

Abstract
At the mammalian skeletal neuromuscular junction, cycling of nicotinic ACh receptors (nAChRs) is critical for the maintenance of a high postsynaptic receptor density. However, the mechanisms that regulate nAChRs recycling in living animals remain unknown. Using in vivo time-lapse imaging, fluorescence recovery after photobleaching, and biochemical pull down assays, we demonstrated that recycling of internalized nAChRs into fully functional and denervated synapses was promoted by both direct muscle stimulation and pharmacologically induced intracellular calcium elevations. Most of internalized nAChRs are recycled directly into synaptic sites. Chelating of intracellular calcium below resting level drastically decreased cycling of nAChRs. Furthermore we found that calcium-dependent AChR recycling is mediated by Ca2+/calmodulin-dependent kinase II (CaMKII). Inhibition of CaMKII selectively blocked recycling and caused intracellular accumulation of internalized nAChRs, whereas internalization of surface receptors remained unaffected. Electroporation of CaMKII-GFP isoforms into the sternomastoid muscle showed that muscle-specific CaMKIIβm isoform is highly expressed at the neuromuscular junction (NMJ) and precisely colocalized with nAChRs at crests of synaptic folds while the CaMKIIγ and δ isoforms are poorly expressed in synaptic sites. These results indicate that Ca2+ along with CaMKII activity are critical for receptor recycling and may provide a mechanism by which the postsynaptic AChR density is maintained at the NMJ in vivo.
Introduction
Efficient synaptic transmission requires maintenance of a high postsynaptic receptor density directly apposed to the nerve terminal. In the CNS, changes in postsynaptic receptor number and/or density are associated with synaptic plasticity (Shepherd and Huganir, 2007; Petrini et al., 2009). Endocytic recycling plays a critical role in such changes by affecting receptors and other accessory proteins that enhance synaptic transmission and modulate structural alterations of synapses (Ehlers, 2000; Malinow and Malenka, 2002; Bredt and Nicoll, 2003; Park et al., 2004; Kennedy and Ehlers, 2006; Lu et al., 2007). In the last decade, insights into how receptors undergo endocytosis and recycling have emerged and several Ca2+ sensor molecules have been shown to play a role in these events. For example, Ca2+/calmodulin-dependent protein kinase II (CaMKII) and Myosin Vb have been shown to be required for the mobilization, fusion and exocytosis of recycling endosomes during LTP (Ehlers and Augustine, 1999; Ehlers, 2000; Hayashi et al., 2000; Wang et al., 2008). Ca2+ influx into postsynaptic excitatory synapses has also been shown to play a key role in synaptic strength.
At the mature neuromuscular junction (NMJ), a synapse that forms between motor neurons and skeletal muscle fibers, recycled nicotinic ACh receptors (nAChRs) contribute to the steady-state postsynaptic receptor density in an activity-dependent manner (Bruneau et al., 2005, 2009; Bruneau and Akaaboune, 2006; Wu et al., 2010). However, the mechanisms that regulate the trafficking and recycling of internalized vesicles containing-AChRs are not known. In an effort to identify factors involved in the cycling of internalized nAChRs, we began the present study by investigating the effect of intracellular calcium on AChR delivery and then we asked whether CaMKII, a serine/threonine protein kinase family member that is activated by calcium and calmodulin is the sensing protein involved in receptor recycling at the NMJ in living mice. CaMKII is best studied in the brain and has been reported to have a key role in regulating synaptic plasticity in brain (Pratt et al., 2003). Mammalian skeletal muscle expresses several isoforms of CaMKII γ, δ and a splice variant of the β isoform (βM), and a nonfunctional kinase variant of the α isoform, anchoring kinase protein (αkap) (Schworer et al., 1993; Bayer et al., 1998).
In the present study we investigated a possible mechanism that regulates the trafficking and recycling of internalized AChR at the NMJ in living mice. We demonstrate that receptor recycling is dependent upon intracellular calcium concentration. By increasing or decreasing intracellular calcium concentration, the muscle cell is able to up or downregulate the cycling of AChR into the postsynaptic membrane. This process is mediated by CaMKII activity as blockade of the enzyme dramatically inhibits receptor recycling. These results establish a role for calcium and calcium activated kinase, CaMKII in the recycling of receptors at the NMJ in vivo.
Materials and Methods
Labeling of nAChR pools and neuromuscular junction live imaging.
In this study we used Non-Swiss Albino adult female mice (6–10 weeks old, 25–30 g) that were purchased from Harlan. All animal usage followed methods approved by the University of Michigan Committee on the Use and Care of Animals. Mice were anesthetized with an intraperitoneal injection of a mixture of ketamine and xylazine (18.38 mg/ml) and the sternomastoid muscle was exposed as described in detail previously (Lichtman et al., 1987; Balice-Gordon and Lichtman, 1994; Turney et al., 1996; Bruneau and Akaaboune, 2006). Briefly, the sternomastoid muscle was bathed with bungarotoxin (BTX)-biotin (5 μg/ml, 1 h; Invitrogen) to label nAChRs, and then with streptavidin conjugated to Alexa 488 (strept-488; green; 10 μg/ml, 3 h; Invitrogen) to saturate all biotin sites as previously described (Bruneau et al., 2005; Bruneau and Akaaboune, 2006). The anesthetized mouse was placed on its back on the stage of a customized epifluorescence microscope, and superficial neuromuscular junctions were viewed under a coverslip with a water-immersion objective (x20 UApo 0.8 NA Olympus BW51; Optical Analysis Corp.) and a digital CCD camera (Retiga EXi, Qimaging) and imaged (IPLAB software, Scanalytics). At the end of an imaging session, the incision was closed and the mouse was allowed to recover until reanesthetized and prepared for a second imaging session.
We used a method of labeling to selectively label recycled receptors that has been previously described in detail (Bruneau et al., 2005). In that study, extensive controls were conducted which demonstrated that (1) α-bungarotoxin (BTX) remains bound to AChR intracellularly, (2) within internal vesicles (likely endosomes), Alexa-labeled streptavidin dissociates rapidly (within hours) from BTX-biotin, and (3) on the extracellular surface the rate of dissociation of Alexa-labeled streptavidin from biotin was too slow to be detected even after several days. Thus we can assess recycling by adding fresh Alexa streptavidin hours or days after an initial saturating dose. Briefly, the sternomastoid muscle was bathed with BTX-biotin (5 μg/ml, 1 h; Invitrogen) to label nAChRs, and then with streptavidin conjugated to an Alexa dye (strept-488; green; 10 μg/ml, 3 h; Invitrogen). To verify that all biotin sites were saturated, a second color of streptavidin-Alexa (typically Alexa-594; red, 10 μg/ml, 10–30 min) was then added to the sternomastoid muscle. Experiments only continued if there was no red labeling at the NMJ indicating that all biotin sites were indeed saturated.
Muscle stimulation.
The sternomastoid muscle was exposed and then directly stimulated for ∼7 h with a Grass SD5 stimulator connected to two platinum wires placed either side of the muscle as described by Akaaboune et al. (1999). The stimulus pulses (3 ms bipolar pulses of 6–9 V at 10 Hz for 1 s duration every 2 s) elicited maximal twitching and therefore action potentials in all muscle fibers. To minimize dehydration, a coverslip was placed over the exposed muscle.
Immunofluorescence.
The sternomastoid muscle was fixed with 2% paraformaldehyde and then permeabilized for 20 min with 1% Triton X-100 and 10% bovine serum albumin in phosphate buffered solution. Muscle fibers were then incubated with goat polyclonal anti-CaMKIIβ (C-20), goat polyclonal anti-CaMKIIγ (C-18), or goat polyclonal anti-CaMKIIδ (A-17) (Santa Cruz Biotechnology), overnight at 4°C overnight or 2 h at room temperature. All these antibodies partially cross react with each (according to data sheet of the manufacturer). After extensive washing with PBS, muscle fibers were incubated with anti-goat-Alexa 488 secondary antibodies (Invitrogen), as well as BTX-Alexa 594 (Invitrogen). Muscles were scanned with a confocal microscope (Leica) and imaged. The z-stacks were then collapsed and the contrast of images was adjusted with Adobe Photoshop.
Pharmacological treatment.
To chelate cytosolic calcium, we used BAPTA-AM (500 μm) (Invitrogen), a membrane-permeant substance that generates the high affinity calcium chelator BAPTA by the hydrolysis of ester bonds (Tsien, 1981; Martinez-Pena y Valenzuela et al., 2005). To inhibit CaMKII activity we used KN93 (10 μm; Sigma) and KN62 (10 μm; Sigma), and we used its inactive analog, KN92, as a control (10 μm; Sigma) (Tokumitsu et al., 1990; Hudmon and Schulman, 2002). To inhibit intracellular calcium (sarcoendoplasmic reticulum Ca2+-ATPase) pumps, we used thapsigargin (1 μm) (Sagara and Inesi, 1991).
Muscle denervation.
