

Abstract
Fragile X syndrome (FXS) is a neurodevelopmental disorder characterized by variable cognitive impairment and behavioral disturbances such as exaggerated fear, anxiety and gaze avoidance. Consistent with this, findings from human brain imaging studies suggest dysfunction of the amygdala. Underlying alterations in amygdala synaptic function in the Fmr1 knock-out (KO) mouse model of FXS, however, remain largely unexplored. Utilizing a combination of approaches, we uncover profound alterations in inhibitory neurotransmission in the amygdala of Fmr1 KO mice. We demonstrate a dramatic reduction in the frequency and amplitude of phasic IPSCs, tonic inhibitory currents, as well as in the number of inhibitory synapses in Fmr1 KO mice. Furthermore, we observe significant alterations in GABA availability, both intracellularly and at the synaptic cleft. Together, these findings identify abnormalities in basal and action potential-dependent inhibitory neurotransmission. Additionally, we reveal a significant neuronal hyperexcitability in principal neurons of the amygdala in Fmr1 KO mice, which is strikingly rescued by pharmacological augmentation of tonic inhibitory tone using the GABA agonist gaboxadol (THIP). Thus, our study reveals relevant inhibitory synaptic abnormalities in the amygdala in the Fmr1 KO brain and supports the notion that pharmacological approaches targeting the GABAergic system may be a viable therapeutic approach toward correcting amygdala-based symptoms in FXS.
Introduction
Fragile X syndrome (FXS) is the most common inherited form of cognitive impairment and a leading genetic cause of autism (Hagerman et al., 2009). FXS results from a loss of function mutation in the FMR1 gene, which encodes the fragile X mental retardation protein (FMRP), an mRNA binding protein essential for the transport and translation of 4–8% of synaptic proteins (Bassell and Warren, 2008). Brains from both humans and the Fmr1 knock-out (KO) animal model of FXS are characterized by the presence of long and thin dendritic spines on excitatory neurons in select brain regions, hypothesized to underlie specific synaptic plasticity defects in the Fmr1 KO mouse (Bassell and Warren, 2008). Studies in Fmr1 KOs have revealed that overactivation of class I metabotropic glutamate receptor signaling is a primary defect in the cerebral cortex and hippocampus (Dolen and Bear, 2007). Interestingly, a number of studies have also implicated alterations in the GABA system in FXS, including dramatic changes in levels of expression of GABA receptors (El Idrissi et al., 2005; D'Hulst et al., 2006). Thus, both excitatory and inhibitory defects, in ways that are not fully understood, may contribute to circuit dysfunction in FXS.
The amygdala is a key brain structure involved in aspects of emotional processing within social and nonsocial behavioral contexts, as well as in the acquisition and storage of innate and acquired fear memories (LeDoux, 2003). Amygdala dysfunction in FXS is supported by structural and functional MRI studies (Gothelf et al., 2008; Watson et al., 2008) as well as animal behavioral studies demonstrating that Fmr1 KOs exhibit abnormal social behavior (McNaughton et al., 2008), consistent with findings in humans (Hagerman et al., 2009). In addition, patients with FMR1 mutations display a high prevalence of phenotypic manifestations suggestive of amygdala dysfunction, such as gaze avoidance and anxious behavior (Hessl et al., 2004; Budimirovic et al., 2006; Hagerman et al., 2009). This is experimentally supported by only two studies of the amygdala in Fmr1 KO mice in which deficiencies in lateral amygdala long-term potentiation were observed (Zhao et al., 2005; Suvrathan et al., 2010). However, despite evidence that amygdala dysfunction is a hallmark characteristic in FXS, the fundamental defects in neurotransmission in the amygdala currently remain unknown.
In the present study, we examined cellular and synaptic defects in the basolateral nucleus of the amygdala (BL) in Fmr1 KO mice. This nucleus is comprised of circuits that regulate fear and anxiety behaviors (Ehrlich et al., 2009). Using a combination of approaches, we demonstrate a marked reduction in inhibitory neurotransmission in BL circuits. Underlying this reduction are fundamental abnormalities in inhibitory fast synaptic and tonic GABAergic transmission that are concurrent with a reduction in the number of inhibitory synapses and GABA availability. In addition, we find that pharmacological augmentation of tonic GABAergic transmission rescues cellular hyperexcitability in the BL in Fmr1 KO mice. Thus, these findings reveal specific defects in GABAergic neurotransmission in the amygdala of Fmr1 KOs and provide strong evidence that pharmacological targeting of the GABAergic system may correct amygdala-based phenotypes in FXS.
