

Abstract
The P2Y12 receptor, a Gi protein-coupled receptor, plays a central role in platelet activation. In this study, we did a mutational analysis of residues possibly involved in the ligand interactions with the human P2Y12 receptor. Mutant receptors were stably expressed in CHO-K1 cells with an HA-tag at the N-terminus. Expression of wild-type and mutant receptors was confirmed by detecting the HA-tag on the cell membrane. Residues in transmembrane helical domains (TMs) 3, 5, 6, and 7, which are homologous to residues important for P2Y1 receptor activation and ligand recognition, were replaced by site-directed mutagenesis. ADP-induced inhibition of forskolin-stimulated cAMP levels in the presence or absence of antagonist AR-C69931MX were investigated for each of the mutant receptors. F104S and S288P significantly increased agonist-induced receptor function without affecting the antagonism by AR-C69931MX. Arg256 in TM6 and Arg 265 in extracellular loop 3 (EL3) are more important for antagonist recognition than effect on agonist-mediated receptor function. Compared to wild-type P2Y12 receptor, mutations in Arg 256 or/and Arg 265 significantly increased the sensitivity to antagonist AR-C69931MX. Our study shows that the cytosolic side of TM3 and the exofacial side of TM5 are critical for P2Y12 receptor function, which is different from P2Y1. Arg 256 in TM6 and Arg265 in EL3 appear to play a role in antagonist recognition rather than effects on agonist-induced receptor function.
Keywords: P2Y12, cyclic AMP, site-directed mutation
1. Introduction
Platelets are a fundamental component of the normal hemostatic process and abnormal platelet activation can cause thrombus formation. Upon activation, platelets change shape, aggregate and secrete granules (Jurk and Kehrel, 2005). Adenosine diphosphate (ADP), which is secreted from platelet dense granule, acts as one of the most important players to amplify the primary responses of platelets and form a stable thrombus together with generated thrombin (De Clerck and Janssen, 1990; Offermanns, 2006; Packham et al., 1987).
In platelets, ADP is an important agonist that activates platelets through Gq-coupled P2Y1 and Gi-coupled P2Y12 receptors (Daniel et al., 1998; Jin et al., 1998). Co-stimulation of P2Y1 and P2Y12 is required for ADP-induced platelet aggregation and thromboxane generation (Jin and Kunapuli, 1998). P2Y12 receptors are able to enhance other agonist-induced dense granule release (Dangelmaier et al., 2001; Storey et al., 2000). The P2Y12 receptor does not contribute to platelet shape change. However, downstream signaling events of P2Y12 receptor are essential for platelet full aggregation and thromboxane generation induced by other agonists (Kim et al., 2004, 2006; Shankar et al., 2006; Trumel et al., 1999). In addition, patients with defective P2Y12 receptor suffer from an abnormal ADP-induced adenylyl cyclase inhibition and platelet aggregation but retain a normal platelet shape change response (Cattaneo et al., 2003). Because of the critical role of P2Y12 in platelet activation, the thienopyridine compounds, such as clopidogrel, which target platelet P2Y12 receptor, were generated and widely used as antithrombotic drugs and have shown better benefits than aspirin in the prevention and treatment of thrombotic events (Yoneda et al., 2004).
P2Y12 is one of eight distinct functional P2Y receptors that are expressed in human tissues (Abbracchio et al., 2006). Among these P2Y receptors, P2Y1 and P2Y2 have been studied using mutagenesis and results showed that positively charged residues near the exofacial side of TM3, TM7 and TM6 of P2Y1 receptor were important for recognition of agonist and positively charged residues of TM6 and TM7 were important for agonist binding to P2Y2 receptor (Erb et al., 1995; Jiang et al., 1997). In addition, charged amino acids in EL2 (Glu209) and EL3 (Arg287) are also important for P2Y1 receptor activation (Hoffmann et al., 1999). P2Y12 and P2Y1 receptors have identical agonists: ADP and 2-methylthio-ADP (2-MeSADP), but they only have about 25% identity of amino acids in human sequences (Takasaki et al., 2001). The differences among P2Y receptors may account for differences in their ability to be recognized and activated by agonists.
In the current study, we characterized the sites for the ligand recognition and receptor activation by site-directed mutagenesis in TM3, TM5, TM6, TM7 and EL3 of the P2Y12 receptor. Inhibition of cAMP level by ADP was used as an indicator of receptor function. 5′-adenylic acid, N-[2-(methylthio) ethyl]-2-[(3,3,3-trifluoropropyl) thio]-, monoanhydride with (dichloromethylene) bis [phosphonic acid] (AR-C69931MX) was used to test the ability of mutant receptors to recognize the antagonist. The goal of this work is to provide information which may be useful in designing more selective ligands based on structural differences between the receptors.
2. Materials and methods
2.1 Materials
FITC-labeled monoclonal antibody (HA.11) against the hemagglutinin epitope (HA-tag) was purchased from Covance Research Products (Berkeley, CA). ADP, ATP, forskolin and cAMP, were purchased from Sigma-Aldrich (St. Louis, MO). 3-isobutyl-1-methylxanthine (IBMX) was purchased from Biomol (Plymouth Meeting, PA). [3H]Adenine was purchased from PerkinElmer Life and Analytical Sciences (Boston, MA). All other reagents were reagent-grade, and deionized water was used throughout. Lipofectamine™ 2000 reagent was purchased from Invitrogen (Carlsbad, CA).
2.2 Human P2Y12 plasmid construction and site-direct mutagenesis
Human platelet P2Y12 receptor (GenBank accession number AF313449) (Hollopeter et al., 2001) with an hemagglutinin (HA) tag (YPYDVPDYA) at its N-terminus was cloned into pcDNA3.1/Hygro(+) as previously described (Ding et al., 2006). All mutations were introduced into pcDNA3.1-HA-P2Y12 using the QuickChange site-directed mutagenesis kit from Stratagene. Using the R256Q mutant receptor as a template and R265W mutation primers, a R256Q/R265W double mutant was generated by the same method. The accuracy of all mutations was confirmed by DNA sequencing.
2.3 Cell culture
Chinese hamster ovary (CHO-K1) cells were grown in Ham's F-12 medium (Mediatech, Herndon, VA) supplemented with 10% fetal bovine serum and 1% penicillin, streptomycin, and amphotericin at 37°C with 5% CO2. CHO-K1 cells stably expressing P2Y12 wild-type or mutant receptors were grown in the same medium supplemented with 500 μg/ml hygromycin B (Mediatech, Herndon, VA).