The sternomastoid muscles were denervated by excising a 5 mm piece of the sternomastoid nerve to prevent reinnervation. Mice denervated 6–8 d earlier were anesthetized and the sternomastoid muscle was exposed and labeled first with BTX-biotin followed by a saturating dose of streptavidin (strept-488). Three days later, the animal was reanesthetized and the sternomastoid muscle was bathed with strept-594 (to label recycled nAChRs) and superficial synapses were imaged. The sternomastoid muscle was then either stimulated without drugs, or stimulated in the presence of BAPTA-AM, KN93, or KN62 for ∼7 h. At the end of the experiment, the sternomastoid muscle was bathed with fresh strept-594 (10 μg/ml, 1 h) to label all receptors that had recycled over the time period of the experiment.
Quantitative fluorescence imaging.
The fluorescence intensity of labeled receptors at NMJs was assayed using a quantitative fluorescence imaging technique, as described by Turney et al. (1996), with minor modifications. This technique incorporates compensation for image variation that may be caused by spatial and temporal changes in the light source and camera between imaging sessions, by calibrating the images with a nonfading reference standard. A key feature of the quantitative imaging approach used in the current study is that it involves repetitive imaging of the same fluorescent ligands. Thus, as long as we verified that the image pixel intensity was not saturated, it was possible to get an accurate quantitative measurement of the relative number of nAChRs. Image analysis was performed by using either a procedure written for IPLAB (Scanalytics) or Matlab (The Mathworks). Background fluorescence was approximated by selecting a boundary region around the junction and subtracting it from the original image, and the mean of the total fluorescence intensity (which corresponds to density) was measured (Turney et al., 1996; Martinez-Pena y Valenzuela et al., 2005).
Fluorescent recovery after photobleaching.
To determine whether newly recycled nAChRs are inserted directly into synaptic sites, the sternomastoid muscle was labeled with BTX-biotin/strept-488 and 5 d later, recycled receptors were labeled with strept-594 and superficial synapses were imaged. Fluorescence from labeled recycled nAChRs was removed by illuminating NMJs with an argon laser passed through a neutral density filter. [We have previously shown that the bleaching of synapses with laser does not damage or change the physiology or the metabolism of AChRs at the postsynaptic membrane (Akaaboune et al., 2002).] The recovery of fluorescence was assayed at stimulated and unstimulated muscles, and the amount of fluorescence recovered at bleached synapses was measured. At the end of the experiment, a second dose of the same fluorescent streptavidin was added to label newly recycled nAChRs.
Electroporation of CaMKII-GFPs into the sternomastoid muscle and confocal microscopy.
Adult female mice (20–28 g; Non-Swiss Albino, Harlan) were anesthetized with an intraperitoneal injection of ketamine and xylazine. The sternomastoid muscle was surgically exposed and 10 μg of plasmid driving exogenous expression of a test protein fused to GFP was added (All constructs were driven by a CMV promoter). The proteins tested were muscle-specific CaMKIIβm-GFP (a generous gift from Dr. Ulrich Bayer, University of Colorado, Denver, CO), CaMKIIβ generous gift from Dr. Tobias Meyer, Stanford University, Stanford, CA), α syntrophin (a generous gift from Dr. Marvin Adams, University of Washington, Seattle, WA), CaMKIIγ a generous gift from Dr. Paul Nghiem, University of Washington, Seattle, WA). CaMKIIδ was purchased from OriGene Technologies. CaMKIIγ-GFP was generated using primers, 5′GGAATTCTGATGGCCACCACCGCCACCTGC (forward) and 5′ GGGGTACCTCACTGCAGCGGTGCGGCAGG (reverse). CaMKIIδ-GFP was generated using primers, 5′ GGAATTCTGATGGCTTCGACCACCACCTGCACC (forward) and 5′ GGGGTACCTCAGATGTTTTGCCACAAAGAGG (reverse). GFP-α syntrophin was generated using 5′ CGCTCGAGCTATGGCGTCAGGCAGGCGC (forward) and 5′ GCTCTAGACTAGGCCAAGAGCCCCAAGC (reverse). The plasmid solution was layered over the muscle surface as described by Bruneau et al. (2009). Gold electrodes were placed parallel to the muscle fibers on either side of the muscle, and eight monopolar square-wave pulses were applied perpendicular to the long axis of the muscle. Following electroporation the mouse was sutured and allowed to recover in a heated recovery chamber. One week later, the animal was reanesthetized and BTX-Alexa 594 was added to the sternomastoid muscle to label nAChRs (1 h) and the animal was perfused transcardially with 2% paraformaldehyde (PFA). The sternomastoid muscle was removed, mounted and scanned with a confocal microscope (Olympus fluoview) and imaged. The z-stacks were then collapsed and the contrast adjusted with Photoshop.
Cell fractionation and biochemical pull down assay.
Non-Swiss Albino adult female mice (6–10 weeks old, 25–30 g) were used in this study. In all experiments, the sternomastoid muscle was labeled with BTX-biotin followed by a saturating dose of either a fluorescent or unlabeled streptavidin. Five days after the initial labeling, the sternomastoid muscle was exposed and subjected to different treatments (see above). At the end of each experiment, the sternomastoid muscle was removed and plasma and internal membranes were separated by minor modifications of the procedures described by Dombrowski et al. (1996). Sternomastoid muscles were removed, minced, and homogenized on ice a buffer containing 10 mm NaHCO3, pH 8.4, 250 mm sucrose, 5 mm NaN3, 1 mm EDTA, 10 mm KCl, 100 μm PMSF and protease inhibitor cocktail (Sigma P8340). Homogenates were centrifuged at 1300 × g for 10 min, giving a supernatant (S1) and a pellet (P1). P1 was homogenized and centrifuged at 1300 × g for 10 min, giving a supernatant (S2) and a pellet (P2). S2 was then combined with S1. P2 was resuspended in 0.5 m LiBr and 50 mm Tris at a concentration of 40 mg/ml and stirred using a magnetic bar on a stirring plate at 4°C for 6 h and was centrifuged at 10,000 × g for 30 min, giving a supernatant (S3) and a pellet (P3). S3 was further centrifuged at 53,000 × g for 1 h, giving a supernatant (S4) and a pellet (P4). S4 was centrifuged at 190,000 × g for 1 h, giving a supernatant S5 and a pellet P5. P5 was saved and used as the internal membrane fraction (IM). S1+S2 were centrifuged at 9000 × g for 10 min, giving a supernatant (S6) and a pellet (P6). P6 was discarded and S6 was spun at 190,000 × g for 90 min, giving a supernatant S7 and a pellet P7. P7 was combined with P4 and saved as the plasma membrane fraction. The plasma and internal membrane fractions were then lysed in RIPA buffer (50 mm Tris, pH 8.4, 150 mm NaCl, 1% NP40 and 0.5% sodium deoxycholate) for 30 min on ice with vigorous shaking and centrifuged at 10,000 rpm for 10 min. The same amounts of proteins (500 μg/ml) of the two fractions were incubated overnight at 4°C with RIPA-prewashed Neutravidin beads (Pierce). The Neutravidin beads were collected by brief centrifugation and washed three times with mRIPA buffer without sodium deoxycholate. The beads were resuspended in 30 μl of 2× LDS buffer (Invitrogen) and heated at 85°C for 10 min and released proteins were separated by SDS-NuPage gel electrophoresis on 4–12% (Invitrogen) and transferred onto PVDF membrane. The membranes were bathed in blocking solution for 1 h, incubated in rat AChR-α-subunit (MAB210, MMS-609R; Covance); or mouse AChR α-subunit (clone 26, BD Biosciences), antibody diluted at 1:5000 and 1:1000 respectively in PBST at 4°C overnight. The membranes were then incubated for 1 h in HRP-conjugated goat anti-rat or donkey anti-mouse secondary antibody at 1:20,000 (Jackson ImmunoResearch). After extensive washing, the membranes were incubated with SuperSignal West Femto Maximum Sensitivity Substrate (Pierce).
In another series of experiments, we arranged conditions so that only the internal receptors would be detected. Just before removing the muscle, it was bathed with unlabeled streptavidin to saturate all recycled nAChRs containing BTX-biotin that were on the cell surface. In this case only internalized nAChRs containing BTX-biotin should be pulled down using Neutravidin beads.
