

Abstract

A protocol for forming a highly active Pd(0) catalyst from Pd(OAc)2, water, and biaryldialkylphosphine ligands has been developed. This protocol generates a catalyst system, which exhibits excellent reactivity and efficiency in the coupling of a variety of amides and anilines with aryl chlorides.
Over the past decade great progress has been made in improving the efficiency and applicablilty of palladium-catalyzed C-N cross-coupling reactions.1 Despite these recent advances, many limitations of these methods remain. We have previously shown that biaryldialkylphosphines, of which 1 (Figure 1) is prototypical,2 are excellent supporting ligands in C-N cross-coupling processes. We now disclose a procedure that helps to maximize the efficiency of these ligands when used with a Pd(II) precatalyst.
Figure 1.

Dialkylbiarylphosphine Ligands
The formation of an active LnPd(0) complex is most commonly accomplished in one of the following ways: 1) use of a Pd(0) source such as Pd2(dba)3,3 2) use of [(allyl)PdCl]2,4 3) reduction of a Pd(II) salt [e.g., Pd(OAc)2] using PhB(OH)2,2 a tertiary amine,5 a tertiary phosphine,6 or an amine substrate,1 or 4) employment of a single component precatalyst.7 These methods all have deficiencies when used with biaryldialkylphosphine ligands. Although Pd2(dba)3 has been shown to work well in combination with 1 in many instances,2 diminished reactivity is observed due to the coordination of the dba to the palladium; this is a well known consequence of using dba containing precatalysts.8 Attempts to use [(allyl)PdCl]2 have not proven to be productive with dialkylbiarylphosphine ligands. Reduction of Pd(OAc)2 with 1 is slow due to the steric hindrance of the ligand and initial results show that the use of tertiary amines or PhB(OH)2 as reducing agents give slow formation of the active LnPd(0) catalyst. Lastly, reduction of Pd(OAc)2 works well with primary or secondary amine substrates that have β-hydrogens. However, when non-reducing nucleophiles, such as anilines or amides, are used, formation of LnPd(0) is inefficient.
Because of these deficiencies, we set out to develop a protocol that utilized water and 1 to reduce Pd(OAc)2 and generate the active LnPd(0) complex. This type of activation was first reported in 1992 by Ozawa and Hayashi, in which they were able to reduce Pd(OAc)2 in the presence of 3 equivalents of BINAP.9 They disclosed that in the absence of water the reduction did not proceed; however, by adding extra equivalents of water the rate of activation could be accelerated. This showed that water played a direct role in the formation of the Pd(0) catalyst. Amatore and Jutand further delineated a method in which water and several different tertiary phosphines were employed as reducing agents to form a Pd(0) complex that underwent oxidative addition to aryl halides (Scheme 1).10 In their investigations they found that water converted an intermediate phosphonium salt to the corresponding phosphine oxide in the reduction of Pd(OAc)2.
Scheme 1.

Water-Promoted Activation of Pd(OAc)2
We began our studies by seeking out a fast, efficient, and practical protocol for preactivation of Pd(OAc)2. It was found that a highly active catalyst could be generated by heating the Pd(OAc)2 (1 mol %), water (4 mol %), and 1 (3 mol %) for 1 min at 80 °C in 1,4-dioxane. The activation could be monitored visually by color change as shown in Figure 2. The resulting green catalyst solution11 could then be transferred into a reaction vessel containing the substrates and base. Figure 3 shows the reaction of aniline and 4-chloroanisole utilizing several modes of activation. The water-mediated preactivation protocol gave a 99% GC yield, whereas [(allyl)PdCl]2, Pd(OAc)2 / PhB(OH)2, and Pd2(dba)3 all resulted in yields lower than 50%. This demonstrates the efficiency in which water-mediated preactivation forms the active catalyst, without the detrimental effects of impeding ligands (eg., dba).
Figure 2.

Visualization of Water-Mediated Preactivationa
[a] Reaction conditions: Pd(OAc)2 (0.01 mmol), 1 (0.03 mmol), H2O (0.04 mmol), 1 mL 1,4-dioxane, 80 °C.
Figure 3.

Comparison of Water-Mediated Preactivation with Several Pd Sourcesa
[a] Reaction conditions: aniline (1.2 mmol), 4-chloroanisole (1.0 mmol), Pd (1 mol %), 1 (2.2 mol %), 1,4-dioxane (1 mL), 80 °C, 2 min.