Materials and Methods
Animal use.
Fmr1 WT (stock #4828) and Fmr1 KO (stock #4624) mice were on the congenic FVB mouse background and obtained from the Jackson Laboratory, and maintained according to protocols approved by Children's National Medical Center and Georgetown University School of Medicine.
Histology.
Mice were transcardially perfused with either 4% paraformaldehyde (PFA), or 4% PFA-0.2% glutaraldehyde in PBS. Brains were fixed overnight, embedded in 4% Agar (Fisher Scientific), and sectioned coronally on a vibratome (Leica). For immunohistochemistry, the following antibodies were used: mouse anti-FMRP, obtained from the Developmental Studies Hybridoma Bank developed under the auspices of the National Institute of Child Health and Human Development and maintained by The University of Iowa, Department of Biological Sciences, Iowa City, IA; rabbit anti-GABA (Sigma): rabbit anti-GAD65/67 (l-glutamic acid decarboxylase) (Millipore Bioscience Research Reagents), and rabbit anti-vesicular GABA transporter (VGAT, Synaptic Systems). Appropriate secondary antibodies for immunofluorescence (cy3 at 1:200, FITC at 1:50; all from Jackson ImmunoResearch) or peroxidase immunohistochemistry (biotinylated anti-IgG from Jackson ImmunoResearch, PA and Streptavidin from Sigma) followed primary antibody incubation. For the latter technique, immunoreactive structures were visualized using nickel-intensified DAB (3,3′-diaminobenzidine-4HCl; Sigma). Select sections were also stained with cresyl violet solution for Nissl staining. For electron microscopy, mice were transcardially perfused with 4% PFA-0.2% glutaraldehyde in PBS. Brains were fixed overnight in 4% PFA. Vibratome-sectioned coronal sections were also postfixed with 1% osmium tetroxide, and dehydrated in increasing grades of acetone and embedded in araldita (Electron Microscopy Science). Sections were trimmed in the region of interest and ultrathin sections were cut (70 nm) using an ultramicrotome (Leica).
Western blotting.
Finely dissected BLs were homogenized in RIPA lysis buffer (Santa Cruz Biotechnology). Proteins were loaded into 4–20% gradient gels. Gels were electrotransferred to a 0.2 μm nitrocellulose membrane (Millipore Bioscience Research Reagents). Blots were blocked in 5% milk in TBST and incubated with either anti-GAD65/67 (Millipore Bioscience Research Reagents) or anti-VGAT (Synaptic Systems) antibodies. Bands were detected with appropriate HRP-conjugated secondary antibodies, and reacted with chemiluminescent ECL substrate (Pierce). Band intensity was measured using the ImageJ program (National Institutes of Health).
Microscopy.
Photomicrographs were obtained with a Zeiss LSM 510 confocal microscope or with an Olympus BX51 light microscope. Electron microscopic images were taken with JEOL JEM 1200 EX at 100 kV.
Quantification and statistical analyses.
Quantification of the numbers of cells was achieved by nonstereological count methods. Sections (50 μm thick) from bregma level −0.82 mm to bregma level −1.82 mm were analyzed (Paxinos and Franklin, 2001). Cells stained with Nissl or immunostained with GABA were counted in the BL at P21. Quantification of VGAT punctae in the BL was done by using Volocity software. Images of VGAT were thresholded to select objects displaying intensity levels above the mean. Values were expressed as the mean ± SE. Normality of data distribution was verified using the Shapiro–Wilk test. Statistical comparisons were made using the two-tailed Student's t test, or the nonparametric Mann–Whitney U test, when applicable. For electron microscopy quantitative analysis, one section each from 3 WT brains and from 3 Fmr1 KO brains were analyzed. For each section, 12 ultrathin sections then were analyzed. For each ultrathin section, 10 consecutive images at 15,000× were collected. This led to a total of 360 images per genotype that were blindly analyzed. The number of asymmetric and symmetric synapses were identified and counted according to the following criteria: synapses were defined by the presence of a postsynaptic density facing at least three presynaptic vesicles. Asymmetric synapses were recognized by thick postsynaptic densities, while symmetric synapses were recognized by the presence of equally thin presynaptic and postsynaptic specializations.