Results
Direct muscle stimulation or pharmacologically induced elevation of intracellular calcium concentration promotes the recycling of internalized AChR in vivo
In an effort to identify signaling molecules involved in the cycling of internalized nAChRs at the NMJ, we asked whether muscle electrical activity would promote the reinsertion of internalized nAChRs into synaptic sites. To test this idea, nAChRs in the sternomastoid muscle were sequentially labeled with BTX-biotin and then a single saturating dose of (green) strept-488 as described by Bruneau et al. (2005). Five days after the initial labeling (to allow for accumulation of a sizable amount of receptors that had been recycled), the sternomastoid muscle was bathed with strept-594 (red) to specifically label receptors that had lost their initial strept-488 tag while retaining BTX-biotin during the process of internalization and reinsertion, and superficial synapses were imaged immediately (time 0). The sternomastoid muscle was then directly stimulated by placing electrodes at either end of the muscle. A second dose of strept-594 (red) was then added to the sternomastoid muscle to label receptors that had recycled during the stimulation of the muscle, and the synapses were reimaged and their fluorescence intensities were quantified. In stimulated muscle, we found that the fluorescence intensity of recycled receptors at synaptic sites increased to 131 ± 7% (SD; n = 39) of their original fluorescence at time 0 (normalized at 100%) (Fig. 1B). In unstimulated muscles, however, the fluorescence intensity of recycled nAChRs remained unchanged [99 ± 8% (SD), n = 68; p < 0.0001], indicating that any loss of recycled receptors was compensated by an equivalent addition of newly recycled nAChRs (Fig. 1B), consistent with a previous report (Bruneau and Akaaboune, 2006). These results indicate that direct muscle stimulation promotes delivery of nAChRs from an internal pool onto the postsynaptic membrane, thus increasing the postsynaptic receptor density. In contrast, the number of newly synthesized receptors inserted at NMJs of stimulated muscle was not significantly different from unstimulated muscle (data not shown); indicating that muscle stimulation specifically promotes recycling of internalized nAChRs pool into synaptic sites (see Discussion).
Figure 1.

Muscle stimulation promotes receptor recycling of internalized nAChRs at NMJs in vivo. The sternomastoid muscle was labeled with BTX-biotin followed by a saturating dose of strept-488. Five days later, the animal was anesthetized and the sternomastoid muscle bathed with a saturating dose of strept-594 to specifically label the recycled receptors that had been inserted after the initial labeling. Labeled recycled receptors (Alexa 594) at superficial synapses were then imaged immediately (time 0) and the sternomastoid muscle was then stimulated, bathed with BAPTA-AM, or stimulated in the presence of BAPTA-AM for ∼7 h. At the end of the experiment a second saturating dose of strept-594 was added to the muscle to determine the amount of newly recycled nAChRs that have been reinserted during the time of the experiment and the same synapses were reimaged. A, Experimental protocol used in this study. B, Example of mouse neuromuscular junctions (labeled recycled receptors) of unstimulated, stimulated, and stimulated with BAPTA-AM that were imaged at time 0 and 8 h later. The total fluorescence intensity of labeled recycled AChRs (a measure of the total number of recycled AChRs) was expressed as 100% at the time 0. The fluorescence intensity at subsequent view was compared with the total fluorescence intensity of the junction at time 0. Pseudocolor images provided a linear representation of the density of recycled AChRs (white-yellow, high density; red-black, low density). C, Graph summarizing the recycled AChR insertion percentage obtained by the approach shown in B. Note that the number/density of recycled nAChRs at postsynaptic sites remains nearly constant over the 8 h in unstimulated innervated synapse (Bruneau et al., 2006) and that muscle stimulation increased significantly the reinsertion of internalized recycled receptors into the postsynaptic membrane of the NMJ whereas chelating intracellular calcium drastically decreased receptor recycling in both stimulated and unstimulated muscles. Each data point represents the mean percentage of fluorescence intensity (±SE) at each view. Unstim, Unstimulated; Stim, stimulated.
Because increase of electrical activity of the muscle triggers a significant elevation of intracellular Ca2+ (Chin and Allen, 1996; Posterino et al., 2000), we next asked whether the increase in AChR recycling is mediated by intracellular calcium. To test this, the sternomastoid muscle was labeled as usual and recycled nAChRs at superficial synapses were immediately imaged (time 0). The sternomastoid muscle was then bathed for ∼7 h with calcium-binding BAPTA-AM ester (to buffer intracellular calcium without depleting calcium outside the cells) (Tsien, 1981; Lee et al., 2009). At the end of the experiment a second dose of strep-Alexa 594 was added to label recycled nAChRs that had been inserted during the time period of the experiment and their fluorescence intensity was assayed. Following this treatment, we found that the fluorescence intensity of recycled receptors decreased to 80 ± 5% (SD; n = 38, p < 0.0001) of their original fluorescence at time 0 (normalized at 100%), and a similar result was obtained when the sternomastoid muscle was stimulated in the presence of BAPTA-AM (Fig. 1B,C). These results indicate that a change in intracellular calcium is required for the recycling of internalized nAChRs into the postsynaptic membrane.
In a second set of experiments, we monitored receptor recycling in the presence of thapsigargin (to raise cytosolic calcium concentration by blocking the ability of the cell to pump calcium into intracellular calcium stores) for the duration of the experiment (8 h) after which a second dose of red streptavidin was added to label nAChRs that had been inserted during the treatment. We found that fluorescence intensity increased to reach 126 ± 10% (SD; n = 35) of their original fluorescence at time 0 (normalized at 100%). Thus, blocking calcium uptake into intracellular stores mimics the effect of muscle stimulation on AChR recycling.
Most of recycled nAChRs are inserted into synaptic sites
To determine whether recycled receptors are inserted directly into the synaptic zone or into the nonsynaptic zone and subsequently move into the synaptic zone, we performed fluorescence recovery after photobleaching (FRAP) on the sternomastoid muscle of mice, in which recycled receptors-containing BTX-biotin were labeled with strept-594 (red). Superficial junctions were imaged and carefully traced with a low light intensity of an argon laser that was sufficient to cause bleaching of the fluorophore but not photo-unbinding of fluorescent streptavidin from biotin sites (Akaaboune et al., 2002). The muscle was then either directly stimulated or remained unstimulated for the duration of the experiment. If recycled nAChRs are inserted into the nonsynaptic zone, fluorescence recovery should be greater in stimulated compared with unstimulated muscles. We found that the recovery of fluorescence at the bleached junctions in stimulated muscle was very small (in some cases no recovery was seen), 1.5 ± 1% (SD; n = 13), and not significantly different from unstimulated muscles [1.6 ± 1% (SD), n = 10; p = 0.45]. A second dose of the same fluorescent streptavidin was then added at the end of the frame of the experiment. Under these conditions, we found that in stimulated muscle the fluorescence intensity significantly increased [23 ± 5% (SD), n = 13] compared with unstimulated muscles [12 ± 2.3% (SD), n = 9; p < 0.0001] (Fig. 2A,B). These results suggest that the majority of recycled receptors do not move from the non-synaptic zone but are inserted directly at synaptic sites.
Figure 2.
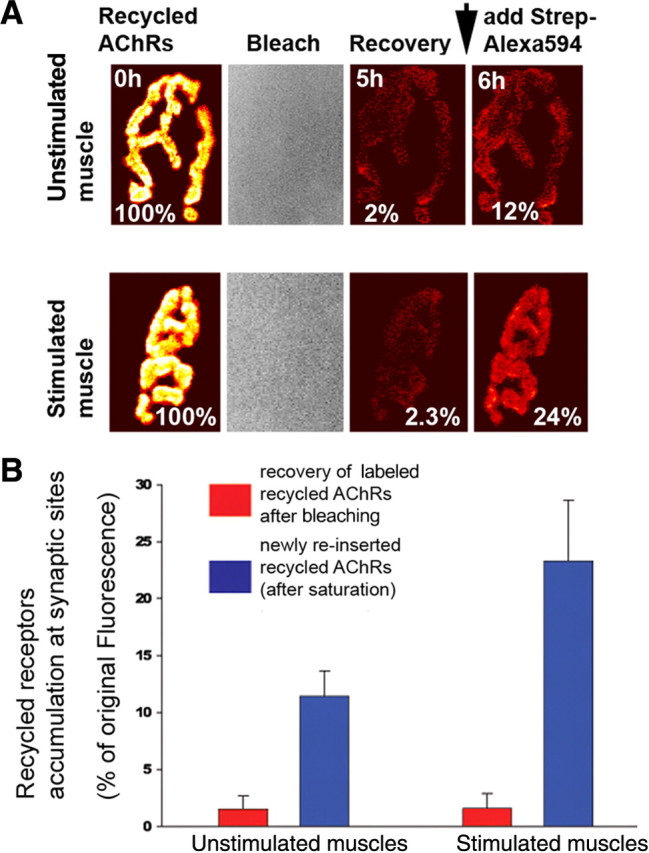
Most of recycled receptors are inserted directly into the postsynaptic sites. The sternomastoid muscle was labeled with BTX-biotin followed by a saturating dose of streptavidin and 5 d later, the animal was anesthetized and the sternomastoid muscle bathed with a saturating dose of strept-594 to specifically label recycled receptors (Bruneau et al., 2005). The fluorescence from individual junctions (labeled recycled receptors) was then removed with a low light intensity of an argon laser (sufficient to cause bleaching but not photo-unbinding) (Akaaboune et al., 2002), and the recovery of fluorescence intensity was monitored over 5 h. At the end of the experiment a second saturating dose of strept-594 was added to label recycled receptors that have been reinserted of the 5 h of the experiment. A, Examples of unstimulated and stimulated NMJs that were imaged at time 0 and immediately bleached and reimaged at 5 and 6 h (after adding a second dose of strept-594 to label recycled nAChRs that were inserted during the experiment). Pseudocolor images provided a linear representation of the density of recycled AChRs. B, Graph summarizes fluorescence recovery of labeled recycled nAChRs at bleached synapses and newly inserted recycled receptors at stimulated and unstimulated muscles. Each data point represents the mean percentage of the original fluorescence intensity before laser illumination (±SD).
