

Abstract
We present the case of a man with Gram-negative sepsis and exposure to oral silica who developed pauci-immune focal necrotizing glomerulonephritis (PI-FNGN) in the setting of a subacute polymicrobial central venous line (CVL) infection. He developed a cytoplasmic antineutrophil cytoplasmic autoantibody (C-ANCA) that was antiproteinase-3 (PR-3) and antimyeloperoxidase (MPO) antibody negative. We believe this is the first reported case of Gram-negative sepsis-associated PI-FNGN. Chronic silica exposure is a leading environmental risk factor in the development of ANCA vasculitis. Oral silica is a common pharmaceutical additive and its bioavailability is being recognized. Oral silica, therefore, may also be a risk for development of autoreactivity. The PI-FNGN resolved with antibiotic therapy alone. The C-ANCA titer declined as the PI-FNGN resolved. The case supports experimental and observational research that environmental exposures act as adjuvants for an immune response and also provide epigenetic triggers for autoreactivity. The C-ANCA was negative for PR-3, its major antigen. C-ANCA antigen specificity may depend on the pathogenesis of the underlying disease, potentially elicited by a cross-reaction of an antibody to foreign and self target antigen sequence homology or alternatively elicited by antigenic epitope spread.
Keywords: gram-negative infection, kidney failure, oral silica, PR3- and MPO-negative pauci-immune focal necrotizing glomerulonephritis, risk factors
Introduction
The evaluation and management of a patient with clinical features of a systemic small vessel vasculitis (SVV) and rapidly progressing kidney failure resulting from acute pauci-immune focal necrotizing glomerulonephritis (PI-FNGN) is a challenge, particularly when findings are atypical and the pathophysiology is not well defined. PI-FNGN commonly results in rapid, irreversible kidney failure. Care decisions are a tenuous balance between benefit and harm. We present the case of a man with rapidly progressive glomerulonephritis (GN) and features of systemic vasculitis. The ultimate diagnosis was established by integrating clinical and renal pathologic findings.
Case report
The patient was a 41-year-old man who had been successfully treated for lumbar osteomyelitis 9 months before admission. He had had lumbar stabilization hardware placed to ease chronic pain that began after a motor vehicle accident. The osteomyelitis occurred perioperatively. He used cocaine but no other illicit drugs. A subclavian port-a-cath that had been placed for the prolonged antibiotic administration after hardware removal was left in place. He had normal kidney function and no history of hypertension, haematuria, proteinuria or inflammatory arthritis; there was no family history of autoimmune disease.
The patient arrived at the hospital with a 2-month history of fever, chills, sweats and a 3-week history of haematuria. He complained of severe generalized arthralgias and myalgias. He had had progressive oedema of his lower extremities, dyspnoea, anorexia, nausea and vomiting. Over the 2 months before admission he used over-the-counter (OTC) pain relievers and anti-inflammatory medications, commonly Stanbacks® (aspirin 650 mg, caffeine 32 mg, salicylamide 200 mg, colloidal silicon dioxide, docusate sodium, potassium chloride), several times daily and had self-treated with intermittent oral antibiotics. Pertinent physical exam findings included a temperature of 98°F, blood pressure of 190/100, oxygen saturation of 96% on ambient air and anasarca. The right-sided port-a-cath showed no erythema or drainage. There was a cardiac friction rub. Examination of the skin revealed multiple scattered petechiae and follicular pustules over a bilateral lower extremities, 1–5-cm-diameter subcutaneous tender nodules involving his lower face and torso and coalesced, suppurating ulcerations in both axillae. Knees, ankles and wrist joints exhibited bilateral swelling and tenderness.
Admission laboratory studies showed the following values: blood urea nitrogen (BUN) 137 mg/dL; creatinine (Cr) 10.7 mg/dL; white cell count (WBC), 31,670/μL, with 97% neutrophils. Urinalysis revealed gross haematuria with 3+ protein, >25 dysmorphic red blood cells (RBC)/high-power field (HPF), leukocyte esterase 3+, >50 WBC/HPF, a few bacteria; many dysmorphic RBC casts and several WBC casts. A 24-h urine Cr clearance (CrCl) was 7 mL/min; total protein excretion was 4.8 grams. Chest X-ray showed interstitial oedema and a small right pleural effusion. The noncontrast chest computed tomographic scan showed a large pericardial effusion, pretracheal adenopathy, small bilateral pleural effusions, peripheral ground glass opacities in the right upper lobe and a 2-cm pulmonary nodule in the right lower lobe. Renal ultrasound showed mildly echogenic kidneys; the right kidney was 13.9 cm, the left 12.9 cm. There were no masses, cysts or hydronephrosis.