Slice preparation for electrophysiological experiments.
Male mice at postnatal days 20–30 (P20–P30) were deeply anesthetized with carbon dioxide (CO2) and then decapitated. Brains were removed and placed for 2–3 min in an ice-cold and oxygenated sucrose slicing solution composed of (in mm): Sucrose (234), Glucose (11), NaHCO3 (26), KCl (2.5), NaH2PO4*H2O (1.25), MgSO4*7H2O (10), and CaCl2*H2O (0.5), and continuously gassed with 95% O2/5% CO2. Coronal slices were prepared by making a cut perpendicular to the rostral-caudal axis at the caudal end of the brain. Brains were then glued cut side-down on a vibratome (Leica) stage and again immersed in ice-cold sucrose slicing solution. Slices containing the BL were cut at a thickness of 250–300 μm. Slices were then incubated for 1 h in preheated (32°C), oxygen-equilibrated, artificial CSF (ACSF), composed of (in mm): NaCl (126), NaHCO3 (26), Glucose (10), KCl (2.5), NaH2PO4*H2O (1.25), MgCl2*7H2O (2), and CaCl2*2H2O (2); pH 7.4.
Electrophysiology.
Slices were visualized with a fixed-staged, upright microscope (Nikon, E600 FN) equipped with a 4× objective and a 60× insulated objective, infrared (IR) illumination, Nomarski optics, and an IR-sensitive video camera (COHU). Recordings were performed at room temperature (21°C-23°C) with continuous perfusion of ACSF at a rate of 2 ml/min. Whole-cell patch-clamp recordings were obtained using glass pipettes with an access resistance of 2.0–3.5 MΩ when filled with intracellular solution. Data were acquired via a Multiclamp amplifier (700A), Digidata (1322A) digitizer, using Clampex 9.2 acquisition software (Molecular Devices). Recordings were obtained from visually identified principal excitatory neurons in the BL. The identities of these cells were confirmed by their responses to brief (600 ms) depolarizing and hyperpolarizing current pulses. Typically, principal excitatory neurons fire broad action potentials (APs) separated by long afterhyperpolarizing potentials (AHPs), and show significant AP frequency adaptation when firing continuously (Sah et al., 2003). When TTX was present in the slice and cells were unable to fire action potentials (APs), excitatory cells were selected based on somatic morphology and their responses to hyperpolarizing current pulses exclusively. For F–I plots (AP frequency for given depolarizing current pulses), the following intracellular solution was used (in mm): K-Gluconate (130), KCl (10), HEPES (10), EGTA (10), and MgCl2 (2); ECl ∼−60 mV, pH 7.3. Membrane potential was adjusted to −60 mV, and brief (600 ms) depolarizing current pulses of increasing amplitude (10 pA interval) were used. AP threshold was defined as the lowest current step for which an action potential was generated. Threshold was measured under control conditions and in the presence of gaboxadol (THIP, 4,5,6,7-tetrahydroisoxazolo[5,4-c]pyridin-3-ol, 10 μm). IPSCs were collected in voltage-clamp (VHold = −60 mV) using one of two high-chloride intracellular solutions. Spontaneous and miniature IPSCs (sIPSCs, mIPSCs) were collected using the following intracellular solution (in mm): K-Gluconate (70), KCl (70), HEPES (10), EGTA (10), MgCl2 (2), Mg-ATP (4), Na-GTP (0.3). Evoked IPSCs, were collected using a cesium-based solution (in mm): CsCl (135), HEPES (10), EGTA (10), QX-314 (5), MgCl2 (2), Mg-ATP (4), and Na-GTP (0.3); pH 7.3, to block downstream effects of postsynaptic GABAB receptor activation. This solution was also used to collect mIPSCs in experiments in which the presynaptic effect of the GABAB receptor agonist Baclofen (1 μm, Tocris Bioscience) on mIPSC frequency was tested. GABAA receptor-mediated inhibition was isolated with the ionotropic glutamate receptor blockers 6,7-dinitroquinoxaline-2,3-dione (DNQX) (20 μm final concentration, Tocris Bioscience) and dl-2-amino-5-phosphonopentanoic acid (APV) (50 μm