Translocation of recycled receptors from the internal pool into the surface of the muscle cell
Next we used cell fractionation as a second method to study changes in intracellular and plasma membrane nAChRs. As a test of the specificity of our separation protocol, we first assessed the distribution of two antigenic markers, Na+/K+-ATPase and EEA1, which have been shown to be predominantly localized to the plasma and internal endosomal vesicle membranes respectively (Takeyasu et al., 1988; Williams et al., 2001; Laifenfeld et al., 2007). The Na+/K+-ATPase marker is highly enriched in the plasma membrane fractions whereas EEA1 is highly enriched in internal endosomal membrane fractions, indicating that there is little, if any contamination between those fractions (Fig. 3A).
Figure 3.

Translocation of internalized nAChRs from the internal pool to the surface muscle cell depends on the rise of intracellular calcium elevation. A, The sternomastoid muscle was labeled with BTX-biotin followed by a saturating dose of streptavidin and 5 d after the initial labeling, the sternomastoid muscle was removed and plasma and internal membranes were separated (see Materials and Methods). To show the specificity of the membrane fractionation procedure, plasma and internal membranes were subjected to immunoblotting using antibodies against markers of plasma (Na+/K+-ATPase) and endosomal internal membranes (EEA1), respectively. As shown in the representative blots, Na+/K+-ATPase is mainly detected in the plasma membrane while EEA1 is specifically detected in internal membrane. Analyses were performed on samples from two different innervated muscles [Inner m1 (innervated muscle1); Inner m2 (innervated muscle 2)]. B, To show biochemically that receptor recycled back into the cell membrane, the sternomastoid muscle was labeled with BTX-biotin followed by a single saturating dose of streptavidin and muscle cells were removed 5 d after the initial labeling. The AChR-BTX-biotin complexes were pulled down using beads coated with streptavidin and plasma and internal membrane fractions (separated as in A) were probed with anti-AChR α subunit antibody. C, The sternomastoid muscle was labeled as described above and 5 d after the initial labeling, the sternomastoid muscle was exposed and either stimulated or bathed with a calcium chelator BAPTA-AM for ∼7 h. At the end of each experiment, the sternomastoid muscle was removed and plasma and internal membranes were separated and probed with rat anti-AChR-α-subunit (MAB210); or mouse anti-AChR-α-subunit (clone 26, BD Biosciences). The input lanes were used to show that the same amount of nAChRs was probed in all experiments. Representative blots show that stimulation and BAPTA control the amount of nAChR in the internal pool. The labels below the lanes indicate whether the muscle was electrically stimulated (stim), treated with BAPTA-AM, or unstimulated (unstim). D, Representative blots show the assessment of the proportion of receptors in the internal pools by a different method. In this experiment, all surface receptors were saturated with unlabeled streptavidin just before extraction, so only internal receptors can be pulled down. At least three independent experiments were performed in each experiment.
To assess AChR translocation from internal to surface domains, the sternomastoid muscle was labeled with BTX-biotin followed by a saturating dose of fluorescent or unlabeled streptavidin and 5 d later the sternomastoid muscle was removed and the plasma membrane fractions (containing the recycled AChR pool) was separated from internal fractions (containing the internalized AChR pool). BTX-biotin-labeled nAChRs were then pulled down with Neutravidin-coated beads, and probed by Western blot with anti-AChR α subunit antibody. As shown in Figure 3B, there were intact α subunits in both the cell surface fraction and internal fraction. Next we examined the effect of directly elicited muscle twitching on translocation of internalized nAChRs onto the postsynaptic membrane. To do this, the labeled sternomastoid of a live animal was directly stimulated for ∼7 h. At the end of the experiment, the muscle was removed and the plasma and internal membranes were separated, and BTX-biotin-labeled nAChRs were then pulled down with Neutravidin-coated beads. As shown in Figure 3C, there is a significant increase in recycled receptors in the plasma membrane fraction, whereas internalized nAChRs were significantly decreased in stimulated muscle by comparison with unstimulated muscle. This result indicates that muscle stimulation induces translocation of internalized nAChRs from the internal pool to the cell surface. Conversely, when muscles were treated with calcium chelator BAPTA-AM, there was significant accumulation of nAChRs in the internal pool, indicating that translocation of nAChRs onto the cell surface is nearly abolished (Fig. 3C).
To further confirm these results, in a second set of experiments nAChRs of the sternomastoid muscle were labeled as usual and 5 d later, the muscle was exposed and directly stimulated for ∼7 h in presence or absence of BAPTA-AM. At the end of the experiment, the sternomastoid muscle was bathed with unlabeled streptavidin to saturate all recycled receptors tagged with BTX-biotin in the postsynaptic membrane. In this way only internalized receptors containing BTX-biotin were pulled down and immunoblotted with receptor antibody. In the stimulated muscle, few internalized receptors were accumulated in the internal pool compared with unstimulated muscles, indicating that muscle activity promoted translocation of receptors from the internal pool to the muscle cell surface. In contrast, when muscle cells were stimulated in the presence of calcium chelator BAPTA-AM, there was more intracellular accumulation of internalized nAChRs (Fig. 3D).
Expression and localization of Ca2+/CaM-dependent kinases at the NMJ
Having found that receptor cycling is dependent upon intracellular calcium concentration, we next asked whether CaMKII, a calcium-sensitive kinase, is involved in this process. We first wanted to know which CaMKII isoforms are expressed in sternomastoid muscle cells. To do this, muscle homogenates from the sternomastoid muscle were immunoblotted with an antibody recognizing all CaMKII isoforms (M176, Santa Cruz Biotechnology). Consistent with published results obtained from other muscle types, we observed four major bands. Based on their mobility, the band at 73 kDa likely corresponds to the muscle-specific β isoform, the band at 55–60 kDa likely corresponding to γ/δ isoforms, and the band migrating at 21–23 kDa, is likely the anchoring protein (αKAP) (Bayer et al., 1998; Damiani et al., 2003) (Fig. 4A).
Figure 4.

Expression and localization of CaMKII at the NMJ. A, Sternomastoid muscle extract proteins were subjected to SDS-PAGE, transferred to nitrocellulose membrane and probed with a pan-polyclonal anti-CaMKII antibody that has been previously shown to be cross-reactive with all isoforms. Three major bands are labeled with Greek letters to indicate the likely molecule recognized (based on the work of Damiani et al., 2003). The fourth band is labeled with an asterisk to indicate either unknown or degradation product of CaMKII isoform. B–D, The sternomastoid muscle was electroporated with CaMKIIβm-GFP, CaMKIIγ-GFP, or α-syntrophin-GFP and 1 week later the mouse was perfused with 2% PFA, and the sternomastoid muscle bathed with BTX-Alexa 594 to label nAChRs. B, Confocal images show that CaMKIIγ-GFP is poorly expressed at synaptic sites. A faint GFP fluorescence was also seen in the perisynaptic zone (dashed white lines), which is illustrated in the enlarged box area. C, A representative image of CaMKIIβm-GFP expressed at the NMJ, where precisely colocalizes with nAChRs at the crests of the junctional folds (see enlarged box). D, A representative NMJ of a sternomastoid muscle electroporated with α-syntrophin-GFP fusion protein, showing that α syntrophin is present in both the troughs and the crests of the junctional folds. Boxed areas are enlarged part of the synapse branch for more details.
Next we asked which of those proteins are localized at the NMJ. To do this, the sternomastoid muscle was fixed, permeabilized, and immunostained with antibody against CaMKIIγ, δ and β isoforms (see Materials and Methods). For all three antibodies, CaMKII labeling was present in both presynaptic nerve terminal and postsynaptic membrane (supplemental Fig. S1, available at www.jneurosci.org as supplemental material), so it would be difficult to determine the precise localization of these isoforms. In addition, these antibodies cross react between each other, which make it difficult to determine which isoforms are concentrated at the postsynaptic cell. To do this, the sternomastoid muscles were electroporated with GFP-fusions of each of the kinases and 1 week later, the sternomastoid muscle was bathed with BTX-Alexa594 to label nAChRs and doubly labeled NMJs were imaged by confocal microscopy. From the imaging of GFP constructs, it appears that CaMKIIγ-GFP (Fig. 4B) and CaMKIIδ-GFP (data not shown) are reasonably well localized at synaptic sites, just not as clearly as muscle-specific CaMKIIβm-GFP (Fig. 4C). Higher resolution images show that CaMKIIβm-GFP is restricted to the crest of folds where it precisely colocalizes with nAChRs in contrast to α syntrophin (Fig. 4D), a protein that is present in both the troughs and the crests of the junctional folds (Kramarcy and Sealock, 2000). When we compared the amounts of recycled receptors at synapses of electroporated and nonelectroporated muscle fibers with CaMKIIβm (highly expressed at the synapse compared with other isoforms) within the same sternomastoid muscle, we found that there was no significant difference in the amount of recycled receptors between electroporated and nonelectroporated muscle fibers (supplemental Fig. S2, available at www.jneurosci.org as supplemental material). This suggests that perhaps there is already a saturating amount of CaMKII at the synapse or perhaps there are other upstream signals to CaMKII that are the rate limiting step.
