less compared with that in the control group. The overall patterns of mutations in the AA- and GA-treated groups, with the exception of high-exposure AA group (only 3-week treatment), differed significantly (p < 0.05) from the controls (Table 2). In addition, there were no significant differences between the spectra for AA- and GA-treated mice at either the low or high exposure concentration. Figure 3 shows the distribution of the mutations over the cII gene (from −18 to 294). In AA- and GA-treated groups, the mutations were distributed throughout the gene. There was a −1/+1 frameshift mutation probably as a hot spot at a homopolymeric run of dGs at base pairs 179–184 for both the controls and the AA- or GA-treated mice. There was also a hot spot mutation at the base pair 64, and this was only found in AA- and GA-treated mice (with five mutations for each chemical). Some mutations were observed at base pairs 125–177 and 215–294 (harboring a homopolymeric run of dAs at base pairs 241–246) with AA and GA treatment, whereas there were no mutations at these sites in control mice.
FIG. 3.
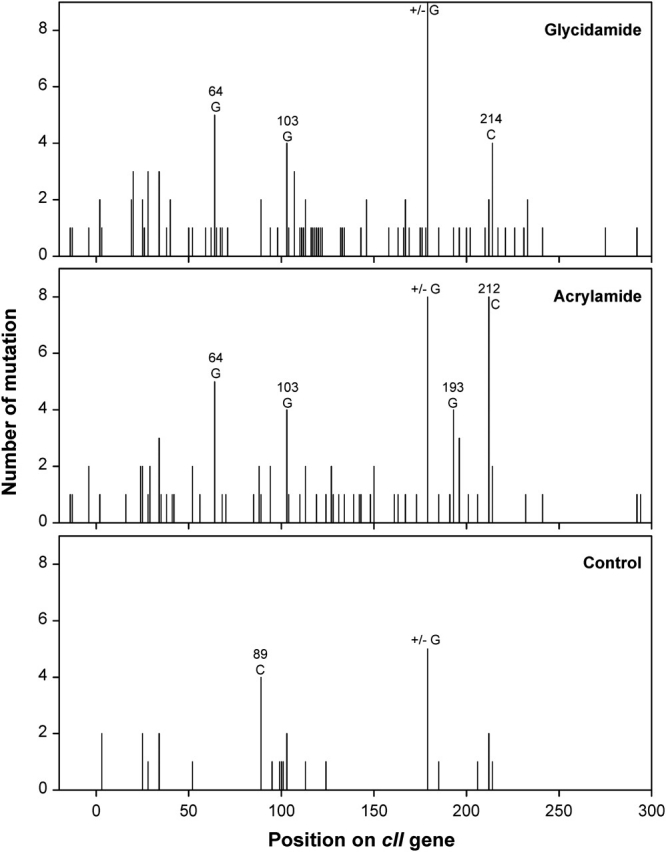
Distribution of mutations in the cII gene of testes from control and AA- or GA-treated mice. The mutations were combined from both low- and high-exposure groups in treated mice. The first 18 bases belong to the promoter region, and the remaining 294 bases represent the protein-coding region.
DISCUSSION
The genotoxicity of AA and its metabolite GA is manifested as both clastogenic and mutagenic events, and AA has proven to be genotoxic to germ cells. Dearfield et al. (1995) reviewed the in vivo and in vitro genotoxicity data and concluded that AA was clastogenic. Favor and Shelby (2005) reviewed seven studies measuring transmitted mutations in the mouse including the heritable translocation test and the specific-locus test and concluded that AA is mutagenic in spermatozoa and spermatid stages of the male germ cell development and is mainly a clastogen in these spermatogenic stages. However, these effects are not necessarily related to mutations at base pairs of sperm DNA, and so far, there was no direct evidence that AA is a gene mutagen in male germ cells, i.e., single base mutations in gene(s) are being monitored.