final, Tocris Bioscience) in the ACSF. mIPSCs were collected in the added presence of the voltage-gated Na+ channel blocker tetrodotoxin (TTX) (1 μm final, Alomone Labs) in the bath. The relative strength of the synaptic GABA transient in WT and Fmr1 KO mice was assessed by bath application of (1,2,5,6-tetrahydropiridin-4-yl)methylphosphonic acid (TPMPA) (50–200 μm, Tocris Bioscience). Averaged sIPSCs and mIPSCs were best fit by the double-exponential function f(t) = Afaste−t/τfast + Aslowe−t/τslow. Fitted IPSCs were used to determine the weighted time constant: τd,w = [(Afast τfast) + (Aslow τslow)]/(Afast + Aslow). Overall inhibitory efficacy was estimated by integrating total sIPSC charge per 1 s interval (Huntsman et al., 1999).
Tonic currents were measured using previously reported methods (Krook-Magnuson et al., 2008). Briefly, 10 s samples were taken from voltage-clamp recordings (Vh = −60 mV) before (IBSLN) and after 100 μm Gabazine (SR-95531, IGBZ), or in 1 μm TTX (ITTX) for ambient GABA source experiments. To minimize bias from phasic events, a Gaussian distribution was fit to the right side of an all-points histogram from each sample from a point 1–3 pA left of the peak. The peak of the distribution determined the mean current for that sample. Total tonic currents were calculated from the difference of baseline and Gabazine mean holding current (IGBZ − IBSLN) and AP-dependent tonic currents were calculated from the difference of baseline and TTX mean holding currents (ITTX − IBSLN).
Evoked responses were elicited via a 25-μm-diameter concentric bipolar Pt-Ir external stimulating electrode (FHC). The electrode was positioned in the BL, adjacent to the external capsule and paired current pulses were delivered. These experiments were conducted in the presence of APV and DNQX to isolate IPSCs, and only experiments in which fast, monosynaptic responses were evoked were included for analysis (average 10–90% rise time of 4.36 ± 0.42 ms, n = 24). For each cell, IPSC threshold was established (the intensity at which failures were observed at a rate of ∼50%), and stimulus intensity was then increased by 10–20%, such that consistent, rapid, monosynaptic responses were generated. Paired stimuli were delivered at an interval of 200 ms, once every 15 s. This interval was determined to be optimal, as it represented the shortest interval tested (of 50, 100, and 200 ms) at which evoked events did not overlap, and was thus the interval at which the maximum short-term plasticity due to repetitive events could be observed. The paired-pulse ratio (PPR) was defined as the peak amplitude of the second response divided by that of the first. The coefficient of variation (CV) was defined as the SD of the amplitude of a series of responses divided by the mean amplitude. PPR and CV measurements were made from 9–13 consecutive paired responses from each cell (mean = 10.6 responses/cell), excluding traces in which events coincided with excessive spontaneous activity.
For post hoc morphological analyses, biocytin (1%, Sigma) was included in the intracellular recording solution. After recording the firing properties of a given cell, biocytin was injected by delivering a series of short depolarizing current pulses (1 nA). After removal from the recording chamber, slices were fixed overnight in 4% paraformaldehyde in 0.1 m PBS. Slices were then incubated in 0.6% H2O2 for 30 min, and then twice in 50% ethanol for 10 min. After washing twice in PBS, slices were incubated for 1 h in PBS containing fluorescein-tagged Avidin D (10 μl/ml, Vector Laboratories), 10% horse serum, 5% Triton X-100, and 0.02 g/ml bovine serum albumin (Sigma). Following three more washes in PBS, slices were mounted and covered with mounting medium (Vectashield). Filled cells were visualized using an Olympus Fluoview laser-scanning confocal microscope.