CaMKII activity promotes receptor recycling at the neuromuscular junction in vivo
Having shown that CaMKII isoforms are expressed in sternomastoid muscle cells and that the CaMKIIβm-GFP splice variant is highly concentrated at synapses, we next investigated whether inhibition of CaMKII activity would prevent AChR recycling. Recycled receptors were relabeled with strept-594 as usual and superficial synapses were immediately imaged (time 0). Muscle fibers were then bathed with KN93, an inhibitor of CaMKII activity, or with the inactive analog KN92 (Hudmon and Schulman, 2002; Tavi et al., 2003) for ∼7 h while being stimulated with extracellular electrodes. At the end of the experiment, a second dose of strept-594 was then added to label recycled receptors and their fluorescence intensity was assayed. As shown in Figure 5, B and C, the fluorescence intensity decreased to 77 ± 12% (SD; n = 39) of original fluorescence at time 0. Similar results were obtained when KN62 (a distinct chemical inhibitor of CaMKII) [82 ± 5% (SD), n = 11)], thapsigargin and KN93 [75 ± 9% (SD), n = 20], or unstimulated and KN93 [80 ± 12% (SD), n = 39] were added to the muscle. However, in muscles treated with derivative KN92 (a molecule very similar to KN93 but with no inhibitory activity), the fluorescence intensity was similar to untreated synapses [98 ± 5% (SD); n = 15].
Figure 5.

Receptor recycling is dependent upon CaMKII activity. Sternomastoid muscles were bathed with BTX-biotin/strep Alexa 488 and relabeled after 5 d with strept-594 to label specifically the recycled nAChRs and then stimulated in presence or absence of CaMKII inhibitor (KN93) or in the presence of KN92 (inactive analog). At the end of the experiment, the sternomastoid muscle was bathed with strept-594 to label recycled nAChRs that have been reinserted during the experiment. A, Schematic diagram of the experimental protocol used in this study. B, Example of labeled recycled receptors with Alexa 594 at NMJs that were imaged immediately (time 0), and reimaged after 8 h of a mouse whose sternomastoid muscle has been stimulated in presence or absence of KN93. The fluorescence intensity of labeled recycled nAChRs was expressed as 100% at the time 0. Fluorescence signals show that electrical muscle stimulation promotes the reinsertion of internalized nAChRs and significantly increases their number and density at the postsynaptic membrane. However, addition of CaMKII inhibitor depresses receptor recycling both in stimulated and unstimulated muscles and thus lowers their number and density at the postsynaptic sites. The views of the same synapse are displayed on the same intensity scale. C, Graph summarizing data obtained from many synapses with approach shown in B. Each data point represents the mean percentage of fluorescence intensity (±SE) at each view.
To examine the effect of KN93 treatment on the removal rate of preexisting receptors (not yet internalized AChRs), the sternomastoid muscle was labeled with a nonsaturating dose of BTX-biotin followed with strept-488 (green) and superficial synapses were imaged immediately (time 0) and reimaged 8 h later and the loss of fluorescence was assayed. We found that the fluorescence intensity of preexisting nAChRs (not yet internalized) decreased to 91 ± 5% (SD; n = 20) of original fluorescence over 8 h and was not significantly different from untreated muscles [94 ± 5% (SD); n = 30)], or those treated with KN92 [94 ± 5% (SD); n = 10)]. These results suggest that CaMKII does not play a role in receptor internalization, and that endocytosis and recycling are controlled by separate mechanisms.
If CaMKII activity promotes AChR translocation to the surface then when this enzyme is inhibited, receptors in the internal pool should be increased. To test this, 5 d after the initial labeling of nAChRs with BTX-biotin/strept the sternomastoid muscle was electrically stimulated in the presence of KN93 for 8 h and at the end of the experiment, the muscle was separated into plasma membrane and internal fractions; nAChRs containing BTX-biotin were pulled down with Neutravidin-coated beads and immunoblotted with anti-AChR subunit α. As shown in Figure 6, the amount of surface-associated recycled nAChRs is markedly reduced by inhibition of CaMKII activity, whereas the number of nAChRs that accumulated intracellularly is markedly increased compared with control (Fig. 6B). These results were further confirmed in the alternative assay that detects only internalized nAChRs (Fig. 6C). Changes in the amount of receptors by the various conditions of treatments were not due to rate of dissociation of BTX-biotin from receptors as the total amount of Biotin-BTX-AChRs in the muscle was not changed (supplemental Fig. S3, available at www.jneurosci.org as supplemental material).
Figure 6.

CaMKII activity promotes the translocation of internalized receptors from the internal pool into the postsynaptic membrane. The sternomastoid muscle of a live animal was first labeled with BTX-biotin followed by a saturating dose of streptavidin and 5 d later the muscle was either exposed to CaMKII inhibitor (KN93) alone, stimulated in the presence of CaMKII inhibitor, or stimulated only for 8 h. At the end of the experiment, muscles were removed and nAChRs-BTX-biotin were pulled down with Neutravidin-coated beads from plasma and internal membrane fractions and subjected to immunoblotting using antibody against AChR subunit α. A, Experimental protocol used in this study. B, Blots show that treated stimulated or unstimulated sternomastoid muscles with CaMKII inhibitor exhibit more intracellular accumulation of internalized nAChRs than untreated stimulated and unstimulated muscles. However, in the plasma membrane fractions the amount of pulled down recycled nAChRs was less in the presence of CaMKII inhibitor than untreated muscles. These results indicate that inhibition of CaMKII causes the accumulation of internalized nAChRs by preventing them from recycling, whereas direct muscle stimulation promotes their delivery onto the surface muscle cell. C, At the end of muscle treatments, the sternomastoid muscle was bathed with unlabeled streptavidin to saturate all recycled AChR pool in the membrane and only internalized nAChRs containing free BTX-biotin were pulled down. Blots show most of internalized nAChRs remain in the internal pool when muscles were treated with CaMKII inhibitor. At least three independent experiments were performed in each experiment.
Muscle activity reroutes internalized nAChRs from the degradative pathway to the recycling pathway in denervated NMJs via CaMKII activity
Finally we examined the effect of CaMKII activity on receptor recycling at denervated synapses where most of the internalized nAChRs are processed for degradation; leaving few of them to recycle back (Akaaboune et al., 1999; Bruneau and Akaaboune, 2006). We asked whether direct muscle stimulation is able to prevent internalized receptors from entering the degradation pathway by switching them to the recycling pathway. To this end, denervated sternomastoid muscle was labeled with BTX-biotin/strept-488 (green) and 3 d later, a second strept-594 (red) was added to label recycled nAChRs. The muscle was then stimulated continuously for ∼7 h and the number of recycled receptors (AChRs that had been reinserted during stimulation) was determined. As expected, in unstimulated muscle few internalized receptors were recycled onto the postsynaptic membrane, as shown by the decrease in fluorescence intensity (Fig. 7B,C), consistent with a previous report (Bruneau and Akaaboune, 2006). However, in stimulated muscles, the fluorescence intensity of recycled nAChRs at synaptic sites was substantially increased to reach 127 ± 6% (SD; n = 25) of fluorescence at time 0 (normalized at 100%) (Fig. 7B,C). This result indicates that muscle stimulation reroutes internalized nAChRs from being degraded and promotes their delivery onto the postsynaptic membrane. In contrast, when denervated sternomastoid muscle was stimulated and bathed with BAPTA-AM for ∼7 h, we found that the fluorescence intensity of recycled receptors decreased to 84 ± 4% (SD; n = 12) of their original fluorescence at time 0. Similarly when denervated sternomastoid muscle was stimulated in the presence of CaMKII inhibitor KN93 the fluorescence intensity of recycled nAChRs decreased to 77% ±13 (SD; n = 25) compared with that of fluorescent nAChRs at the NMJ of stimulated muscles (Fig. 7B,C). These data indicate that calcium-dependent AChR recycling is mediated by CaMKII activity.
Figure 7.