2.4 Stable Expression of Human P2Y12 Wild-Type and Mutant Receptor in CHO-K1 Cells
CHO-K1 cells stably expressed wild-type P2Y12 receptor were generated in our lab as previously described (Ding et al., 2006). The expression constructs for mutant P2Y12 receptors (8 μg) were used to transfect CHO-K1 cells in 60-mm plate using Lipofectamine follow the manufacturer's instruction. The growth medium was replaced after 6 h with fresh medium. Stable transfectants were cultured in medium containing 500 μg/ml hygromycin B and screened for receptor expression by HA-tag detection via immunofluorescence microscopy.
2.5 HA-tag detection in CHO-K1 cells by immunofluorescence microscopy
Cells were cultured in 24 well dishes overnight and washed twice with 1×phosphate buffered saline (PBS). Cells were fixed with 4% paraformaldehyde for 15 minutes and washed 3 times with PBS at room temperature. Cells were then incubated with blocking buffer for 1 hour at room temperature. After washing with PBS, FITC-labeled monoclonal antibody against HA tag (1:1000) was added and incubated in 4°C overnight. Cells were further washed 3 times with 1× PBS and examined under Nikon Eclipse TE300 fluorescence microscope (Melville, NY)
2.6 cAMP Assay
Intracellular cAMP assays were conducted by a modification of a protocol described previously (Ding et al., 2003). In brief, cells were cultured in six-well plates and labeled with 1 μl/ml [3H] adenine (74 kBq/ml) overnight at 37°C. The radiolabeling medium was replaced by fresh growth medium containing 0.4 mM IBMX and incubated for 10 min at 37°C. In the presence of 20 μM forskolin, various concentrations of agonist or/and antagonist were added and incubated at 37°C for 5 min unless otherwise indicated. The reactions were terminated by the addition of 1 ml of stop solution containing 5% trichloroacetic acid, 1 mM ATP, and 1 mM cAMP. Levels of cAMP were determined and cAMP conversion from ATP was calculated using the following formula: cAMP conversion from ATP = [3H]cAMP/([3H]ATP + [3H]cAMP) × 103.
2.7 Statistics
ANOVA followed by the Bonferroni post-test was used to test the significance between wild-type and mutated P2Y12 groups. IC50 value (concentrations causing half-maximal inhibition of cAMP level) was calculated and the difference between different groups are tested by student t-test (Prism 5, Graph Pad, San Diego, CA). P<0.05 or lower was considered as significant.
3. Results
3.1 Construction and stable expression of wild-type and mutant human P2Y12 receptors in CHO-K1 cells
Different mutant constructs of human P2Y12 receptor were prepared as described in MATERIALS AND METHODS and the residues of human P2Y12 receptor selected for site-directed mutagenesis are shown in Fig. 1. CHO-K1 cells were transfected with the P2Y12 wild-type receptor and mutants, and clones growing from single cells which were resistant to 500 μg/ml hygromycin B, were selected. We screened the clones by detecting membrane HA-tag expression using a FITC-labeled monoclonal antibody against HA-tag located at the N-terminus of the receptor. Cell lines, which showed similar expression of the HA tag compared with cells expressing the wild-type P2Y12 receptor (Table 1), were selected for later experiments.
Fig. 1.
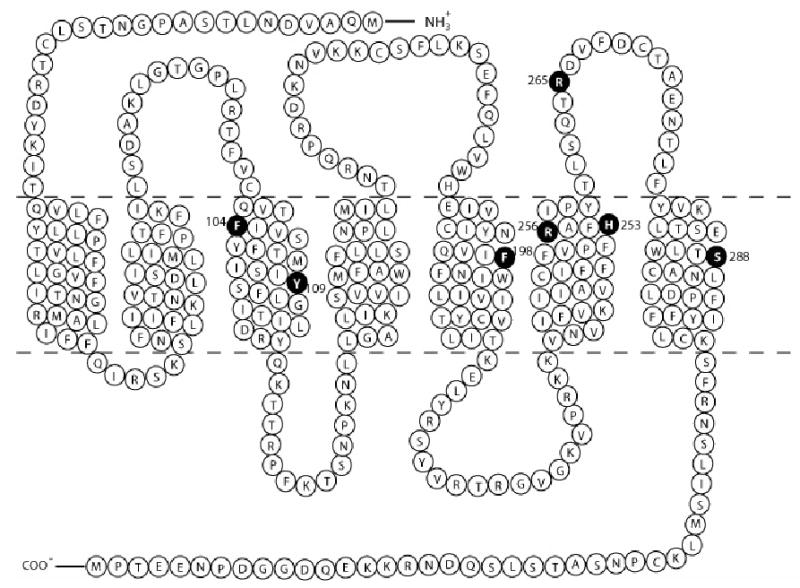
Topology of human P2Y12 receptors showing mutation sites replacing native amino acids with serine or other amino acids. Mutated amino acids are marked as solid circles.
Table 1.
Inhibition by ADP and expression levels of wild-type and mutant P2Y12 receptor constructs expressed in CHO-K1 cells.
| Construct | Max. inhibition by ADP (% of control) | IC50 ADP(μM) | Expression levels (% of wild-type) |
|---|---|---|---|
| Wild-type | 85.37 ± 2.14 (3) | 0.25±0.04 | 100 ± 1.09 (14) |
| F104S | 77.7 ± 1.12 (3) | 0.06±0.01a | 103.21 ± 1.78 (12) |
| Y109S | 31.96 ± 0.15b (3) | 0.17±0.09 | 120.87 ± 0.7 (13) |
| F198P | 40.37 ± 10.64b(3) | 0.15±0.05 | 143.46 ± 0.75 (15) |
| H253S | 62.21 ± 1.93 (3) | 0.35±0.21 | 105.39 ± 0.99 (24) |
| R256T | 79.53 ± 4.83 (3) | 0.29±0.05 | 123.28 ± 0.75 (26) |
| R256Q | 75.32 ± 6.91 (3) | 0.57±0.03a | 106.19 ± 0.72 (11) |
| R265W | 58.01 ± 3.08a (3) | 0.45±0.21 | 87.38 ± 0.90 (8) |
| R256QR265W | 56.68 ± 6.32a (3) | 6.46±0.19b | 148.85 ± 0.81 (20) |
| S288P | 68.89 ± 6.24 (3) | 0.04±0.01a | 120.17 ± 0.97 (16) |
Mean ± S.E.M. of (n) experiments.
P<0.01,
P<0.001, significant differences vs. values determined at wild-type P2Y12 receptors (student t-test). Expression levels were estimated by averaging fluorescence values from the membrane regions of cells after immunofluorescence staining with a monoclonal antibody against the HA-tag.