Physiological recordings were analyzed off-line (Clampfit v. 9.2, Molecular Devices or MiniAnalyis v. 6.0.7, Synaptosoft,) All values are expressed as the mean ± SE. Statistical comparisons were made using the two-tailed Student's t test, or the nonparametric Mann–Whitney U test, when applicable (Origin v 7.0552, OriginLab).
Results
FMRP expression in BL inhibitory neurons
FMRP is an mRNA binding protein highly expressed in most neurons in the CNS (Bakker et al., 2000). Numerous studies have demonstrated the presence of FMRP in glutamatergic (excitatory) neurons (Huber et al., 2002; Antar et al., 2004; Tervonen et al., 2009) and in nonneuronal cell types such as GFAP+ or NG2+ glia (Pacey and Doering, 2007). In contrast, very little attention has been focused on the study of FMRP expression within GABAergic cells. To examine FMRP expression in the amygdala, we performed single and dual immunohistochemical analyses of FMRP and the GABAergic inhibitory neuron marker GAD65/67 in the BL at postnatal day 21 (P21) (Fig. 1A). Our immunohistochemical analyses showed strong FMRP expression in the cytoplasm of most cells in the BL (Fig. 1B,C), consistent with similar observations in the cerebral cortex and cerebellum (Abitbol et al., 1993; Devys et al., 1993; Bakker et al., 2000). Interestingly, our results also showed remarkably higher FMRP expression in BL cells compared with other nuclei of the amygdala such as the central nucleus (Fig. 1B). Furthermore, colocalization analysis showed that 74.6% (332 of 445) of GAD65/67+ cells also expressed FMRP (Fig. 1C–E). This novel finding that FMRP normally is expressed in GABAergic cells in the BL suggests potential function of FMRP in this neuronal population.
Figure 1.

FMRP is expressed in the majority of interneurons in the BL. A, Schematic of a coronal view of a brain at the level of the amygdala with the BL highlighted in red. B, Image of FMRP immunostaining at P21 showing the intense and high levels of FMRP expression in the L and BL in contrast to the much lesser immunostaining in other regions of the amygdala such as the central nucleus. C–E, The majority of GAD65/67-positive neurons also show FMRP expression in the BL at P21 (arrowheads). Ce, Central nucleus of the amygdala; ec, external capsule; EnD, endopiriform nucleus; L, lateral nucleus of the amygdala; Pir, piriform cortex. Scale bars: B, E (for C–E), 10 μm.
Diminished inhibitory transmission in Fmr1 KOs
The amygdala is characterized by strong inhibitory tone, suggesting that GABAergic neurotransmission is a major determinant of network activity in the amygdala (Sah et al., 2003). Since most of the GABAergic cells of the BL express FMRP we reasoned that inhibitory drive might be impaired in Fmr1 KO mice, which may have profound implications for amygdala function. We first examined two forms of GABAA receptor-mediated inhibition in BL excitatory principal neurons in Fmr1 KO mice: fast synaptic inhibition, and tonic inhibition, which relies on high-affinity extrasynaptic receptors (Semyanov et al., 2004; Farrant and Nusser, 2005). Whole-cell electrophysiological recordings were collected from excitatory principal neurons in the BL (supplemental Fig. 1A, available at www.jneurosci.org as supplemental material). Principal neurons were identified primarily by their large, pyramidal-like somata and their AP firing patterns in response