We next investigated the application of this protocol to amidation reactions of aryl chlorides. Our research group recently reported an efficient catalyst system for this transformation utilizing the combination of Pd2(dba)3 and ligand 2 (Figure 1). It has been proposed that with biaryldialkylphosphine ligands the rate-limiting step in amidation reactions is the “transmetallation” (amide binding and/or deprotonation).12 We postulated that by replacing the Pd2(dba)3 with a preactivation of Pd(OAc)2 a more active catalyst system could be achieved, because there would be no dba present in the reaction to compete for binding at the Pd center. By heating Pd(OAc)2 (1 mol %), water (4 mol %), and 2 (3 mol %) for 1.5 min at 110 °C in t-BuOH a highly active catalyst was generated for these reactions. For example, whereas the coupling of either cyclohexanecarboxamide or acetamide with 4-chloroanisole using Pd2(dba)3 and 2 previously proceeded in 24 h, water-mediated preactivation decreased the reaction times to 3 h, and provided the products in excellent yields (Table 1, entries 1 and 2).12
Table 1.
Coupling of Amides With Aryl Chlorides
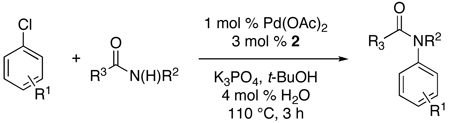
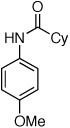
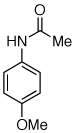
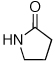
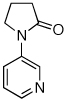
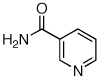
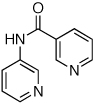
Reaction conditions: 1.5 min preactivation at 110 °C with 4 mol % H2O, 1 mol % Pd(OAc)2, and 3 mol % 2 in 2 mL/mmol of t-BuOH; 1.0 equiv aryl chloride, 1.2 equiv amide, and 1.4 equiv K2CO3.
Yields represent isolated yields (average of 2 runs).
1.5 equiv of amide.
We next looked at cross-couplings reactions of amides with 3-chloropyridine, which have been difficult in the past.11,13 When the preactivation protocol was employed, formamide, nicotinamide, and 2-pyrrolidinone were all successfully coupled with 3-chloropyridine, in 3 h, using 1 mol% Pd (Table 1, entries 3, 4, and 5). Previously, the reactions with formamide and 2-pyrrolidinone had required the use of 2 mol % Pd and reaction times of 812 and 24 h,13 respectively. Additionally, the latter case required the addition of Et3B to achieve good yields.
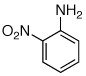
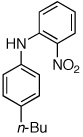
Having established that the combination of water and 2 efficiently converted Pd(OAc)2 to an active catalyst, we sought to expand the protocol to include reactions of electron-deficient anilines. Electron-deficient anilines have been difficult coupling partners due to their modest nucleophilicity and intolerance of strong base.7a,14 By performing a preactivation of Pd(OAc)2, 1, and water at 110 °C for 1 min, electron deficient 4-nitroaniline and 2-nitroaniline were successfully coupled with 4-n-butylchlorobenzene in excellent yields (Table 2, entries 1 and 2). Other electron-deficient anilines containing an ester or a cyano group were coupled as well in excellent yields using the newly developed protocol (Table 2, entries 3 and 4).
Table 2.
Coupling of Electron-Deficient Anilines
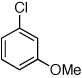
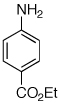
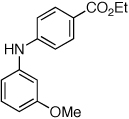
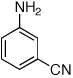
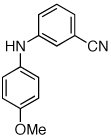
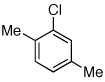
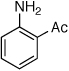
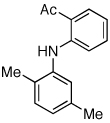
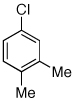
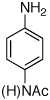
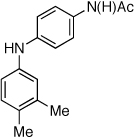
Reaction conditions: 1 min preactivation at 110 °C with 4 mol % H20, 1 mol % Pd(OAc)2, and 3 mol % 1 in 2 mL/mmol of t-BuOH; 1.0 equiv aryl chloride, 1.2 equiv amine, and 1.4 equiv K2CO3.
Yields represent isolated yields (average of 2 runs).
Reaction time of 2 h.
2.5 equiv of K2CO3 was used.