3.2 Effect of wild-type and mutant P2Y12 receptors on agonist recognition
Agonist-induced inhibition of cAMP was measured to determine whether mutant receptors were functionally active. CHO-K1 cells, which stably expressed wild-type human P2Y12 receptors, were used as control to compare with the effects of mutation and ADP (10 μM) caused about 85% of cAMP inhibition (IC50 = 0.25±0.04 μM).
In human P2Y1 receptors, it has been already reported that residues in TM3, TM6 and TM7 are critical for receptor activation. Agonist-induced P2Y1 activation was almost completely abolished when Arg128, Arg310 or Ser314 were mutated (Jiang et al., 1997).
According to the homology between P2Y receptors (Takasaki et al., 2001), we generated P2Y12 mutant receptors by individually replacing Phe104 and Tyr 109 in TM3, Phe 198 in TM5, His253 and Arg 256 in TM6 and Ser 288 in TM7 with other amino acids.
In contrast to homologous residues in TM3 of P2Y1 receptor, replacing Phe104 with the uncharged amino acid Ser increased potency of activation by ADP by 5-fold (Fig. 2A), whereas, mutation of Tyr109 with uncharged amino acid Ser diminished receptor function and only about 20% of inhibition was observed at 10 μM ADP (Fig. 2A). These data indicated that different amino acids in the same TM may have different effects on receptor activation. The charge on Phe104 may be critical for receptor recognition. Tyr109, on the other hand, is essential for receptor activity. Similarly, Phe198, another amino acid located in TM5, is also critical for receptor function. ADP only caused about maximal 40% inhibition of cAMP level in the F198P mutant receptor as compared with 85% inhibition in P2Y12 wild-type receptor when stimulated by 10 μM ADP (Fig. 2B). In addition to F104S, the mutant S288P receptor also showed an intermediate left shift of ADP concentration curve (about 4-fold greater) (Fig. 2C).
Fig. 2. Concentration-response curves of P2Y12 receptors with mutated residues in TM3, TM5 and TM7.
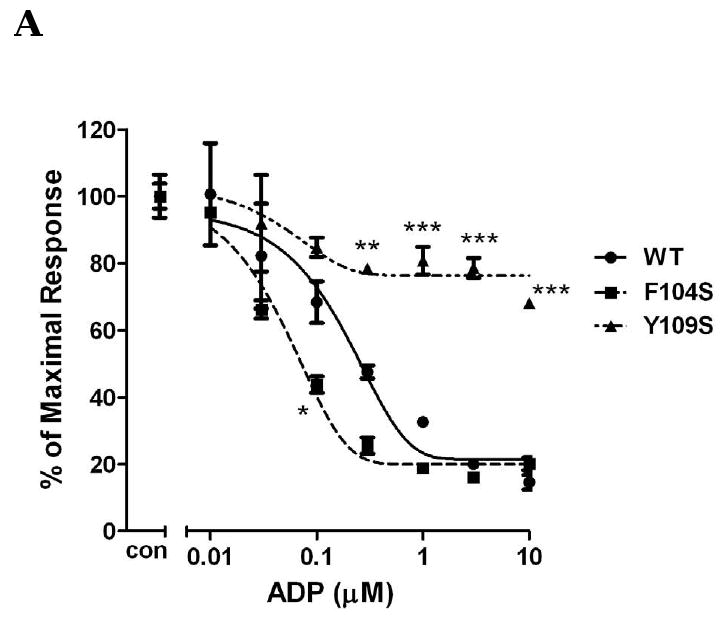
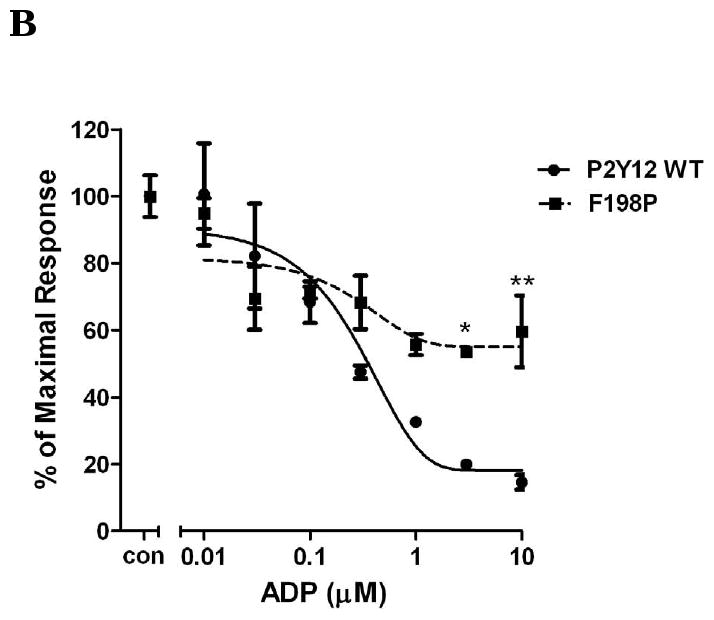
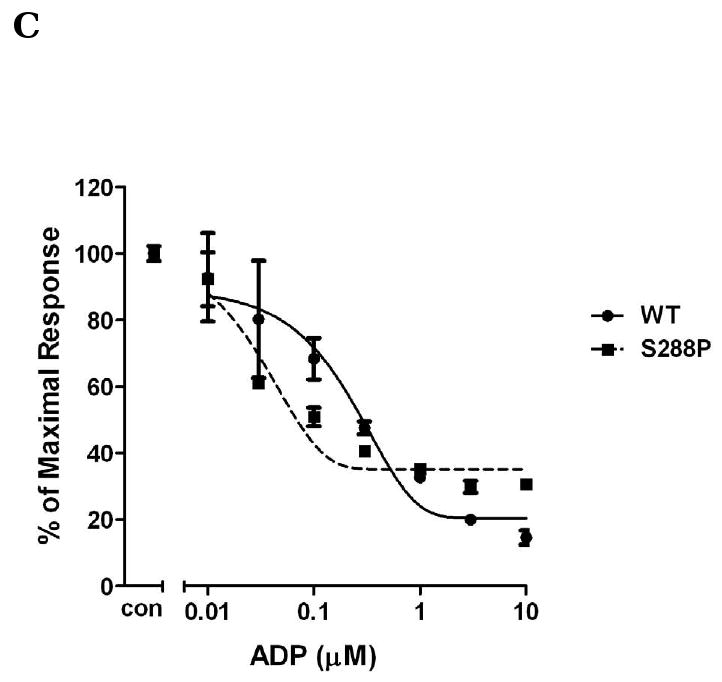
Human wild-type P2Y12 receptor or mutant receptors, in which Phe104 was mutated to Ser and Tyr109 was mutated to Ser (panel A), Phe198 was mutated to Pro (panel B) and Ser288 was mutated to Pro (panel C), were stably expressed in CHO-K1 cells. Inhibition of cAMP was measured in the presence of different concentrations of ADP (0.01-10 μM) in cells stimulated for 5 min with 20 μM forskolin. Data were normalized to the maximal response obtained in the absence of agonist, which served as control (mean ± S.E.M., n=3).*P<0.05, **P<0.01, ***P<0.001, significant differences vs. respective wild-type control (ANOVA followed by the Bonferroni post-test).