Male germ cells go through an intricate differentiation process in an orderly progression, and the entire process takes about 42 days in mice to proceed from spermatogonial stem cells to mature sperm (Adler, 1996; Ashby et al., 1997; Ghanayem et al., 2005; Shelby and Tindall, 1997; Singer et al., 2006). Using Big Blue mice, it has been demonstrated that the spermatogonial stem cells are more sensitive to ethylnitrosourea (ENU)-induced mutagenesis than the spermatocytes, spermatids, and spermatozoa, based on ENU-induced MFs using pooled vas deferens sperm or the caudal epididymal sperm from six to nine mice collected at 3, 14, 22, or 93 days after treatment (Katoh et al., 1997). In addition, the MF detected in whole testes was similar to that detected in the vas deferens sperm or caudal epididymal sperm (Katoh et al., 1997). In this study, we collected testes 3 weeks after 4 weeks of treatment (Fig. 1). Our results revealed that the mutation frequencies produced by the AA and GA treatments were higher than controls (Table 1), suggesting that all the induced mutations recovered in the testes originated in spermatogonial stem cells because those from later stages at the time of treatment would have exited the testes (Fig. 1). Therefore, it is possible to evaluate mutagenesis of spermatogonial stem cells in whole testes in situ. The spermatogonial stem cells are mitotically active and DNA repair proficient and persist through the reproductive life of males.
Whole testicular tissue was assayed in this study as that contained a mixed population of germ cells at different stages. With the exposure concentrations and treatment regimen used, both AA and GA significantly increased the mutation frequencies in testes of Big Blue mice (Table 1), and the cII mutations were confirmed by DNA sequencing (Table 2). This mutagenic effect was found at exposure concentrations of AA and GA (1.4mM), where no testicular atrophy was observed (Fig. 2), indicating that the mutagenic effect is independent of the reproductive toxicity of AA or GA. Also, we noticed that the Big Blue mouse may be more sensitive for detecting the mutations in testes caused by AA and GA than the Big Blue rat as there was no increase in the MFs in testes of rats treated with AA or GA at 1.4mM for 60 days and killed after 60-day treatment (Mei et al., 2010). Although the same exposure concentration (1.4mM) was used in both male mouse and rat studies, the average daily received dose of 7.7 mg/kg bw in rats (Mei et al., 2010) was less than half of the average daily dose in this mouse study (∼19 mg/kg bw), and by the treatment regimen in rats, the targeting sperm cells were all stages of germinal cells including spermatogonia, spermatocytes, spermatids, and spermatozoa. Therefore, the received effective doses could account for the sensitivity of mice relative to rats to the mutagenicity of AA. It has been reported that 100 mg/kg bw of AA induced specific-locus mutations in spermatogonial stem cells of mice, i.e., six mutations in offspring with the conception occurring between 43 and 436 days posttreatment (Ehling and Neuhauser-Klaus, 1992).
Because of the neurotoxicity of AA (i.e., hind-leg paralysis and sluggish movement), the group of mice treated with the high dose of AA was halted after 3 weeks of treatment and killed along with the mice in other groups (i.e., 4 weeks after 3-week treatment; Manjanatha et al., 2006). In this study, we observed that there was no significant difference in cII mutation frequencies (Table 1) and mutation spectra (Table 2) between low and high exposure concentrations of AA. However, surprisingly, there was also no significant difference in mutation spectra between high exposure concentration of AA group and control group (Table 2), whereas the mutation spectrum from low exposure concentration of AA group differed significantly from those of control group. The reason could be that the premutational damages can be repaired or eliminated in damaged cells because of the additional week of sampling time (or manifestation time) compared with the low-AA exposure group. After treatment, the part of spermatogonial stem cells treated with AA during 3 weeks were now at the stage of differentiating spermatogonia in spermatogenic cycle, which are DNA repair–proficient germ cell stages.
It has been reported that about 60% of AA is converted to the reactive epoxide GA in mice according to the urinary metabolites (Fennell et al., 2003). Similarly, the DNA adduct levels in liver produced by oral dosing with AA were ∼70% of that produced by dosing with equimolar amount of GA (Doerge et al., 2005b). However, we found that AA and GA have a similar potential for mutation induction in testes (Table 1) when they are administered under the same conditions, i.e., equimolar exposure concentration (1.4mM) and the same length of treatment (4 weeks). Furthermore, both chemicals share similar patterns of mutation that were significantly different from those observed in the control mice (Table 2). Although it is not clear why AA with about 60–70% conversion to GA resulted in similar mutation frequencies when compared with GA treatment in the testes in this study and similar MFs in the livers and lymphocytes in the previous study (Manjanatha et al., 2006), all these results confirm the hypothesis that AA produces the mutagenic effect through metabolic conversion to its epoxide metabolite GA, which can covalently bind to DNA and induce genotoxicity (Besaratinia and Pfeifer, 2007; Gamboa da Costa et al., 2003; Solomon et al., 1985).