Cultures resulted in the following: blood (three sets of three), Enterococcus, Stenotrophomonas maltophilia, Klebsiella; urine culture, Klebsiella; porta-cath tip, Stenotrophomonas maltophilia, Klebsiella; left axillary skin ulcer, methicillin-resistant Staphylococcus aureus (MRSA). Biopsy of the skin and subcutaneous nodules were negative for vasculitis; it showed metastatic calcinosis cutis with transepidermal elimination. Hepatitis C virus (HCV) antibody was present. Complement (C) studies revealed C3 of 57 mg/dL (normal range 79–152), C4 of 11.4 mg/dL (normal range 16–38) and CH50 of 73 units/mL (normal range ≥110). Serum cryoglobulins were negative, as were serologic tests for hepatitis B, human immunodeficiency virus type 1, antinuclear antibodies and anti-glomerular basement membrane antibodies. The rheumatoid factor (RF) was 260 IU/mL. Serology for ANCA revealed cytoplasmic antineutrophil cytoplasmic autoantibody (C-ANCA) 1:160 and perinuclear-ANCA (P-ANCA) <1:20. No antiproteinase-3 (PR-3) antibody or antimyeloperoxidase (MPO) activity was detected. An echocardiogram showed a pericardial effusion, but no vegetations or tamponade.
A percutaneous kidney biopsy was performed on hospital day 15.
Kidney biopsy
Light microscopy sections contained 19 glomeruli, 4 of which were globally sclerotic, appearing to be compressed by fibrous crescents. Cellular and fibrocellular crescents involved 37% of the glomeruli (Figure 1A and B). Four glomeruli had cellular crescents with necrotizing lesions, one glomerulus without a crescent had a segmental necrotizing lesion and three additional glomeruli contained fibrocellular crescents. The remaining glomeruli showed segmental mild mesangial expansion without hypercellularity. Tubulointerstitial oedema and interstitial inflammation with prominent tubulitis were noted. Interstitial fibrosis was estimated at 20–30%. Blood vessels were unremarkable.
Fig. 1.
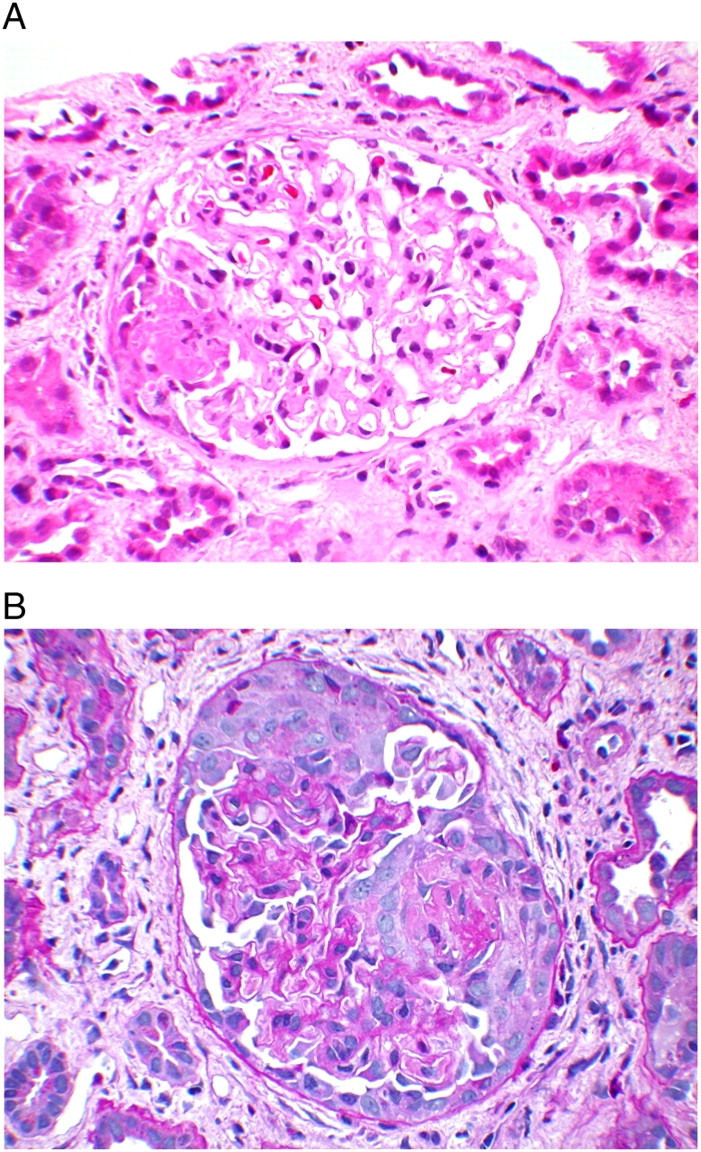
(A) Glomerulus with small segmental necrotizing lesion. Note the absence of mesangial or endocapillary proliferation in the rest of the glomerulus. (H&E; original magnification ×200). 173 × 130 mm (300 × 300 DPI) (B) Glomerulus with large cellular crescent. (Periodic Acid Schiff-Hematoxylin stain; original magnification ×200). 173 × 130 mm (300 × 300 Dots per inch)
Immunofluorescence examination revealed diffuse mesangial staining for IgM (2+), C3 (2+) and C1q (1+) on a 0 to 4 scale. There was no staining for IgG, IgA, albumin or light chains. There was no staining in the vessels or the tubular basement membranes.