Different results were achieved when mutating amino acids in TM6. Replacing Arg256 with uncharged amino acid Thr did not show any changes of receptor activation, but a slight decrease of potency was observed when replacing with another uncharged amino acid Gln. Replacement of His253 with the uncharged amino acid Ser and the mutant R265W in EL3 both displayed a lesser potency to ADP than the wild-type. However, these two mutants still had a good response to agonist and 10 μM ADP caused about 60% inhibition of cAMP (Fig. 3).
Fig. 3. Concentration-response curves of P2Y12 receptors with mutated residues inTM6 (R256T, R256Q, H253S) and EL3 (R265W).
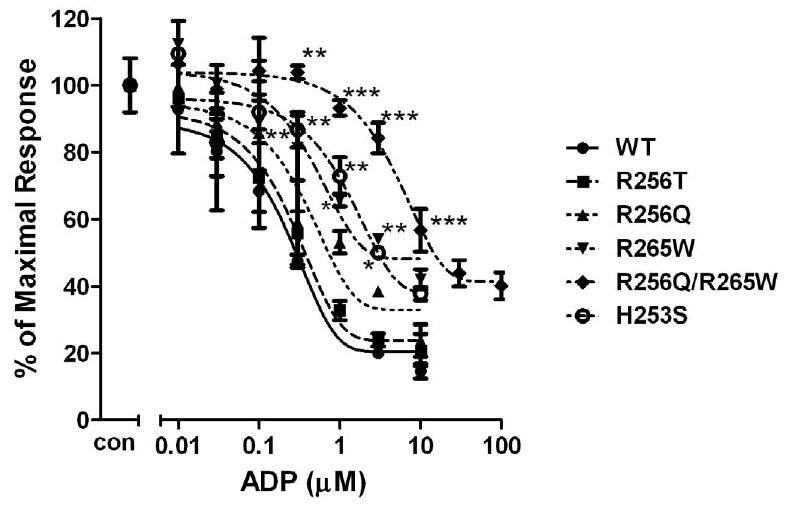
Human wild-type P2Y12 receptor or mutant receptors, in which Arg256 was mutated to Thr or Gln, Arg265 was mutated to Trp and His253 was mutated to Ser, were stably expressed in CHO-K1 cells. Inhibition of cAMP was measured in the presence of different concentrations of ADP (0.01-10 μM) in cells stimulated for 5 min with 20 μM forskolin. Data were normalized to the maximal response obtained in the absence of agonist, which served as control (mean ± S.E.M., n=3). *P<0.05, **P<0.01, ***P<0.001, significant differences vs. respective wild-type control (ANOVA followed by the Bonferroni post-test).
In addition, we also generated a double mutant P2Y12 receptor: R256Q/R265W mutant based on the report that a patient with this mutation in his P2Y12 receptor had an abnormal response to ADP (Cattaneo et al., 2003). Our data showed that the potency of this double mutation (IC50: 6.46±0.19 μM) was dramatically decreased compared to wild-type receptor (IC50: 0.25±0.04 μM). However, it still could achieve maximum activation (about 20% less than wild-type) under higher agonist concentration (30 μM of ADP) (Fig. 3).
3.3 Recognition of antagonist by the wild-type and mutant P2Y12 receptors
According to our results, mutant F104S and S288P cause a left shift of ADP-induced cAMP inhibition compared to wild-type, which indicated that these two mutations increased receptor sensitivity to agonist. Here, we further investigated the effect of antagonists on these mutations receptors. Fig. 4 shows that compared to wild-type transfected cells, different concentrations of AR-C69931MX cause a similar concentration-dependent response in F104S and S288P mutants in the presence of 10 μM ADP. To further confirm this result, we also use sub-maximum concentration of AR-C69931MX (30 nM). It shows that under low concentration of antagonist, all of them shift similarly the concentration curve of ADP to the right compared to wild-type (IC50 for wild-type is 2.39 ± 0.22 μM; IC50 for F104S is 2.02 ± 0.35 μM; IC50 for S288P is 0.79 ± 0.48 μM.). AR-C69931MX (30 nM) inhibit ADP-induced inhibition of cAMP only when ADP concentration is lower than 3 μM (Fig. 5). All these data indicated that these two mutations only affected agonist-induced receptor activation; instead, neither of them affected antagonist-induced inhibition of receptor function.
Fig. 4. Concentration-response curves of P2Y12 receptors with mutated residues in TM3 (F104S) and TM7 (S288P) in antagonist analysis.
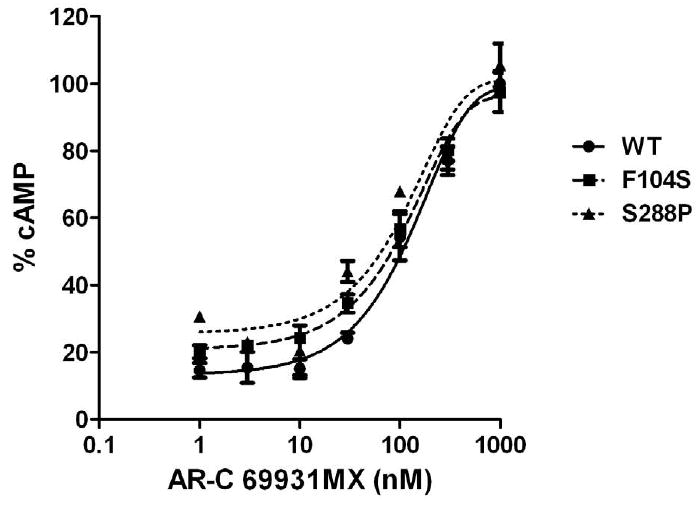
Cells were treated with 10μM ADP in the absence or the presence of (0.001-1 μM) AR-C69931MX. Data were normalized to the response in the presence of 20μM forskolin alone taken as 100%. (mean ± S.E.M., n=3).
Fig. 5. The effect of submaximum AR-C69931MX on ADP-induced concentration-response curves of wild-type P2Y12 receptors (panel A) and mutants with mutated residues in TM3 (F104S) (panel B) and TM7 (S288P) (panel C) in antagonist analysis.