In our previous study, we observed that only MFs in the liver of male mice treated with high exposure concentration (7.0mM, equivalent to daily dose of ∼98 mg/kg bw) of AA and GA were significantly increased (Manjanatha et al., 2006). However, using the same animals in this study, the mutagenicity of AA and GA in testicular tissue was shown to occur at both low (1.4mM, equivalent to daily dose of ∼19 mg/kg bw) and high exposure concentrations. The reasons for the discrepancy in the mutagenic effect between the two tissues are not known. It has been reported that AA- and GA-induced DNA adducts were detected in both testes and livers of mice, with DNA adduct levels in testes being about half of those in livers (Doerge et al., 2005a). Both AA and GA are equally distributed systemically and would have access to the germ cell compartments (Doerge et al., 2005b). Mice exposed by gavage with 50 mg/kg bw had a maximal AA serum concentration of 450μM at 0.5 h and had a maximal GA concentration of 190μM at 2 h (Doerge et al., 2005a). On the other hand, it is obvious that the background MF was much lower in testes, being only 13% of that in liver (3.8 × 10−6 in testes vs. 28.4 × 10−6 in livers), and this might account for part of the difference. Furthermore, we statistically analyzed the mutation spectra in the two tissues (Fig. 4). In control mice, the mutation spectra were similar in both testes and liver. However, after treatment with 7.0mM of AA or GA, the mutation patterns were significantly different between the two tissues (p < 0.001). As mentioned above, there was also no significant difference in mutation spectra between AA (7.0mM) and control groups in testes. These results may suggest a different mechanistic route for DNA damage and/or repair in these tissues. In testes, the treatments with AA and GA induced substantial increases in A:T → G:C transition and G:C → C:G transversion, whereas in liver, the predominant types of mutations were G:C → T:A transversions and −1/+1 frameshifts (Manjanatha et al., 2006). Using early-passage embryonic fibroblasts from embryos of Big Blue mice, AA or GA resulted in more A:T → G:C transitions and G:C → C:G transversions than those in control cells and GA also induced more G:C → T:A transversions (Besaratinia and Pfeifer, 2004). Administration to mice with a single ip dose of either AA or an equimolar amount of GA resulted in N7-GA-guanine and N3-GA-adenine DNA adduct formation in all tissues examined including liver and testis (Doerge et al., 2005a). If the proportions of the different DNA adducts formed by AA or GA are not the same in testes and liver, or if these adducts are repaired in a tissue-specific manner, this could account for at least part of the observed difference in the mutation spectra between the two tissues.
FIG. 4.

Comparison of cII mutation spectra between testes and liver of control and AA- or GA-treated (7mM of exposure concentration) mice. The testis data were from Table 2 and the liver data from a previous study (Manjanatha et al. 2006). There were significant differences in the mutation spectra between testes and livers of Big Blue mice treated with AA or GA (p < 0.001), whereas there was no significant difference between two control groups of testes and liver.
Although a recently completed mouse 2-year cancer bioassay indicated that the target tissues for AA carcinogenesis in mice were lung, harderian gland, mammary gland (females), and forestomach, but not testes (F. A. Beland, personal communication), our study is the first comparative study of the mutagenicity of AA and GA as gene mutagens in testes, originated in spermatogonial stem cells of mice. The similar mutagenic effects, including the mutation frequencies and the types of cII mutations, induced by AA and GA treatments indicate that GA is the major genotoxic metabolite of AA. Mutagenic events occurring in spermatogonial stem cells would indicate long-term risk and are of great concern to human health as they may result in genetically determined diseases in subsequent generations.
FUNDING
This research was supported in part by Interagency Agreement 224-07-0007 between National Center for Toxicological Research (NCTR)/U.S. Food and Drug Administration and the National Institute for Environmental Health Sciences/National Toxicology Program. This research was partly supported by appointments (R.-S.W.) to the Research Program at the NCTR administered by the Oak Ridge Institute for Science and Education through an interagency agreement between the U.S. Department of Energy and the U.S. Food and Drug Administration.
Acknowledgments
We thank Drs Frederick A. Beland, Deborah K. Hansen, Robert H. Heflich, and Martha M. Moore for their helpful discussions and comments. The views presented in this paper do not necessarily reflect those of the U.S. Food and Drug Administration.