The chemoselectivity of the system was also briefly explored. Using 2’-aminoacetophenone ketone α-arylation was completely suppressed and exclusive C-N coupling was observed (Table 2, entry 5). This selectivity can be attributed to the high activity of the catalyst achieved utilizing water-mediated preactivation, which allows for the use of K2CO3.15 The reaction of 4-aminoacetanilide also showed complete selectivity for arylation at the free aniline (Table 2, entry 6).2 This catalyst system showed both high activity and excellent functional group compatibility.
Last, we wanted to examine the applicability of this protocol for the coupling of anilines at low catalyst loadings. There are currently very few examples of couplings with anilines and aryl chlorides below 0.5 mol % catalyst loading.7a,7b,16 Using the preactivation protocol several examples were performed at 0.05 mol % catalyst loading in 20 min (Table 3). For example, electrondeficient 3-aminobenzotrifluoride and 4-fluoroaniline were successfully coupled with electron-rich aryl chlorides within 20 min, affording the products in excellent yields (Table 3, entries 3 and 4).
Table 3.
Couplings of Anilines at 0.05 mol % Catalyst Loading
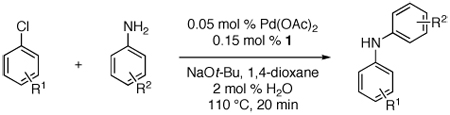
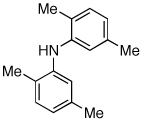
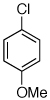
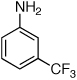
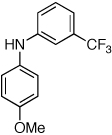
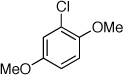
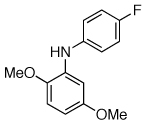
Reaction conditions: 1 min preactivation at 80 °C with 2 mol % H2O, 0.05 mol % Pd(OAc)2, and 0.15 mol % 1 in 1 mL/mmol of 1,4-dioxane; 1.0 equiv aryl chloride, 1.2 equiv amine, and 1.2 equiv NaOt-Bu.
Yields represent isolated yields (average of 2 runs).
In conclusion, an efficient protocol for C-N cross-coupling reactions has been described. An active catalyst was formed by water-mediated preactivation of readily available Pd(OAc)2 using biaryldialkylphosphine ligands. This new protocol allowed lower catalyst loadings, shorter reactions times, and exclusion of additives, such as Et3B, in couplings of amides with aryl chlorides. It also gives access to a system that exhibits both high activity and excellent catalyst stability in couplings of electrondeficient anilines, and couplings of anilines at low catalyst loadings. Further studies of other applications of this protocol are currently ongoing in our laboratories.
Supplementary Material
Experimental procedures and spectral data for all compounds. This material is free of charge via the Internet at http://pubs.acs.org.
Acknowledgment
We thank the National Institutes of Health (GM 58160) for financial support of this project. We thank Merck and BASF [Pd(OAc)2] for additional support. The Varian NMR instrument used was supported by the NSF (CHE 9808061 and DBI 9729592).
References
- 1.a) Hartwig JF, Shekhar S, Shen Q, Barrios-Landeros F. In: The Chemistry of Anilines Part 1. Rappoport Z, editor. Weinheim: WILEY-VCH; 2007. p. 455. [Google Scholar]; b) Buchwald SL, Mauger C, Mignani G, Scholz U. Adv. Synth. Catal. 2006;348:23. [Google Scholar]; c) Schlummer B, Scholz U. Adv. Synth. Catal. 2004;346:1599. [Google Scholar]; d) Jiang L, Buchwald SL. In: Metal-Catalyzed Cross-Coupling. 2nd ed. de Meijere A, Diederich F, editors. Weinheim: WILEY-VCH; 2004. p. 699. [Google Scholar]; e) Hartwig JF. In: Handbook on Organopalladium Chemistry for Organic Synthesis. Negishi E, editor. Weinheim: WILEY-VCH; 2002. p. 1051. [Google Scholar]
- 2.Huang X, Anderson KW, Zim D, Jiang L, Klapars A, Buchwald SL. J. Am. Chem. Soc. 2003;125:6653. doi: 10.1021/ja035483w. [DOI] [PubMed] [Google Scholar]