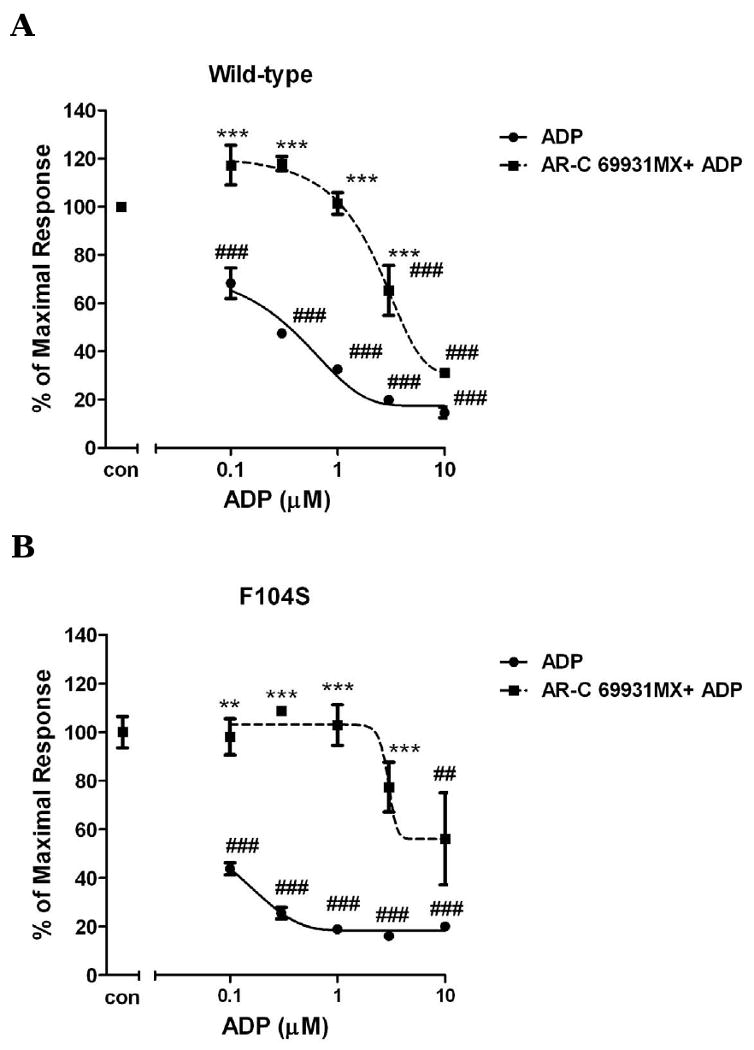
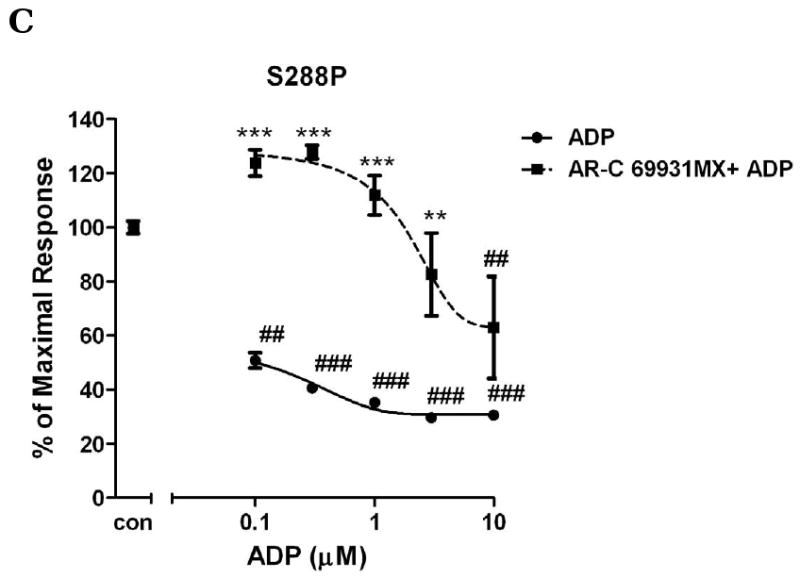
Inhibition of cAMP was measured after incubation of cells with different concentrations of ADP (0.1-30 μM) and stimulated for 5 min with or without 30 nM AR-C69931MX in presence of 20 μM forskolin. Data were normalized to the maximal response obtained in the absence of agonist and antagonist, which served as control (mean ± S.E.M., n=3). ## P<0.01, ### P<0.001, significant differences vs. respective control without agonist and antagonist. ** P<0.01, *** P<0.001, significant differences vs. respective values in the absence of the antagonist. (ANOVA followed by the Bonferroni post-test).
Arg256 and Arg265 have been reported to be important for P2Y12 receptor function (Cattaneo et al., 2003). Our results show that a change of Arg256 to a different amino acid or/and change of Arg265 had partially effect on agonist-induced receptor activation. We further investigated whether these mutations would have any effect on antagonist-induced inhibition of receptor function. For wild-type, it is required to use 1 μM AR-C69931MX to completely inhibit 10 μM ADP-induced inhibition of cAMP and the antagonism effect did not occur if the concentration of AR-C69931MX was decreased to 10 nM. However, R256T, which had a similar effect on different concentration of ADP when compared to wild-type (Fig. 3), causes a significant left shift compared to wild-type when different concentrations of AR-C69931MX were applied in the presence of 10μM ADP and 100nM AR-C69931MX is enough to block ADP (10 μM) -induced activation, however, it is necessary to use 1μM AR-C69931MX for wild-type (Fig. 6B). When Arg256 was changed to Gln, it also caused a leftward shift (Fig. 6A). R265W, which is located in EL3, caused an even more leftward shift compared to R256Q and R256T. For R265W, it was enough to block ADP-induced activation by using 30 nM AR-C69931MX (Fig. 6C). Similarly, double mutation R256QR265W also showed a leftward shift of the antagonist concentration curve as expected (Fig. 6D). All these data indicate that mutations in TM5 and EL3 have a more significant effect on antagonist recognition compared to their effect on agonist recognition.
Fig. 6. Concentration-response curves of P2Y12 receptors with mutated residues in TM6 and EL3 (R265W).