References
- Adams WT, Skopek TR. Statistical test for the comparison of samples from mutational spectra. J. Mol. Biol. 1987;194:391–396. doi: 10.1016/0022-2836(87)90669-3. [DOI] [PubMed] [Google Scholar]
- Adler ID. Comparison of the duration of spermatogenesis between male rodents and humans. Mutat. Res. 1996;352:169–172. doi: 10.1016/0027-5107(95)00223-5. [DOI] [PubMed] [Google Scholar]
- Adler ID, Baumgartner A, Gonda H, Friedman MA, Skerhut M. 1-Aminobenzotriazole inhibits acrylamide-induced dominant lethal effects in spermatids of male mice. Mutagenesis. 2000;15:133–136. doi: 10.1093/mutage/15.2.133. [DOI] [PubMed] [Google Scholar]
- Adler ID, Reitmeir P, Schmoller R, Schriever-Schwemmer G. Dose response for heritable translocations induced by acrylamide in spermatids of mice. Mutat. Res. 1994;309:285–291. doi: 10.1016/0027-5107(94)90103-1. [DOI] [PubMed] [Google Scholar]
- Ashby J, Gorelick NJ, Shelby MD. Mutation assays in male germ cells from transgenic mice: overview of study and conclusions. Mutat. Res. 1997;388:111–122. doi: 10.1016/s1383-5718(96)00107-6. [DOI] [PubMed] [Google Scholar]
- Besaratinia A, Pfeifer GP. Genotoxicity of acrylamide and glycidamide. J. Natl. Cancer Inst. 2004;96:1023–1029. doi: 10.1093/jnci/djh186. [DOI] [PubMed] [Google Scholar]
- Besaratinia A, Pfeifer GP. A review of mechanisms of acrylamide carcinogenicity. Carcinogenesis. 2007;28:519–528. doi: 10.1093/carcin/bgm006. [DOI] [PubMed] [Google Scholar]
- Bull PJ, Brooke RK, Cocker J, Jones K, Warren N. An occupational hygiene investigation of exposure to acrylamide and the role for urinary S-carboxyethyl-cysteine (CEC) as a biological marker. Ann. Occup. Hyg. 2005;49:683–690. doi: 10.1093/annhyg/mei028. [DOI] [PubMed] [Google Scholar]
- Bull RJ, Robinson M, Laurie RD, Stoner GD, Greisiger E, Meier JR, Stober J. Carcinogenic effects of acrylamide in Sencar and A/J mice. Cancer Res. 1984a;44:107–111. [PubMed] [Google Scholar]
- Bull RJ, Robinson M, Stober JA. Carcinogenic activity of acrylamide in the skin and lung of Swiss-ICR mice. Cancer Lett. 1984b;24:209–212. doi: 10.1016/0304-3835(84)90138-1. [DOI] [PubMed] [Google Scholar]
- Cariello NF, Piegorsch WW, Adams WT, Skopek TR. Computer program for the analysis of mutational spectra: application to p53 mutations. Carcinogenesis. 1994;15:2281–2285. doi: 10.1093/carcin/15.10.2281. [DOI] [PubMed] [Google Scholar]
- Collins BW, Howard DR, Allen JW. Kinetochore-staining of spermatid micronuclei: studies of mice treated with X-radiation or acrylamide. Mutat. Res. 1992;281:287–294. doi: 10.1016/0165-7992(92)90023-b. [DOI] [PubMed] [Google Scholar]
- Dearfield KL, Abernathy CO, Ottley MS, Brantner JH, Hayes PF. Acrylamide: its metabolism, developmental and reproductive effects, genotoxicity, and carcinogenicity. Mutat. Res. 1988;195:45–77. doi: 10.1016/0165-1110(88)90015-2. [DOI] [PubMed] [Google Scholar]
- Dearfield KL, Douglas GR, Ehling UH, Moore MM, Sega GA, Brusick DJ. Acrylamide: a review of its genotoxicity and an assessment of heritable genetic risk. Mutat. Res. 1995;330:71–99. doi: 10.1016/0027-5107(95)00037-j. [DOI] [PubMed] [Google Scholar]
- Doerge DR, da Costa GG, McDaniel LP, Churchwell MI, Twaddle NC, Beland FA. DNA adducts derived from administration of acrylamide and glycidamide to mice and rats. Mutat. Res. 2005a;580:131–141. doi: 10.1016/j.mrgentox.2004.10.013. [DOI] [PubMed] [Google Scholar]