Two glomeruli were examined by electron microscopy. No electron-dense deposits were identified in the mesangium or along the capillary loops. The foot processes showed extensive effacement.
The final diagnosis was PI-FNGN.
Clinical course
After 4 weeks of antibiotic therapy the patient was nonoliguric and symptomatically improving. However, he had persistent gross haematuria and remained dialysis dependent. After discharge he was lost to follow-up until readmission 1 month later with cocaine-induced stroke, fever and gross haematuria. He had missed dialysis for 2 weeks. Admission findings showed the following values: BUN, 54 mg/dL; Cr, 3.5 mg/dL; C-ANCA, 1:40; P-ANCA, <1:20; normal C; RF, 119 IU/mL; a 24-h urine collection showed a CrCl of 25 mL/min. Dialysis was discontinued, and antibiotics were restarted. Laboratory studies by the end of hospitalization showed a 24-h CrCl of 45 mL/min; BUN, 22 mg/dL; Cr, 1.8 mg/dL.
Discussion
This is an unusual case of a patient who presented with C-ANCA-positive but PR-3 and MPO antibody-negative PI-FNGN in the setting of subacute Gram-negative sepsis, polymicrobial bacteria central venous line (CVL) infection and suppurative MRSA skin lesions. He had been using oral OTC pain medications containing colloidal silicon dioxide and aspirin several times daily for 2 months prior to admission. We believe this is the first reported case of Gram-negative sepsis-associated PI-FNGN that resolved with antibiotic therapy alone. The C-ANCA titer declined as the PI-FNGN resolved. PI-FNGN has been reported in subacute endocarditis but not with a subacute gram-negative CVL-associated sepsis.
The two diagnostic considerations for the severe renal inflammation, both having similar clinical presentations, were PI-FNGN and an immune complex-mediated GN. This distinction is crucial since PI-FNGN often results in irreversible kidney failure; infection-associated GN, in contrast, is frequently reversible. The patient had a fimbriated Gram-negative CVL-associated sepsis and HCV. There was no residual osteomyelitis by clinical and radiographic exam. An immune complex-mediated kidney injury can be caused by a bacterial CVL infection and an HCV-associated mixed cryoglobulinaemia. However, the negative immunofluorescence and electron microscopic findings ruled out an immune complex-mediated injury.