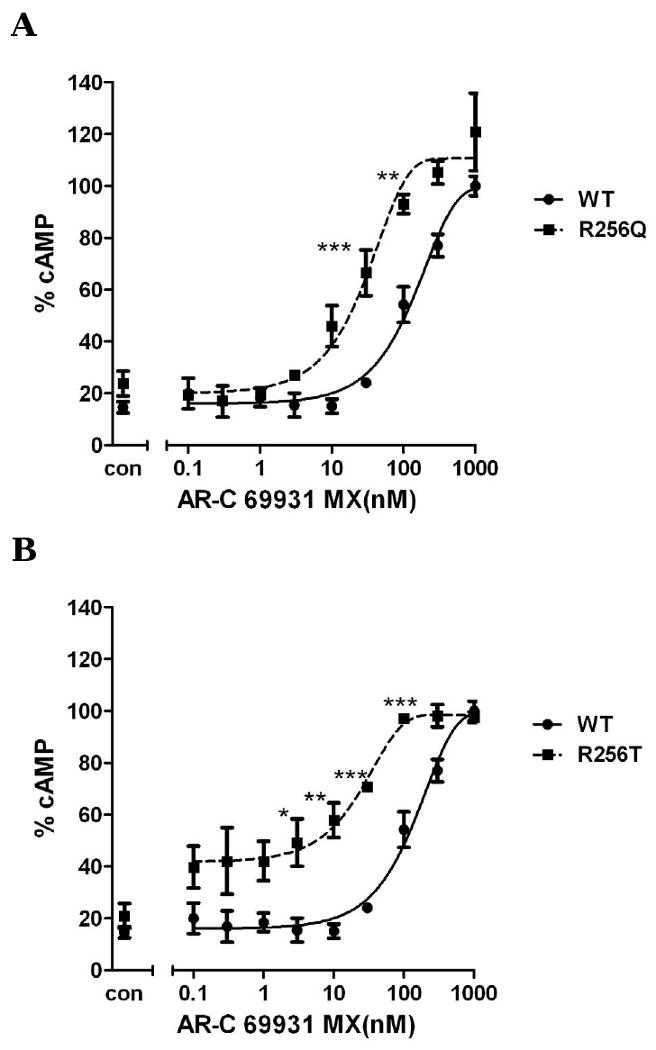
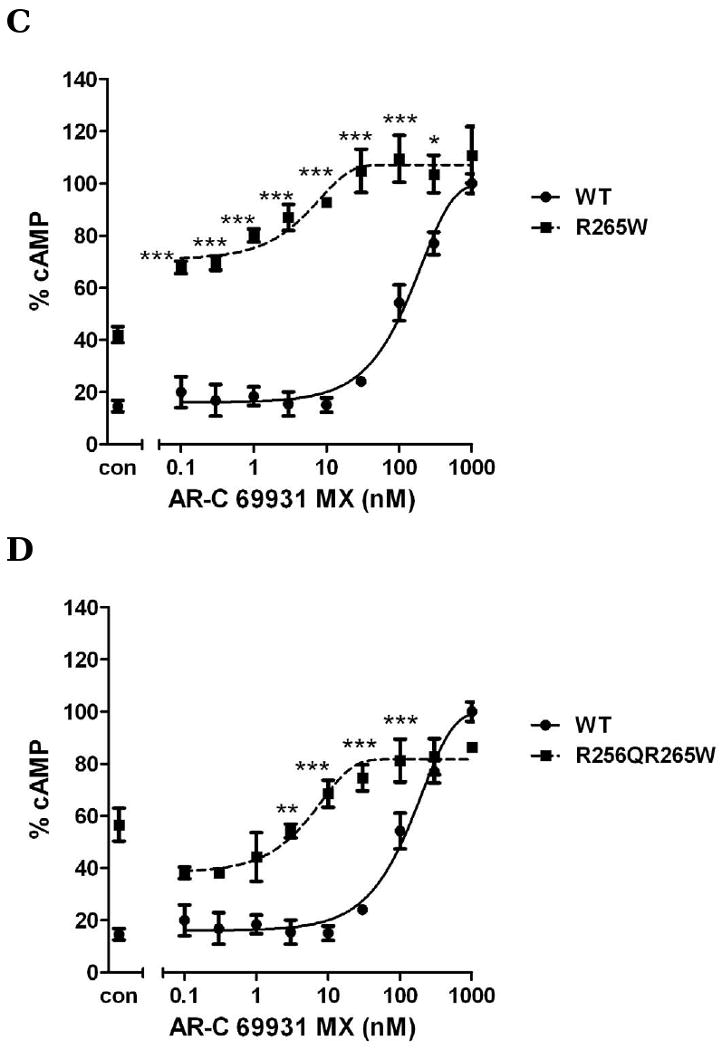
Wild-type and mutants R256Q (panel A), R256T (panel B) R265W (panel C) and R256Q/R265W (panel D) were treated with 10μM ADP in the absence or the presence of (0.0001-1 μM) AR-C69931MX. Data were normalized to the response in the presence of 20μM forskolin alone taken as 100%. Cells treated with 10 μM ADP alone marked as control (mean ± S.E.M., n=3). * P<0.05, ** P<0.01, *** P<0.001, significant differences vs. respective wild-type control (ANOVA followed by the Bonferroni post-test).
4. Discussion
In the present study, we investigated residues within the TMs and EL3 of human P2Y12 receptor that may be critical for agonist binding and receptor activation. Since P2Y1 and P2Y12 share the same agonist and both receptors are important for platelet activation, we generated site-direct mutant P2Y12 receptors by modifying certain amino acids, which have been shown critical for receptor function based on studies of the P2Y1 receptor (Costanzi et al., 2004; Jiang et al., 1997).
Based on the docking model of P2Y1, the automatic docking of ADP and P2Y12 showed a similar binding mode to P2Y1 (Costanzi et al., 2004). According to this theoretical model, the upper part of TM3, TM6 and TM7 and closed by EL2 are also critical for P2Y12-agonist binding. In our study, the F104S in TM3 and S288P in TM7 mutant receptors potentiated receptor binding affinity as compared with wild-type P2Y12 receptor, which is in contrast to the role of these homologous amino acids in the P2Y1 receptor (Jiang et al., 1997). In the P2Y1 receptor, mutation of Arg128 and Ser314, located at the same site as Phe104 and Ser288 in P2Y12 receptor, to alanine completely abolished receptor activation (Jiang et al., 1997). The sequence differences between these two receptors may explain these contrasting results. It has been reported that ADP is a more potent agonist for P2Y1 activation than for P2Y12 activation (Hall and Hourani, 1993; Hourani and Hall, 1996). It is possible that replacement of Phe104 or Ser288 will make P2Y12 receptor ligand binding domain more similar to P2Y1 and increase the binding affinity for ADP.
Our study clearly demonstrates that amino acids located proximal to the cytosolic side of TM3 and the exofacial side of TM5 are critical for receptor activation. The mutant Y109S resulted in a nonfunctional P2Y12 receptor. Another possible important amino acid for P2Y12 receptor activation is found in TM5. The mutant F198P receptor shows a 50% lesser activity as compared with wild-type P2Y12 receptor. In fact, the essential amino acids for P2Y1 receptor activation are located in the more exofacial side of TM3 and TM7 instead of the cytosolic side of TM3 and the exofacial side of TM5.