- Doerge DR, Young JF, McDaniel LP, Twaddle NC, Churchwell MI. Toxicokinetics of acrylamide and glycidamide in B6C3F1 mice. Toxicol. Appl. Pharmacol. 2005b;202:258–267. doi: 10.1016/j.taap.2004.07.001. [DOI] [PubMed] [Google Scholar]
- Ehling UH, Neuhauser-Klaus A. Reevaluation of the induction of specific-locus mutations in spermatogonia of the mouse by acrylamide. Mutat. Res. 1992;283:185–191. doi: 10.1016/0165-7992(92)90106-r. [DOI] [PubMed] [Google Scholar]
- Favor J, Shelby MD. Transmitted mutational events induced in mouse germ cells following acrylamide or glycidamide exposure. Mutat. Res. 2005;580:21–30. doi: 10.1016/j.mrgentox.2004.09.010. [DOI] [PubMed] [Google Scholar]
- Fennell TR, Snyder RW, Krol WL, Sumner SC. Comparison of the hemoglobin adducts formed by administration of N-methylolacrylamide and acrylamide to rats. Toxicol. Sci. 2003;71:164–175. doi: 10.1093/toxsci/71.2.164. [DOI] [PubMed] [Google Scholar]
- Friedman M. Chemistry, biochemistry, and safety of acrylamide. A review. J. Agric. Food Chem. 2003;51:4504–4526. doi: 10.1021/jf030204+. [DOI] [PubMed] [Google Scholar]
- Friedman MA, Dulak LH, Stedham MA. A lifetime oncogenicity study in rats with acrylamide. Fundam. Appl. Toxicol. 1995;27:95–105. doi: 10.1093/toxsci/27.1.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamboa da Costa G, Churchwell MI, Hamilton LP, Von Tungeln LS, Beland FA, Marques MM, Doerge DR. DNA adduct formation from acrylamide via conversion to glycidamide in adult and neonatal mice. Chem. Res. Toxicol. 2003;16:1328–1337. doi: 10.1021/tx034108e. [DOI] [PubMed] [Google Scholar]
- Generoso WM, Sega GA, Lockhart AM, Hughes LA, Cain KT, Cacheiro NL, Shelby MD. Dominant lethal mutations, heritable translocations, and unscheduled DNA synthesis induced in male mouse germ cells by glycidamide, a metabolite of acrylamide. Mutat. Res. 1996;371:175–183. doi: 10.1016/s0165-1218(96)90106-8. [DOI] [PubMed] [Google Scholar]
- Ghanayem BI, Witt KL, El-Hadri L, Hoffler U, Kissling GE, Shelby MD, Bishop JB. Comparison of germ cell mutagenicity in male CYP2E1-null and wild-type mice treated with acrylamide: Evidence supporting a glycidamide-mediated effect. Biol. Reprod. 2005;72:157–163. doi: 10.1095/biolreprod.104.033308. [DOI] [PubMed] [Google Scholar]
- Hashimoto K, Tanii H. Mutagenicity of acrylamide and its analogues in Salmonella typhimurium. Mutat. Res. 1985;158:129–133. doi: 10.1016/0165-1218(85)90075-8. [DOI] [PubMed] [Google Scholar]
- Johnson KA, Gorzinski SJ, Bodner KM, Campbell RA, Wolf CH, Friedman MA, Mast RW. Chronic toxicity and oncogenicity study on acrylamide incorporated in the drinking water of Fischer 344 rats. Toxicol. Appl. Pharmacol. 1986;85:154–168. doi: 10.1016/0041-008x(86)90109-2. [DOI] [PubMed] [Google Scholar]
- Katoh M, Horiya N, Valdivia RP. Mutations induced in male germ cells after treatment of transgenic mice with ethylnitrosourea. Mutat. Res. 1997;388:229–237. doi: 10.1016/s1383-5718(96)00121-0. [DOI] [PubMed] [Google Scholar]
- Lahdetie J, Suutari A, Sjoblom T. The spermatid micronucleus test with the dissection technique detects the germ cell mutagenicity of acrylamide in rat meiotic cells. Mutat. Res. 1994;309:255–262. doi: 10.1016/0027-5107(94)90100-7. [DOI] [PubMed] [Google Scholar]
- LoPachin RM. The changing view of acrylamide neurotoxicity. Neurotoxicology. 2004;25:617–630. doi: 10.1016/j.neuro.2004.01.004. [DOI] [PubMed] [Google Scholar]