to depolarizing current steps (Fig. 2A1,B1). Additionally, post hoc morphological analysis of biocytin-filled cells (n = 8) revealed a prominent apical dendrite and dense dendritic spines, both characteristic of BL excitatory neurons (Sah et al., 2003) (supplemental Fig. 1B, available at www.jneurosci.org as supplemental material). This analysis revealed major reductions in synaptic and tonic inhibition in Fmr1 KO mice (Fig. 2). Specifically, we observed significant decreases in the frequency (Fig. 2D; WT: 7.27 ± 1.23 Hz, n = 7; Fmr1 KO: 3.58 ± 0.37 Hz, n = 19, p = 0.0007) and amplitude (Fig. 2C,E; WT: 40.49 ± 11.04 pA, n = 7; Fmr1 KO: 23.24 ± 2.05 pA, n = 19, p = 0.026) of sIPSCs in Fmr1 KOs. In addition, we observed a slight prolongation of the sIPSC decay time constant (Fig. 2C,F; WT: 12.64 ± 1.08 ms, n = 7; Fmr1 KO: 16.10 ± 0.41 ms, n = 19, p = 0.001). Even with prolonged decay kinetics, diminished amplitude and frequency contribute to a significant reduction in overall inhibitory efficacy in Fmr1 KOs (Fig. 2G; WT: 3706 ± 1501 pC/s, n = 7; Fmr1 KO: 1398 ± 181 pC/s, n = 19, p = 0.020). Further, we recorded mIPSCs, which are AP-independent events. As observed with sIPSCs, there were significant reductions in mIPSC frequency (Fig. 2D; WT: 3.91 ± 0.28 Hz, n = 21; Fmr1 KO: 3.01 ± 0.25, n = 23; p = 0.019), amplitude (Fig. 2E; WT: 17.56 ± 0.67 pA, n = 21; Fmr1 KO: 14.68 ± 0.87 pA, n = 23; p = 0.0041), and efficacy (Fig. 2G; WT: 939 ± 87 pC/s, n = 21; Fmr1 KO: 650 ± 73 pC/s, n = 23; p = 0.0015), as well as a trend toward prolonged decay (Fig. 2F; WT: 13.76 ± 0.37 ms, n = 21; Fmr1 KO: 14.78 ± 0.48 ms, n = 23; p = 0.11).
Figure 2.

Phasic and tonic inhibition are impaired in Fmr1 KO mice. A1, B1, Current-clamp traces identifying cells from WT (A1) and Fmr1 KOs (B1) as excitatory principal neurons. Repetitive AP firing in response to a 40 pA depolarizing current step is shown. A2, B2, Continuous voltage-clamp traces from the above cells showing sIPSCs recorded from these same cells in the presence of APV (50 μm) and DNQX (20 μm). C, Overlay of averaged sIPSCs from cells in A1–B2. D–G, Averaged group data, showing reductions in sIPSC and mIPSC frequency (D), amplitude (E), and inhibitory efficacy (G), as well as a significant prolongation of sIPSC weighted tau (F). Note the similarity between frequency of mIPSCs in WT and sIPSCs in Fmr1 KOs. These differences were not related to differences in intrinsic membrane properties (Vm: WT, −61.1 ± 0.7 mV; Fmr1 KO, −60.7 ± 0.8 mV; p = 0.95; Rm: WT, 223 ± 13 MΩ; Fmr1 KO, 217 ± 13 MΩ; p = 0.55). H, I, Voltage-clamp traces from representative WT and Fmr1 KO principal neurons (Vhold = −60 mV) indicating the difference between the average baseline holding current and average holding current in locally applied 100 μm Gabazine, as determined by fitted Gaussian curve (see Materials and Methods). J, Total inhibitory tonic current is significantly reduced in Fmr1 KO principal neurons. K, Voltage-clamp trace from a WT principal neuron illustrating its AP-dependent tonic GABAergic current and total tonic current. L, Averaged group data, showing that the primary source of tonic inhibitory current in both WT and Fmr1 KO principal neurons is AP-dependent GABA release. *p < 0.05; ***p < 0.005. Calibration: A1, B1, 20 mV, 200 ms; A2, B2, 100 pA, 500 ms; C, 20 pA, 20 ms; H, I, 20 pA, 5 s; K, 10 pA, 5 s.