- 3.Ito TS, Hasegawa S, Takahashi Y, Ishii Y. J. Organomet. Chem. 1974;73:401. [Google Scholar]
- 4.Viciu MS, Germaneau RF, Navarro-Fernandez O, Stevens ED, Nolan SP. Organometallics. 2002;21:5470. [Google Scholar]
- 5.Trzeciak AM, Ciunik Z, Ziolkowski JJ. Organometallics. 2002;21:132. [Google Scholar]
- 6.Amatore C, Jutand A, M’Barki MA. Organometallics. 1992;11:3009. [Google Scholar]
- 7.a) Biscoe MR, Fors BP, Buchwald SL. J. Am. Chem. Soc. 2008;130:6686. doi: 10.1021/ja801137k. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Marion N, Navarro O, Mei J, Stevens ED, Scott NM, Nolan SP. J. Am. Chem. Soc. 2006;128:4101. doi: 10.1021/ja057704z. [DOI] [PubMed] [Google Scholar]; c) Zim D, Buchwald SL. Org. Lett. 2003;5:2413. doi: 10.1021/ol034561h. [DOI] [PubMed] [Google Scholar]; d) Stambuli JP, Kuwano R, Hartwig JF. Angew. Chem. Int. Ed. 2002;41:4746. doi: 10.1002/anie.200290036. [DOI] [PubMed] [Google Scholar]; e) Schnyder A, Indolese AF, Studer M, Blaser H. Angew. Chem. Int. Ed. 2002;41:3668. doi: 10.1002/1521-3773(20021004)41:19<3668::AID-ANIE3668>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]; f) Li GY, Zheng G, Noonan AF. J. Org. Chem. 2001;66:8677. doi: 10.1021/jo010764c. [DOI] [PubMed] [Google Scholar]; g) Andreu MG, Zapf A, Beller M. Chem. Comm. 2000:2475. [Google Scholar]; h) Riermeier TH, Zapf A, Beller M. Topics in Catalysis. 1997;4:301. [Google Scholar]
- 8.a) Fairlamb IJS, Kapdi AR, Lee AF, McGlacken GP, Weissburger F, de Vries AHM, de Vondervoort LS. Chem. Eur. J. 2006;12:8750. doi: 10.1002/chem.200600473. [DOI] [PubMed] [Google Scholar]; b) Mace Y, Kapdi AR, Fairlamb IJS, Jutand A. Organometallics. 2006;25:1795. [Google Scholar]; c) Amatore C, Jutand A. Coord. Chem. Rev. 1998;178–180:511. [Google Scholar]
- 9.Ozawa F, Kubo A, Hayashi T. Chem. Lett. 1992:2177. [Google Scholar]
- 10.a) Amatore C, Jutand A, Khalil F. Arkivoc. 2006;4:38. [Google Scholar]; b) Amatore C, Jutand A. J. Organomet. Chem. 1999;576:254. [Google Scholar]; c) Amatore C, Carre E, Jutand A, M’Barki MA. Organometallics. 1995;14:1818. [Google Scholar]
- 11.a) Initial 31P NMR experiments were inconclusive as to the structure of contained Pd species. b) The activated catalyst solution could be stored under argon for 24 h at −25 °C with minimal loss in catalyst activity. c) When water was added directly to the reaction mixture containing all of the components (i.e., without preactivation), little or no activity was observed.
- 12.Ikawa T, Barder TE, Biscoe MR, Buchwald SL. J. Am. Chem Soc. 2007;129:13001. doi: 10.1021/ja0717414. [DOI] [PubMed] [Google Scholar]
- 13.Shen Q, Hartwig JF. J. Am. Chem. Soc. 2007;129:7734. doi: 10.1021/ja0722473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hartwig JF. Inorg. Chem. 1936;46 doi: 10.1021/ic061926w. [DOI] [PubMed] [Google Scholar]
- 15.a) It has been shown that α-arylations of ketones usually require a strong base, except for reactions of particularly acidic carbonyl compounds. Culkin DA, Hartwig JF. Ace. Chem. Res. 2003;36:234. doi: 10.1021/ar0201106. b) For an exception using K3P04 (not K2C03) see: Fox JM, Huang X, Chieffi A, Buchwald SL. J. Am. Chem. Soc. 2000;122:1360.
- 16.a) Xu C, Gong J, Wu Y. Tetrahedron Lett. 2007;48:1619. [Google Scholar]; b) Rataboul F, Zapf A, Jackstell R, Harkal S, Riermeier T, Monsees A, Dingerdissen U, Beller M. Chem. Eur. J. 2004;10:2983. doi: 10.1002/chem.200306026. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Experimental procedures and spectral data for all compounds. This material is free of charge via the Internet at http://pubs.acs.org.