The pathogenesis of PI SVV is unclear. Associations of environmental exposures, both infectious and toxins with PI-FNGN, have been suggested since its description in the 1930s. Reports of the association of PI-FNGN with Gram-negative bacterial endocarditis and discitis [1–4] and reversible kidney injury not only led to the hypothesis that environmental factors can trigger a PI-FNGN in genetically susceptible individuals [5] but also led to new hypotheses for the triggering of primary PI-FNGN and to uncertainty regarding appropriate management of secondary PI-FNGN. Efforts in the field of epigenetics have revitalized the study of how foreign antigens modify DNA and histones, thereby altering gene expression and leading to autoimmunity [6,7]. The detection of ANCA in many cases of PI-FNGN established it as an autoimmune disease [8,9] and began a new focus for clinical and basic science research [10–12]. Clinical reports describe variable presentations of ANCA-associated disease and variable ANCA specificities, including Gram-negative bacterial subacute endocarditis, PR-3-positive and PR-3-negative C-ANCA; Gram-negative discitis, ANCA negative; HCV infection [13], MPO-positive P-ANCA, a single case of Gram-negative bacterial endocarditis, and PR-3-negative C-ANCA [14] with PI-FNGN. The presentations were subacute, many cases were not PR-3 and MPO antibody specific and ANCA titers became negative with antibiotics with and without immunosuppressive therapy [14,15]. Clinical and observational studies have associated respiratory silica exposure with the development of SVV since the 1960s and with ANCA-associated SVV since the 1990s [5,16,17]. In addition to Gram-negative sepsis, this patient had chronic exposure to colloidal silicon dioxide via daily ingestion of an OTC pain medication. Oral silica is a common pharmaceutical additive and its bioavailability is being recognized [18]. Oral silica, therefore, may also be a risk for the development of autoreactivity.
Hypotheses of pathogenetic mechanisms that lead to vasculitis include activated neutrophils, bacterial superantigen-induced autoantibody induction, and microbial antigens and host proteins cross-reaction-triggered autoimmunity. Whether autoantibodies have a role in the development of vasculitis is not known [19–22]. PR-3 and MPO antigens have been best studied because of their close association with Wegener’s granulomatosis and microscopic polyangiitis, respectively, but a pathogenetic role has not yet been clearly defined. Other components of the neutrophil and monocyte granules have been shown to be ANCA targets in patients with PI-FNGN and have been termed minor ANCA antigens. Minor antigens that can give a false C-ANCA test include enolase, lysozyme, possibly bactericidal/permeability increasing protein (BPI) and rarely MPO. These and other atypical ANCA antigen specificities, such as human lysosomal-associated membrane protein 2 (h-LAMP-2) ANCA, are not being tested for in the clinical arena. How they might help to improve our understanding of the pathogenesis of SVV in infection and autoimmunity in PI-FNGN is an ongoing topic of discussion between clinicians and biologists. But as yet, ANCA tests specific for minor antigens have not entered the clinical arena and are not done in clinical laboratories.
The pathogenesis of antigen-specific ANCA is being investigated. Bacterial infections have long been postulated in the provocation of autoantibody synthesis. Even in his original report, Wegener suggested that bacteria may be involved. This idea was reiterated by others but the expected sequence homologies that could provide evidence for a cross-reactive immune response between bacterial proteins and the antigen targets were not found for Wegener’s granulomatosis or microscopic polyangiitis. An alternate ANCA to h-LAMP-2 has been detected in many patients with PI-FNGN. H-LAMP-2 is a type 1 heavily glycosylated membrane protein expressed by neutrophils and endothelial cells as well as by many other cell types. It is involved in cellular adhesion and homeostasis. It has sequence homology with a bacterial adhesin, FimH, on fimbriated Gram-negative bacteria. In vitro, the h-LAMP-2 autoantibodies injure human microvascular endothelium and when injected into rats, induce PI-FNGN. FimH, when injected into rats, induced antibodies to h-LAMP-2 and a PI-FNGN [23,24]. This alternate antigen target is found in 90% of patients with PI-FNGN and is being studied for its potential role in the mechanism of autoimmunity induction, potentially via a molecular mimicry mechanism where the monoclonal antibodies recognize both microbial and self antigens. Sequence similarities between self and foreign proteins may result in antibodies to the foreign antigen to cross-react with a self protein. The ANCA in this case report is specific for an unidentified alternate antigen target in the setting of a Gram-negative, polymicrobial CVL infection. Multiple antigen specificities may occur as a result of epitope spread. The decline of the autoantibody with antibiotic therapy is consistent with a theory of ANCA induction by a subacute infection and a potential role for ANCA in pathogenesis of PI-FNGN.