Muscle stimulation enhances recycled nAChRs at denervated NMJs. Denervated sternomastoid muscles (6–8 d after denervation) were labeled with BTX-biotin/strept-488 and 3 d later the sternomastoid muscle was exposed and bathed with a saturating dose of strept-594 to label recycled nAChRs that had been inserted after the initial labeling. Labeled recycled receptors with Alexa 594 at NMJs were imaged immediately and the sternomastoid muscle was either stimulated, or stimulated in the presence of CaMKII inhibitors. At the end of the experiment, a second dose of strept-594 was added to label recycled receptors that had been reinserted during the experiment and the same synapses were reimaged and their fluorescence intensity was assayed. A, The experimental protocol used in this study. B, Example of mouse NMJs that have been imaged before and after treatment. After 8 h, fluorescence intensity of labeled recycled receptors in denervated muscle was less than innervated synapses; however, when denervated muscle was chronically stimulated, the fluorescence was largely increased, indicating that stimulation enhances significantly the recycling of internalized nAChRs. The increased recycled receptors were abolished in the presence of CaMKII inhibitor. C, Graph summarizing the fluorescence measurements obtained from many synapses. D, E, Blots showing that most internalized nAChRs are shifted from the intracellular pool into the postsynaptic membrane when the muscle is stimulated, but remain in the internal pool when muscles were treated with CaMKII inhibitor. Each data point represents the mean percentage of fluorescence intensity (±SE) at each view. For Western blots, at least two independent experiments were performed for each experiment.
The effect of muscle stimulation on recycled receptors at denervated synapses was confirmed by pull down assays. Denervated sternomastoid muscles were labeled with BTX-Biotin/strept-Alexa and 3 d later recycled nAChRs in the plasma membrane were separated from internalized AChR-BTX-biotin in the internal pool and probed with anti-AChR antibody. When the denervated muscle was directly stimulated for 8 h, most of the internalized nAChRs were translocated back into the postsynaptic membrane while few internalized nAChRs remained in the internal pool. Conversely, when denervated muscles were stimulated in the presence of CaMKII inhibitors, most receptors were retained in the internal fraction (Fig. 7D,E). These data indicate that CaMKII activity is required for recycling of internalized nAChRs into the postsynaptic membrane. CaMKII inhibition has the same effect on both stimulated denervated and innervated muscles, suggesting that inhibition of CaMKII in the presynaptic nerve terminal is unlikely to be involved in receptor recycling in the postsynaptic cell.
Discussion
The work presented here brings new insight into the signaling mechanisms involved in the cycling of nAChRs at the NMJ in vivo. We demonstrate that, following internalization, reinsertion of internalized nAChRs into the postsynaptic membrane is sensitive to a rise of intracellular calcium. This calcium-dependent recycling pathway is mediated by CaMKII activity. Specifically, when intracellular calcium is chelated or CaMKII activity is inhibited, receptor recycling is depressed. In addition, this work demonstrates that of all CaMKII isoforms expressed by the muscle, only CaMKIIβm is highly expressed at the postsynaptic site and tightly clustered with synaptic nAChRs.
The dynamics of AChRs at the neuromuscular junction was extensively studied using different methods of labeling. When fluorescent bungarotoxin was used as a ligand to monitor receptor turnover rates at fully functioning synapses, the half-life was quite long (∼9–14 d) (Akaaboune et al., 1999). However, when synaptic activity was totally blocked, receptor half-life decreased substantially to several hours. These estimates were based on the assumption that once AChRs are internalized, they are targeted to degradation pathway, and did not consider the possibility those receptors can continuously recycle back to the postsynaptic membrane with their BTX tag. With the discovery that a significant amount of internalized AChRs recycle back and intermingle with preexisting receptors at the same synapse, the lifetimes of preexisting AChR was estimated to 5 d and recycled AChRs to 1 d (Bruneau and Akaaboune, 2006; Bruneau et al., 2009). However, it remains unclear whether the binding of BTX to receptors, which also silences them, can affect the turnover rate of receptor pools at the synapse. One argument against this possibility is that when few receptors are labeled (as long as the neuromuscular junction activity is not totally blocked and the threshold of its safety factor is not exceeded) the turnover rate of receptors is very slow. At present there are no available means to track receptor dynamics without blocking them with the ligand bungarotoxin.
In the current studies, the existence of an intracellular reserve pool, which is apparently available to recycle into the postsynaptic membrane in response to synaptic activity, could constitute a source for the steady-state of the postsynaptic receptor density and therefore the maintenance of a high safety factor for synaptic transmission. While 20–30% change in the number of recycled AChRs over ∼7 h at healthy synapses (which have large safety factor) might not significantly affect synaptic transmission, they can have a dramatic effect on the efficacy of synaptic responses when receptor number is compromised by diseases. Since a significant number of recycled nAChRs contributes to the postsynaptic density, it is possible that a defect in the receptor recycling pathway can have a dramatic effect on the efficacy of synaptic transmission by lowering the safety factor of the NMJ. Consistent with this, recent work has shown that impairment of recycling of junctional receptors leads to movement disorders in dilute mice (Röder et al., 2008). It would be of interest to know whether other NMJ components also recycle to synaptic sites or whether recycling is restricted to AChR. Since there is no way to distinguish between recycled, preexisting, and newly synthesized proteins, it is not possible to determine whether the recycling of these proteins with nAChRs occur at the NMJ. This issue warrants to be further investigated.
Although a rise in intracellular calcium levels is critical for promoting the recycling of vesicles containing internalized nAChRs into the postsynaptic membrane, the insertion rate of newly synthesized nAChRs remains largely unaffected. These observations suggest the presence of an intracellular reservoir of internalized receptors that can allow the cell to maintain a high postsynaptic receptor density. Indeed the existence of an intracellular receptor pool distinct from newly synthesized nAChRs has been described in cultured myotubes (Devreotes and Fambrough, 1975; Kumari et al., 2008). The clamping of intracellular calcium with BAPTA-AM significantly attenuates the recycling of internalized receptors (Figs. 1, 3). This observation is consistent with previous reports showing that recycling from the endocytic compartment to the plasma membrane of other cell types is enhanced by elevations of intracellular calcium concentration reference. It has been reported that recycling endosomes into spines by myosin Vb is dependent on intracellular Ca2+ (Wang et al., 2008). Such mechanism has been shown to play a critical role during synaptic plasticity at central synapses (Bear and Malenka, 1994; Ehlers, 2000; Park et al., 2004). Calcium is also involved in the formation of agrin-induced receptor clusters on the surface of cultured muscle cells (Megeath and Fallon, 1998).
Our present study suggests that the majority of recycled nAChRs are directly inserted into synaptic sites. This implies that the contribution of recycled receptors from the nonsynaptic zone to synaptic sites is limited (Fig. 2), which raises the question of why internalized nAChRs from synaptic sites are predominantly reinserted into the same sites. It is possible that internalized nAChRs may have a tag-memory that allows them to recycle to sites from where they were removed. Consistent with this idea, it has been reported for example that P2X receptors' memory for extracellular Ca2+ can speed their recovery from desensitization (Cook et al., 1998). It is also conceivable that-specific cues in the synaptic region of the basal lamina might exist and guide recycled receptors to their original synaptic sites, whereas molecules that are concentrated in the extrasynaptic zone of the basal lamina might repel them. Consistent with this idea, it has been shown that axons regenerate to reinnervate denervated skeletal muscle fibers precisely at original synaptic sites (Sanes et al., 1978) and internalized nAChRs are recycled at synaptic sites of denervated muscle cells (Fig. 7). Analogously, a similar theoretical model was proposed for AMPA receptor cycling at mammalian synapses involving CaMKII (Lisman and Zhabotinsky, 2001).
We report that calcium exerts its effect on the cycling of nAChRs through CaMKII. In many situations, CaMKII does not need signals other than calcium for its activation (De Koninck and Schulman, 1998). In muscle cells, CaMKII can be activated by calcium from different sources, including influx and Ca2+ released from internal stores (He et al., 2000). In addition, it has been shown that direct elicited muscle twitching increases CaMKII activity (Tavi et al., 2003). In this work we demonstrate that CaMKII inhibitors KN62 or KN93 but not the inactive analog KN92 depress receptor recycling and promote accumulation of internalized nAChRs in the intracellular pool. While the mechanism by which CaMKII activity affects AChR recycling is not known, it is possible that CaMKII may modulate proteins involved in the sorting, targeting, and/or fusion of endocytic vesicles with the postsynaptic membrane. In fact, calmodulin antagonists were able to block endosome fusion in a macrophage cell line (Colombo et al., 1997) and suppress transferrin receptor recycling without affecting receptor-mediated endocytosis in rat reticulocytes (Grasso et al., 1990). Similar to what we have observed at the NMJ, changes in intracellular Ca2+ levels in the postsynaptic cell at central synapses have been shown to induce the activation of CaMKII (Malenka et al., 1989; Yamagata et al., 2009), which in turn causes an enhancement of membrane receptor trafficking and recycling (Derkach et al., 1999; Malinow and Malenka, 2002).