The docking model of P2Y12 also showed that Arg 256 was the only common amino acid among three basic amino acids involved in phosphate coordination in both P2Y1 and P2Y12 (Costanzi et al., 2004). Lys280 of P2Y1, which corresponds to Arg256 in the P2Y12 receptor, was reported to be critical for agonist recognition (Jiang et al., 1997; Van Rhee et al., 1995). Cattaneo et al reported that Arg256 in TM6 and Arg265 in EL3 are critical for P2Y12 activation because: 1) platelets from a patient with defective P2Y12 receptor had an abnormal response to ADP, and 2) there was a significantly decrease in ADP inhibited forskolin-induced increase of cAMP in transfected cell lines without change of binding affinity (Cattaneo et al., 2003). Recent studies on the P2Y12 receptor also showed that Arg 256 is important for interaction with a negatively charged domain of the agonist (Hoffmann et al., 2008). Here our studies showed that replacing Arg256 with Gln may change the structure of receptor binding site and make it more difficult for ligand recognition. R265W, another mutation in a patient's P2Y12 receptor located in EL3 clearly decreased the potency to ADP but had less effect on receptor maximal activation. Our result was consistent with the finding in P2Y1 receptor. Arg287 of P2Y1 is the homologous residue of Arg265 in the P2Y12. Although this residue was not tolerated by removal or changing the charge of amino acid side chain, the R287K mutant still could retain receptor activity with a lower potency (Hoffmann et al., 1999). It is possible that the same positive charge of Arg265 in P2Y12 also could directly interact with the phosphate group of adenine nucleotides as reported for the P2Y1 receptor (Jiang et al., 1997) and this interaction could be inhibited by changing the residue to an amino acid with an aromatic R group, which was not tested in the previous P2Y1 study.
To mimic the defective patient P2Y12 receptor (Cattaneo et al., 2003), a double mutation R256Q/R265W was constructed. Our results showed that double mutation only caused decreased potency of agonist-induced receptor activation. The maximal effect was still evident at higher concentrations of ADP. These data indicated that mutations on these two sites might only decrease the binding affinity of receptor but did not dramatically affect receptor activation. Another important residue was His253. Similar to the homologous residue in P2Y1, replacement of His253 with uncharged amino acid Ser caused a clear right shift of ADP concentration-response curve but less inhibition of receptor activation (maximal inhibition by 60%) compared to wild-type P2Y12 (maximal inhibition by 85%). These data support the idea that amino acids in TM6 and EL3 are more important for receptor binding than receptor activation. Low binding affinity for ligand may be compensated by increasing concentrations of the ligand.
In addition, antagonist analysis studies showed a different result compared to agonist analysis. High concentrations of AR-C69931MX were able to reverse the decreased cAMP level induced by ADP in both wild-type and mutant receptors. However, the recognition ability of these mutations is different. F104S and S288P recognize the antagonist similarly compared to the wild-type receptor, although they have a higher potency for agonist recognition. R256Q, R256T, R265W and double mutation R256Q/R265W have a much higher sensitivity for antagonist recognition compared to wild-type receptor. It is possible that the amino acids in P2Y12, which are responsible for identifying agonist and antagonist, are different. From our data, it is indicated that F104 and S288P are more important for recognition of agonist; however, R256 and R265 are more important for antagonist recognition.
Although P2Y1 and P2Y12 share the same agonists, they have a relatively low identity of amino acids in their sequence (Takasaki et al., 2001). In previous studies, all models of P2Y12 were on the analogy of studies on P2Y1 receptor. The findings in this study provide evidence that the role of specific amino acids in P2Y12 receptor is different from P2Y1 receptor. Our data show that mutation of F104 inTM3 is related to P2Y12 receptor lower-affinity for agonist as compared with P2Y1 receptor. Y109 in TM3 and F198 in TM5 of the P2Y12 receptor are more critical for receptor function than the amino acids in TM6 and TM7.
Taken together, these data provide information to propose a more accurate model for studying P2Y12 receptor activation. Furthermore, this study revealed differences between P2Y1 and P2Y12 receptors that will help to characterize the structure-function differences among P2Y receptors. The present site-directed mutagenesis will help us to understand P2Y12 structure better and contribute to identification of more potent and specific ligands.
Acknowledgments
This work is supported by Grants HL80444 and HL60683 from National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abbracchio MP, Burnstock G, Boeynaems JM, Barnard EA, Boyer JL, Kennedy C, Knight GE, Fumagalli M, Gachet C, Jacobson KA, Weisman GA. International Union of Pharmacology LVIII: update on the P2Y G protein-coupled nucleotide receptors: from molecular mechanisms and pathophysiology to therapy. Pharmacol Rev. 2006;58:281–341. doi: 10.1124/pr.58.3.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cattaneo M, Zighetti ML, Lombardi R, Martinez C, Lecchi A, Conley PB, Ware J, Ruggeri ZM. Molecular bases of defective signal transduction in the platelet P2Y12 receptor of a patient with congenital bleeding. Proc Natl Acad Sci U S A. 2003;100:1978–1983. doi: 10.1073/pnas.0437879100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costanzi S, Mamedova L, Gao ZG, Jacobson KA. Architecture of P2Y nucleotide receptors: structural comparison based on sequence analysis, mutagenesis, and homology modeling. J Med Chem. 2004;47:5393–5404. doi: 10.1021/jm049914c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dangelmaier C, Jin J, Smith JB, Kunapuli SP. Potentiation of thromboxane A2-induced platelet secretion by Gi signaling through the phosphoinositide-3 kinase pathway. Thromb Haemost. 2001;85:341–348. [PubMed] [Google Scholar]