To our knowledge, this is the first reported case of a PI-FNGN affiliated with a Gram-negative sepsis in the setting of subacute polymicrobial CVL infection and oral silica exposure. The patient had a C-ANCA that was PR-3 and MPO negative. Alternate antigen specificity detection assays remain in the research domain and were not done. This report illustrates the association of an infection with a Gram-negative fimbriated bacteria and a PR-3-negative C-ANCA PI-FNGN in an environment where Gram-negative bacteria have become increasingly common causes of CVL infection. Furthermore, it demonstrates that PI-FNGN may resolve with antibiotic therapy alone since the C-ANCA titer declined and the kidney function improved without immunosuppressive therapy. The resolution of haematuria, like that seen in infection-induced immune complex GN, was delayed. We conclude that PR-3-negative ANCA in the setting of infection may have pathogenetic significance and as such a potential future research. Adjuvants for induction of autoimmunity are not well understood or characterized. The mechanism whereby silica and infection induce autoimmunity needs further research. H-LAMP-2 antibody may have clinical utility in cases of atypical ANCA specificity and may shed light on how infection links with autoimmunity.
Acknowledgments
S.S.C. was supported by the National Institutes of Health grant (5R01DK077073).
Conflict of interest statement. None declared.
References
- 1.Chirinos JA, Corrales-Medina VF, Garcia S, et al. Endocarditis associated with antineutrophil cytoplasmic antibodies: a case report and review of the literature. Clin Rheumatol. 2007;26:590–595. doi: 10.1007/s10067-005-0176-z. [DOI] [PubMed] [Google Scholar]
- 2.Kishimoto N, Mori Y, Yamahara H, et al. Cytoplasmic antineutrophil cytoplasmic antibody positive pauci-immune glomerulonephritis associated with infectious endocarditis. Clin Nephrol. 2006;66:447–454. doi: 10.5414/cnp66447. [DOI] [PubMed] [Google Scholar]
- 3.Fukuda M, Motokawa M, Usami T, et al. PR3-ANCA-positive crescentic necrotizing glomerulonephritis accompanied by isolated pulmonic valve infective endocarditis, with reference to previous reports of renal pathology. Clin Nephrol. 2006;66:202–209. doi: 10.5414/cnp66202. [DOI] [PubMed] [Google Scholar]
- 4.Peel R, Sellars L, Long ED, et al. A man with backache and renal failure. Am J Kidney Dis. 2003;41:E1. doi: 10.1053/ajkd.2003.50019. [DOI] [PubMed] [Google Scholar]
- 5.Tervaert JW, Stegeman CA, Kallenberg CG. Silicon exposure and vasculitis. Curr Opin Rheumatol. 1998;10:12–17. doi: 10.1097/00002281-199801000-00003. [DOI] [PubMed] [Google Scholar]
- 6.Richardson B. Primer: epigenetics of autoimmunity. Nat Clin Pract Rheumatol. 2007;3:521–527. doi: 10.1038/ncprheum0573. [DOI] [PubMed] [Google Scholar]
- 7.Pisetsky DS. The role of innate immunity in the induction of autoimmunity. Autoimmun Rev. 2008;8:69–72. doi: 10.1016/j.autrev.2008.07.028. [DOI] [PubMed] [Google Scholar]
- 8.Davies DJ, Moran JE, Niall JF, et al. Segmental necrotising glomerulonephritis with antineutrophil antibody: possible arbovirus aetiology? Br Med J (Clin Res Ed) 1982;285:606. doi: 10.1136/bmj.285.6342.606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.van der Woude FJ, Rasmussen N, Lobatto S, et al. Autoantibodies against neutrophils and monocytes: tool for diagnosis and marker of disease activity in Wegener’s granulomatosis. Lancet. 1985;1:425–429. doi: 10.1016/s0140-6736(85)91147-x. [DOI] [PubMed] [Google Scholar]