Another possible mechanism for CaMKII at the NMJ is that it could act directly on cytoskeletal proteins that are involved in delivery of receptors to synaptic sites. The presence of muscle-specific CaMKIIβm, and to some extent γ and δ isoforms at synaptic sites, reinforces the idea that they may act as scaffolding proteins by tethering and arranging synaptic proteins into signaling complexes. Confocal images show that CaMKIIβm is precisely colocalized with nAChRs receptors at the crests of the junctional folds (Fertuck and Salpeter, 1976) (Fig. 4D,E), similar to rapsyn distribution, but different from α syntrophin, which is equally distributed in both the troughs and the crests of the junctional folds (Kramarcy and Sealock, 2000). This observation indicates that CaMKII isoforms may play a role in receptor stability at synaptic sites in addition to playing a role in receptor recycling. It is possible that CaMKII at synapses could phosphorylate receptors and/or substrate proteins that are involved in anchoring and/or clustering receptors in the right position directly opposite to active zones (transmitter release sites). Consistent with this, at the NMJ in Drosophila, CaMKII has been shown to regulate synaptic structure and function (Koh et al., 1999; Beumer et al., 2002; Haghighi et al., 2003; Pratt et al., 2003) and at central synapses, CaMKII interacts with many proteins of the postsynaptic apparatus, including ion channels such as NMDA receptor and voltage-sensitive calcium channels (Hudmon et al., 2005).
Our work has relevance for neuromuscular diseases such as myasthenia gravis that are characterized by a low density and number of nAChRs in the postsynaptic membrane and have devastating effects on synaptic transmission. It is tempting to speculate that mechanisms for enhancing receptor recycling (through muscle stimulation and CaMKII activation, for instance) could provide valuable means for shifting receptors into the postsynaptic density and attenuating the deleterious effects that might arise from receptor loss.
Footnotes
This work was supported by the National Institutes of Health (M.A.) and partially by the National Science Foundation (M.A). We thank the members of the M. Akaaboune, J. Kuwada, R. Hume, C. Collins, and H. Xu laboratories for thoughtful comments and discussions. We are also grateful to Drs. Amy Chang, Richard Hume, Orie Shafer, and Hans Brenner for the input and comments on this manuscript.
References
- Akaaboune M, Culican SM, Turney SG, Lichtman JW. Rapid and reversible effects of activity on acetylcholine receptor density at the neuromuscular junction in vivo. Science. 1999;286:503–507. doi: 10.1126/science.286.5439.503. [DOI] [PubMed] [Google Scholar]
- Akaaboune M, Grady RM, Turney S, Sanes JR, Lichtman JW. Neurotransmitter receptor dynamics studied in vivo by reversible photo-unbinding of fluorescent ligands. Neuron. 2002;34:865–876. doi: 10.1016/s0896-6273(02)00739-0. [DOI] [PubMed] [Google Scholar]
- Balice-Gordon RJ, Lichtman JW. Long-term synapse loss induced by focal blockade of postsynaptic receptors. Nature. 1994;372:519–524. doi: 10.1038/372519a0. [DOI] [PubMed] [Google Scholar]
- Bayer KU, Harbers K, Schulman H. alphaKAP is an anchoring protein for a novel CaM kinase II isoform in skeletal muscle. EMBO J. 1998;17:5598–5605. doi: 10.1093/emboj/17.19.5598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bear MF, Malenka RC. Synaptic plasticity: LTP and LTD. Curr Opin Neurobiol. 1994;4:389–399. doi: 10.1016/0959-4388(94)90101-5. [DOI] [PubMed] [Google Scholar]
- Beumer K, Matthies HJ, Bradshaw A, Broadie K. Integrins regulate DLG/FAS2 via a CaM kinase II-dependent pathway to mediate synapse elaboration and stabilization during postembryonic development. Development. 2002;129:3381–3391. doi: 10.1242/dev.129.14.3381. [DOI] [PubMed] [Google Scholar]
- Bredt DS, Nicoll RA. AMPA receptor trafficking at excitatory synapses. Neuron. 2003;40:361–379. doi: 10.1016/s0896-6273(03)00640-8. [DOI] [PubMed] [Google Scholar]
- Bruneau EG, Akaaboune M. The dynamics of recycled acetylcholine receptors at the neuromuscular junction in vivo. Development. 2006;133:4485–4493. doi: 10.1242/dev.02619. [DOI] [PubMed] [Google Scholar]
- Bruneau E, Sutter D, Hume RI, Akaaboune M. Identification of nicotinic acetylcholine receptor recycling and its role in maintaining receptor density at the neuromuscular junction in vivo. J Neurosci. 2005;25:9949–9959. doi: 10.1523/JNEUROSCI.3169-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruneau EG, Esteban JA, Akaaboune M. Receptor-associated proteins and synaptic plasticity. FASEB J. 2009;23:679–688. doi: 10.1096/fj.08-107946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin ER, Allen DG. The role of elevations in intracellular [Ca2+] in the development of low frequency fatigue in mouse single muscle fibres. J Physiol. 1996;491:813–824. doi: 10.1113/jphysiol.1996.sp021259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colombo MI, Beron W, Stahl PD. Calmodulin regulates endosome fusion. J Biol Chem. 1997;272:7707–7712. doi: 10.1074/jbc.272.12.7707. [DOI] [PubMed] [Google Scholar]
- Cook SP, Rodland KD, McCleskey EW. A memory for extracellular Ca2+ by speeding recovery of P2X receptors from desensitization. J Neurosci. 1998;18:9238–9244. doi: 10.1523/JNEUROSCI.18-22-09238.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damiani E, Sacchetto R, Salviati L, Margreth A. Two splice variants of CaMKII-anchoring protein are present in the sarcoplasmic reticulum of rabbit fast-twitch muscle. Biochem Biophys Res Commun. 2003;302:73–83. doi: 10.1016/s0006-291x(03)00110-4. [DOI] [PubMed] [Google Scholar]
- De Koninck P, Schulman H. Sensitivity of CaM kinase II to the frequency of Ca2+ oscillations. Science. 1998;279:227–230. doi: 10.1126/science.279.5348.227. [DOI] [PubMed] [Google Scholar]
- Derkach V, Barria A, Soderling TR. Ca2+/calmodulin-kinase II enhances channel conductance of alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionate type glutamate receptors. Proc Natl Acad Sci U S A. 1999;96:3269–3274. doi: 10.1073/pnas.96.6.3269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devreotes PN, Fambrough DM. Acetylcholine receptor turnover in membranes of developing muscle fibers. J Cell Biol. 1975;65:335–358. doi: 10.1083/jcb.65.2.335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dombrowski L, Roy D, Marcotte B, Marette A. A new procedure for the isolation of plasma membranes, T tubules, and internal membranes from skeletal muscle. Am J Physiol. 1996;270:E667–E676. doi: 10.1152/ajpendo.1996.270.4.E667. [DOI] [PubMed] [Google Scholar]
- Ehlers MD. Reinsertion or degradation of AMPA receptors determined by activity-dependent endocytic sorting. Neuron. 2000;28:511–525. doi: 10.1016/s0896-6273(00)00129-x. [DOI] [PubMed] [Google Scholar]
- Ehlers MD, Augustine GJ. Cell signalling. Calmodulin at the channel gate. Nature. 1999;399:105, 107–108. doi: 10.1038/20079. [DOI] [PubMed] [Google Scholar]
- Fertuck HC, Salpeter MM. Quantitation of junctional and extrajunctional acetylcholine receptors by electron microscope autoradiography after 125I-alpha-bungarotoxin binding at mouse neuromuscular junctions. J Cell Biol. 1976;69:144–158. doi: 10.1083/jcb.69.1.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grasso JA, Bruno M, Yates AA, Wei LT, Epstein PM. Calmodulin dependence of transferrin receptor recycling in rat reticulocytes. Biochem J. 1990;266:261–272. doi: 10.1042/bj2660261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haghighi AP, McCabe BD, Fetter RD, Palmer JE, Hom S, Goodman CS. Retrograde control of synaptic transmission by postsynaptic CaMKII at the Drosophila neuromuscular junction. Neuron. 2003;39:255–267. doi: 10.1016/s0896-6273(03)00427-6. [DOI] [PubMed] [Google Scholar]
- Hayashi Y, Shi SH, Esteban JA, Piccini A, Poncer JC, Malinow R. Driving AMPA receptors into synapses by LTP and CaMKII: requirement for GluR1 and PDZ domain interaction. Science. 2000;287:2262–2267. doi: 10.1126/science.287.5461.2262. [DOI] [PubMed] [Google Scholar]