- Manjanatha MG, Aidoo A, Shelton SD, Bishop ME, McDaniel LP, Lyn-Cook LE, Doerge DR. Genotoxicity of acrylamide and its metabolite glycidamide administered in drinking water to male and female Big Blue mice. Environ. Mol. Mutagen. 2006;47:6–17. doi: 10.1002/em.20157. [DOI] [PubMed] [Google Scholar]
- Mei N, Arlt VM, Phillips DH, Heflich RH, Chen T. DNA adduct formation and mutation induction by aristolochic acid in rat kidney and liver. Mutat. Res. 2006;602:83–91. doi: 10.1016/j.mrfmmm.2006.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mei N, McDaniel LP, Dobrovolsky VN, Guo X, Shaddock JG, Mittelstaedt RA, Azuma M, Shelton SD, McGarrity LJ, Doerge DR, et al. The genotoxicity of acrylamide and glycidamide in Big Blue rats. Toxicol. Sci. 2010;115:412–421. doi: 10.1093/toxsci/kfq069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice JM. The carcinogenicity of acrylamide. Mutat. Res. 2005;580:3–20. doi: 10.1016/j.mrgentox.2004.09.008. [DOI] [PubMed] [Google Scholar]
- Rosen J, Hellenas KE. Analysis of acrylamide in cooked foods by liquid chromatography tandem mass spectrometry. Analyst. 2002;127:880–882. doi: 10.1039/b204938d. [DOI] [PubMed] [Google Scholar]
- Sega GA, Alcota RP, Tancongco CP, Brimer PA. Acrylamide binding to the DNA and protamine of spermiogenic stages in the mouse and its relationship to genetic damage. Mutat. Res. 1989;216:221–230. doi: 10.1016/0165-1161(89)90008-3. [DOI] [PubMed] [Google Scholar]
- Shelby MD, Tindall KR. Mammalian germ cell mutagenicity of ENU, IPMS and MMS, chemicals selected for a transgenic mouse collaborative study. Mutat. Res. 1997;388:99–109. doi: 10.1016/s1383-5718(96)00106-4. [DOI] [PubMed] [Google Scholar]
- Sickles DW, Sperry AO, Testino A, Friedman M. Acrylamide effects on kinesin-related proteins of the mitotic/meiotic spindle. Toxicol. Appl. Pharmacol. 2007;222:111–121. doi: 10.1016/j.taap.2007.04.006. [DOI] [PubMed] [Google Scholar]
- Singer TM, Lambert IB, Williams A, Douglas GR, Yauk CL. Detection of induced male germline mutation: correlations and comparisons between traditional germline mutation assays, transgenic rodent assays and expanded simple tandem repeat instability assays. Mutat. Res. 2006;598:164–193. doi: 10.1016/j.mrfmmm.2006.01.017. [DOI] [PubMed] [Google Scholar]
- Solomon JJ, Fedyk J, Mukai F, Segal A. Direct alkylation of 2′-deoxynucleosides and DNA following in vitro reaction with acrylamide. Cancer Res. 1985;45:3465–3470. [PubMed] [Google Scholar]
- Sumner SC, Fennell TR, Moore TA, Chanas B, Gonzalez F, Ghanayem BI. Role of cytochrome P450 2E1 in the metabolism of acrylamide and acrylonitrile in mice. Chem. Res. Toxicol. 1999;12:1110–1116. doi: 10.1021/tx990040k. [DOI] [PubMed] [Google Scholar]
- Tareke E, Rydberg P, Karlsson P, Eriksson S, Tornqvist M. Analysis of acrylamide, a carcinogen formed in heated foodstuffs. J. Agric. Food Chem. 2002;50:4998–5006. doi: 10.1021/jf020302f. [DOI] [PubMed] [Google Scholar]
- Tyl RW, Friedman MA. Effects of acrylamide on rodent reproductive performance. Reprod. Toxicol. 2003;17:1–13. doi: 10.1016/s0890-6238(02)00078-3. [DOI] [PubMed] [Google Scholar]
- Tyl RW, Marr MC, Myers CB, Ross WP, Friedman MA. Relationship between acrylamide reproductive and neurotoxicity in male rats. Reprod. Toxicol. 2000;14:147–157. doi: 10.1016/s0890-6238(00)00066-6. [DOI] [PubMed] [Google Scholar]