- Daniel JL, Dangelmaier C, Jin J, Ashby B, Smith JB, Kunapuli SP. Molecular basis for ADP-induced platelet activation. I. Evidence for three distinct ADP receptors on human platelets. J Biol Chem. 1998;273:2024–2029. doi: 10.1074/jbc.273.4.2024. [DOI] [PubMed] [Google Scholar]
- De Clerck FF, Janssen PA. Amplification mechanisms in platelet activation and arterial thrombosis. J Hypertens. 1990 8:S87–93. [PubMed] [Google Scholar]
- Ding Z, Kim S, Dorsam RT, Jin J, Kunapuli SP. Inactivation of the human P2Y12 receptor by thiol reagents requires interaction with both extracellular cysteine residues, Cys17 and Cys270. Blood. 2003;101:3908–3914. doi: 10.1182/blood-2002-10-3027. [DOI] [PubMed] [Google Scholar]
- Ding Z, Kim S, Kunapuli SP. Identification of a potent inverse agonist at a constitutively active mutant of human P2Y12 receptor. Mol Pharmacol. 2006;69:338–345. doi: 10.1124/mol.105.014654. [DOI] [PubMed] [Google Scholar]
- Erb L, Garrad R, Wang Y, Quinn T, Turner JT, Weisman GA. Site-directed mutagenesis of P2U purinoceptors. Positively charged aminoacids in transmembrane helices 6 and 7 affect agonist potency and specificity. J Biol Chem. 1995;270:4185–4188. doi: 10.1074/jbc.270.9.4185. [DOI] [PubMed] [Google Scholar]
- Hall DA, Hourani SM. Effects of analogues of adenine nucleotides on increases in intracellular calcium mediated by P2T-purinoceptors on human blood platelets. Br J Pharmacol. 1993;108:728–733. doi: 10.1111/j.1476-5381.1993.tb12869.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann C, Moro S, Nicholas RA, Harden TK, Jacobson KA. The role of amino acids in extracellular loops of the human P2Y1 receptor in surface expression and activation processes. J Biol Chem. 1999;274:14639–14647. doi: 10.1074/jbc.274.21.14639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann K, Sixel U, Di Pasquale F, von Kugelgen I. Involvement of basic amino acid residues in transmembrane regions 6 and 7 in agonist and antagonist recognition of the human platelet P2Y(12)-receptor. Biochem Pharmacol. 2008;76:1201–1213. doi: 10.1016/j.bcp.2008.08.029. [DOI] [PubMed] [Google Scholar]
- Hollopeter G, Jantzen HM, Vincent D, Li G, England L, Ramakrishnan V, Yang RB, Nurden P, Nurden A, Julius D, Conley PB. Identification of the platelet ADP receptor targeted by antithrombotic drugs. Nature. 2001;409:202–207. doi: 10.1038/35051599. [DOI] [PubMed] [Google Scholar]
- Hourani SM, Hall DA. P2T purinoceptors: ADP receptors on platelets. Ciba Found Symp. 1996;198:53–64. doi: 10.1002/9780470514900.ch3. discussion 64-70. [DOI] [PubMed] [Google Scholar]
- Jiang Q, Guo D, Lee BX, Van Rhee AM, Kim YC, Nicholas RA, Schachter JB, Harden TK, Jacobson KA. A mutational analysis of residues essential for ligand recognition at the human P2Y1 receptor. Mol Pharmacol. 1997;52:499–507. doi: 10.1124/mol.52.3.499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin J, Daniel JL, Kunapuli SP. Molecular basis for ADP-induced platelet activation. II. The P2Y1 receptor mediates ADP-induced intracellular calcium mobilization and shape change in platelets. J Biol Chem. 1998;273:2030–2034. doi: 10.1074/jbc.273.4.2030. [DOI] [PubMed] [Google Scholar]
- Jin J, Kunapuli SP. Coactivation of two different G protein-coupled receptors is essential for ADP-induced platelet aggregation. Proc Natl Acad Sci U S A. 1998;95:8070–8074. doi: 10.1073/pnas.95.14.8070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurk K, Kehrel BE. Platelets: physiology and biochemistry. Semin Thromb Hemost. 2005;31:381–392. doi: 10.1055/s-2005-916671. [DOI] [PubMed] [Google Scholar]
- Kim S, Jin J, Kunapuli SP. Akt activation in platelets depends on Gi signaling pathways. J Biol Chem. 2004;279:4186–4195. doi: 10.1074/jbc.M306162200. [DOI] [PubMed] [Google Scholar]
- Kim S, Jin J, Kunapuli SP. Relative contribution of G-protein-coupled pathways to protease-activated receptor-mediated Akt phosphorylation in platelets. Blood. 2006;107:947–954. doi: 10.1182/blood-2005-07-3040. [DOI] [PubMed] [Google Scholar]
- Offermanns S. Activation of platelet function through G protein-coupled receptors. Circ Res. 2006;99:1293–1304. doi: 10.1161/01.RES.0000251742.71301.16. [DOI] [PubMed] [Google Scholar]
- Packham MA, Kinlough-Rathbone RL, Mustard JF. Thromboxane A2 causes feedback amplification involving extensive thromboxane A2 formation on close contact of human platelets in media with a low concentration of ionized calcium. Blood. 1987;70:647–651. [PubMed] [Google Scholar]
- Shankar H, Garcia A, Prabhakar J, Kim S, Kunapuli SP. P2Y12 receptor-mediated potentiation of thrombin-induced thromboxane A2 generation in platelets occurs through regulation of Erk1/2 activation. J Thromb Haemost. 2006;4:638–647. doi: 10.1111/j.1538-7836.2006.01789.x. [DOI] [PubMed] [Google Scholar]
- Storey RF, Sanderson HM, White AE, May JA, Cameron KE, Heptinstall S. The central role of the P(2T) receptor in amplification of human platelet activation, aggregation, secretion and procoagulant activity. Br J Haematol. 2000;110:925–934. doi: 10.1046/j.1365-2141.2000.02208.x. [DOI] [PubMed] [Google Scholar]
- Takasaki J, Kamohara M, Saito T, Matsumoto M, Matsumoto S, Ohishi T, Soga T, Matsushime H, Furuichi K. Molecular cloning of the platelet P2T(AC) ADP receptor: pharmacological comparison with another ADP receptor, the P2Y(1) receptor. Mol Pharmacol. 2001;60:432–439. [PubMed] [Google Scholar]
- Trumel C, Payrastre B, Plantavid M, Hechler B, Viala C, Presek P, Martinson EA, Cazenave JP, Chap H, Gachet C. A key role of adenosine diphosphate in the irreversible platelet aggregation induced by the PAR1-activating peptide through the late activation of phosphoinositide 3-kinase. Blood. 1999;94:4156–4165. [PubMed] [Google Scholar]
- Van Rhee AM, Fischer B, Van Galen PJ, Jacobson KA. Modelling the P2Y purinoceptor using rhodopsin as template. Drug Des Discov. 1995;13:133–154. [PMC free article] [PubMed] [Google Scholar]
- Yoneda K, Iwamura R, Kishi H, Mizukami Y, Mogami K, Kobayashi S. Identification of the active metabolite of ticlopidine from rat in vitro metabolites. Br J Pharmacol. 2004;142:551–557. doi: 10.1038/sj.bjp.0705808. [DOI] [PMC free article] [PubMed] [Google Scholar]