- He X, Yang F, Xie Z, Lu B. Intracellular Ca(2+) and Ca(2+)/calmodulin-dependent kinase II mediate acute potentiation of neurotransmitter release by neurotrophin-3. J Cell Biol. 2000;149:783–792. doi: 10.1083/jcb.149.4.783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudmon A, Schulman H. Structure-function of the multifunctional Ca2+/calmodulin-dependent protein kinase II. Biochem J. 2002;364:593–611. doi: 10.1042/BJ20020228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudmon A, Schulman H, Kim J, Maltez JM, Tsien RW, Pitt GS. CaMKII tethers to L-type Ca2+ channels, establishing a local and dedicated integrator of Ca2+ signals for facilitation. J Cell Biol. 2005;171:537–547. doi: 10.1083/jcb.200505155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy MJ, Ehlers MD. Organelles and trafficking machinery for postsynaptic plasticity. Annu Rev Neurosci. 2006;29:325–362. doi: 10.1146/annurev.neuro.29.051605.112808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh YH, Popova E, Thomas U, Griffith LC, Budnik V. Regulation of DLG localization at synapses by CaMKII-dependent phosphorylation. Cell. 1999;98:353–363. doi: 10.1016/s0092-8674(00)81964-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramarcy NR, Sealock R. Syntrophin isoforms at the neuromuscular junction: developmental time course and differential localization. Mol Cell Neurosci. 2000;15:262–274. doi: 10.1006/mcne.1999.0823. [DOI] [PubMed] [Google Scholar]
- Kumari S, Borroni V, Chaudhry A, Chanda B, Massol R, Mayor S, Barrantes FJ. Nicotinic acetylcholine receptor is internalized via a Rac-dependent, dynamin-independent endocytic pathway. J Cell Biol. 2008;181:1179–1193. doi: 10.1083/jcb.200709086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laifenfeld D, Patzek LJ, McPhie DL, Chen Y, Levites Y, Cataldo AM, Neve RL. Rab5 mediates an amyloid precursor protein signaling pathway that leads to apoptosis. J Neurosci. 2007;27:7141–7153. doi: 10.1523/JNEUROSCI.4599-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SJ, Escobedo-Lozoya Y, Szatmari EM, Yasuda R. Activation of CaMKII in single dendritic spines during long-term potentiation. Nature. 2009;458:299–304. doi: 10.1038/nature07842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lichtman JW, Magrassi L, Purves D. Visualization of neuromuscular junctions over periods of several months in living mice. J Neurosci. 1987;7:1215–1222. doi: 10.1523/JNEUROSCI.07-04-01215.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisman JE, Zhabotinsky AM. A model of synaptic memory: a CaMKII/PP1 switch that potentiates transmission by organizing an AMPA receptor anchoring assembly. Neuron. 2001;31:191–201. doi: 10.1016/s0896-6273(01)00364-6. [DOI] [PubMed] [Google Scholar]
- Lu J, Helton TD, Blanpied TA, Rácz B, Newpher TM, Weinberg RJ, Ehlers MD. Postsynaptic positioning of endocytic zones and AMPA receptor cycling by physical coupling of dynamin-3 to Homer. Neuron. 2007;55:874–889. doi: 10.1016/j.neuron.2007.06.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malenka RC, Kauer JA, Perkel DJ, Mauk MD, Kelly PT, Nicoll RA, Waxham MN. An essential role for postsynaptic calmodulin and protein kinase activity in long-term potentiation. Nature. 1989;340:554–557. doi: 10.1038/340554a0. [DOI] [PubMed] [Google Scholar]
- Malinow R, Malenka RC. AMPA receptor trafficking and synaptic plasticity. Annu Rev Neurosci. 2002;25:103–126. doi: 10.1146/annurev.neuro.25.112701.142758. [DOI] [PubMed] [Google Scholar]
- Martinez-Pena y Valenzuela I, Hume RI, Krejci E, Akaaboune M. In vivo regulation of acetylcholinesterase insertion at the neuromuscular junction. J Biol Chem. 2005;280:31801–31808. doi: 10.1074/jbc.M502874200. [DOI] [PubMed] [Google Scholar]
- Megeath LJ, Fallon JR. Intracellular calcium regulates agrin-induced acetylcholine receptor clustering. J Neurosci. 1998;18:672–678. doi: 10.1523/JNEUROSCI.18-02-00672.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park M, Penick EC, Edwards JG, Kauer JA, Ehlers MD. Recycling endosomes supply AMPA receptors for LTP. Science. 2004;305:1972–1975. doi: 10.1126/science.1102026. [DOI] [PubMed] [Google Scholar]
- Petrini EM, Lu J, Cognet L, Lounis B, Ehlers MD, Choquet D. Endocytic trafficking and recycling maintain a pool of mobile surface AMPA receptors required for synaptic potentiation. Neuron. 2009;63:92–105. doi: 10.1016/j.neuron.2009.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Posterino GS, Lamb GD, Stephenson DG. Twitch and tetanic force responses and longitudinal propagation of action potentials in skinned skeletal muscle fibres of the rat. J Physiol. 2000;527:131–137. doi: 10.1111/j.1469-7793.2000.t01-2-00131.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pratt KG, Watt AJ, Griffith LC, Nelson SB, Turrigiano GG. Activity-dependent remodeling of presynaptic inputs by postsynaptic expression of activated CaMKII. Neuron. 2003;39:269–281. doi: 10.1016/s0896-6273(03)00422-7. [DOI] [PubMed] [Google Scholar]
- Röder IV, Petersen Y, Choi KR, Witzemann V, Hammer JA, 3rd, Rudolf R. Role of Myosin Va in the plasticity of the vertebrate neuromuscular junction in vivo. PLoS One. 2008;3:e3871. doi: 10.1371/journal.pone.0003871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sagara Y, Inesi G. Inhibition of the sarcoplasmic reticulum Ca2+ transport ATPase by thapsigargin at subnanomolar concentrations. J Biol Chem. 1991;266:13503–13506. [PubMed] [Google Scholar]
- Sanes JR, Marshall LM, McMahan UJ. Reinnervation of muscle fiber basal lamina after removal of myofibers. Differentiation of regenerating axons at original synaptic sites. J Cell Biol. 1978;78:176–198. doi: 10.1083/jcb.78.1.176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schworer CM, Rothblum LI, Thekkumkara TJ, Singer HA. Identification of novel isoforms of the delta subunit of Ca2+/calmodulin-dependent protein kinase II. Differential expression in rat brain and aorta. J Biol Chem. 1993;268:14443–14449. [PubMed] [Google Scholar]
- Shepherd JD, Huganir RL. The cell biology of synaptic plasticity: AMPA receptor trafficking. Annu Rev Cell Dev Biol. 2007;23:613–643. doi: 10.1146/annurev.cellbio.23.090506.123516. [DOI] [PubMed] [Google Scholar]
- Takeyasu K, Tamkun MM, Renaud KJ, Fambrough DM. Ouabain-sensitive (Na+ + K+)-ATPase activity expressed in mouse L cells by transfection with DNA encoding the alpha-subunit of an avian sodium pump. J Biol Chem. 1988;263:4347–4354. [PubMed] [Google Scholar]
- Tavi P, Allen DG, Niemelä P, Vuolteenaho O, Weckström M, Westerblad H. Calmodulin kinase modulates Ca2+ release in mouse skeletal muscle. J Physiol. 2003;551:5–12. doi: 10.1113/jphysiol.2003.042002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tokumitsu H, Chijiwa T, Hagiwara M, Mizutani A, Terasawa M, Hidaka H. KN-62, 1-[N,O-bis(5-isoquinolinesulfonyl)-N-methyl-l-tyrosyl]-4-phenylpiperazine, a specific inhibitor of Ca2+/calmodulin-dependent protein kinase II. J Biol Chem. 1990;265:4315–4320. [PubMed] [Google Scholar]
- Tsien RY. A non-disruptive technique for loading calcium buffers and indicators into cells. Nature. 1981;290:527–528. doi: 10.1038/290527a0. [DOI] [PubMed] [Google Scholar]
- Turney SG, Culican SM, Lichtman JW. A quantitative fluorescence-imaging technique for studying acetylcholine receptor turnover at neuromuscular junctions in living animals. J Neurosci Methods. 1996;64:199–208. doi: 10.1016/0165-0270(95)00135-2. [DOI] [PubMed] [Google Scholar]
- Wang Z, Edwards JG, Riley N, Provance DW, Jr, Karcher R, Li XD, Davison IG, Ikebe M, Mercer JA, Kauer JA, Ehlers MD. Myosin Vb mobilizes recycling endosomes and AMPA receptors for postsynaptic plasticity. Cell. 2008;135:535–548. doi: 10.1016/j.cell.2008.09.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams MW, Resneck WG, Kaysser T, Ursitti JA, Birkenmeier CS, Barker JE, Bloch RJ. Na,K-ATPase in skeletal muscle: two populations of beta-spectrin control localization in the sarcolemma but not partitioning between the sarcolemma and the transverse tubules. J Cell Sci. 2001;114:751–762. doi: 10.1242/jcs.114.4.751. [DOI] [PubMed] [Google Scholar]
- Wu H, Xiong WC, Mei L. To build a synapse: signaling pathways in neuromuscular junction assembly. Development. 2010;137:1017–1033. doi: 10.1242/dev.038711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamagata Y, Kobayashi S, Umeda T, Inoue A, Sakagami H, Fukaya M, Watanabe M, Hatanaka N, Totsuka M, Yagi T, Obata K, Imoto K, Yanagawa Y, Manabe T, Okabe S. Kinase-dead knock-in mouse reveals an essential role of kinase activity of Ca2+/calmodulin-dependent protein kinase IIalpha in dendritic spine enlargement, long-term potentiation, and learning. J Neurosci. 2009;29:7607–7618. doi: 10.1523/JNEUROSCI.0707-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]