- 10.Hagen EC, Ballieux BE, van Es LA, et al. Antineutrophil cytoplasmic autoantibodies: a review of the antigens involved, the assays, and the clinical and possible pathogenetic consequences. Blood. 1993;81:1996–2002. [PubMed] [Google Scholar]
- 11.Kallenberg CG, Brouwer E, Weening JJ, et al. Anti-neutrophil cytoplasmic antibodies: current diagnostic and pathophysiological potential. Kidney Int. 1994;46:1–15. doi: 10.1038/ki.1994.239. [DOI] [PubMed] [Google Scholar]
- 12.Hagen EC, Daha MR, Hermans J, et al. Diagnostic value of standardized assays for anti-neutrophil cytoplasmic antibodies in idiopathic systemic vasculitis. EC/BCR Project for ANCA Assay Standardization. Kidney Int. 1998;53:743–753. doi: 10.1046/j.1523-1755.1998.00807.x. [DOI] [PubMed] [Google Scholar]
- 13.Igaki N, Nakaji M, Moriguchi R, et al. A case of hepatitis C virus-associated glomerulonephropathy presenting with MPO-ANCA-positive rapidly progressive glomerulonephritis. Nippon Jinzo Gakkai Shi. 2000;42:353–358. [PubMed] [Google Scholar]
- 14.Peel R, Sellars L, Long ED, et al. A man with backache and renal failure. Am J Kidney Dis. 2003;41:E1. doi: 10.1053/ajkd.2003.50019. [DOI] [PubMed] [Google Scholar]
- 15.Choi HK, Lamprecht P, Niles JL, et al. Subacute bacterial endocarditis with positive cytoplasmic antineutrophil cytoplasmic antibodies and anti-proteinase 3 antibodies. Arthritis Rheum. 2000;43:226–231. doi: 10.1002/1529-0131(200001)43:1<226::AID-ANR27>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 16.Hogan SL, Cooper GS, Savitz DA, et al. Association of silica exposure with anti-neutrophil cytoplasmic autoantibody small-vessel vasculitis: a population-based, case-control study. Clin J Am Soc Nephrol. 2007;2:290–299. doi: 10.2215/CJN.03501006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hogan SL, Satterly KK, Dooley MA, et al. Silica exposure in anti-neutrophil cytoplasmic autoantibody-associated glomerulonephritis and lupus nephritis. J Am Soc Nephrol. 2001;12:134–142. doi: 10.1681/ASN.V121134. [DOI] [PubMed] [Google Scholar]
- 18.Flythe JE, Rueda JF, Riscoe MK, et al. Silicate nephrolithiasis after ingestion of supplements containing silica dioxide. Am J Kidney Dis. 2009;54:127–130. doi: 10.1053/j.ajkd.2008.10.042. [DOI] [PubMed] [Google Scholar]
- 19.Jennette JC, Wilkman AS, Falk RJ. Diagnostic predictive value of ANCA serology. Kidney Int. 1998;53:796–798. doi: 10.1038/ki.1998.36. [DOI] [PubMed] [Google Scholar]
- 20.Rarok AA, Limburg PC, Kallenberg CG. Neutrophil-activating potential of antineutrophil cytoplasm autoantibodies. J Leukoc Biol. 2003;74:3–15. doi: 10.1189/jlb.1202611. [DOI] [PubMed] [Google Scholar]
- 21.Jennette JC, Xiao H, Falk RJ. Pathogenesis of vascular inflammation by anti-neutrophil cytoplasmic antibodies. J Am Soc Nephrol. 2006;17:1235–1242. doi: 10.1681/ASN.2005101048. [DOI] [PubMed] [Google Scholar]
- 22.Beauvillain C, Delneste Y, Renier G, et al. Antineutrophil cytoplasmic autoantibodies: how should the biologist manage them? Clin Rev Allergy Immunol. 2008;35:47–58. doi: 10.1007/s12016-007-8071-9. [DOI] [PubMed] [Google Scholar]
- 23.Kain R, Matsui K, Exner M, et al. A novel class of autoantigens of anti-neutrophil cytoplasmic antibodies in necrotizing and crescentic glomerulonephritis: the lysosomal membrane glycoprotein h-lamp-2 in neutrophil granulocytes and a related membrane protein in glomerular endothelial cells. J Exp Med. 1995;181:585–597. doi: 10.1084/jem.181.2.585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kain R, Exner M, Brandes R, et al. Molecular mimicry in pauci-immune focal necrotizing glomerulonephritis. Nat Med. 2008;14:1088–1096. doi: 10.1038/nm.1874. [DOI] [PMC free article] [PubMed] [Google Scholar]