In addition to AP-dependent and -independent phasic inhibition described above, a third form of inhibitory neurotransmission comes from tonically active GABAA receptors located at perisynaptic and extrasynaptic locations (Farrant and Nusser, 2005). Since tonic inhibition is a major source of inhibitory tone in the CNS (Semyanov et al., 2004), we also measured tonic inhibitory currents in principal neurons. Tonic current was measured as the difference between the average holding current before and after local application of the GABAA receptor antagonist Gabazine (Fig. 2H–J), and this current was found to be significantly reduced in Fmr1 KO neurons (WT: 20.0 ± 5.2 pA, n = 10; Fmr1 KO: 4.9 ± 1.4 pA, n = 8; p = 0.021). Previous studies implicate AP-dependent release as a major source of ambient GABA required to act on perisynaptic and extrasynaptic tonic GABA receptors to produce the tonic current (Brickley et al., 1996; Bright et al., 2007; Glykys and Mody, 2007). We therefore tested whether tonic inhibition in the BL similarly depends on AP-dependent GABA release. Bath application of TTX (1 μm) removed nearly all the tonic inhibitory current in principal neurons in both WT and Fmr1 KO mice (Fig. 2K,L; WT: 102.95 ± 17.97%, n = 6; Fmr1 KO: 96.13 ± 9.25%, n = 3; p = 0.81), such that subsequent addition of Gabazine had nearly no effect on holding current. Together, these findings indicate a global reduction in inhibitory neurotransmission in the BL of Fmr1 KO mice compared with WT.
Reduced levels of GAD65/67 in Fmr1 KOs
Considering the global reduction in inhibitory transmission in the BL of Fmr1 KOs, we next examined the status of GABA levels. To accomplish this, we performed immunohistochemical and immunoblotting analyses of the major isoforms of the rate-limiting enzyme for GABA synthesis, GAD65/67 at P21. In WT brains, strong GAD65/67 immunostaining was observed in the entire amygdala, including the BL (Fig. 3A,C). Compared with WT, we observed a qualitative decrease in GAD65/67 immunostaining in the BL of the Fmr1 KOs (Fig. 3B,D). This was characterized as less immunostaining in cell bodies, puncta rings around presumptive cell bodies, and neuropil. Consistent with these findings, our immunoblotting revealed a significant decrease in the protein levels of GAD65/67 in the BL of Fmr1 KOs (Fig. 3E,F).
Figure 3.
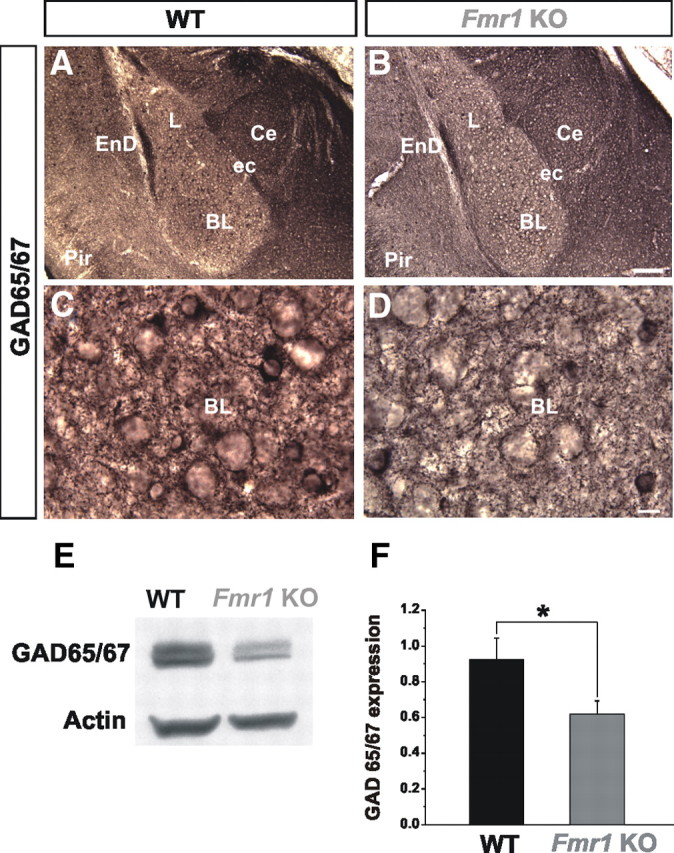
GAD65/67 levels are reduced in Fmr1 KOs. A, B, Normal gross morphology and positioning of GAD65/67+ inhibitory neurons in Fmr1 KO mice (B) compared with WT (A) in the BL is shown at P21 at low-power magnification. C, D, Qualitative decrease in GAD65/67 immunostaining is observed in Fmr1 KOs compared with WT as shown at high-power magnification. E, F, Western blot analyses reveal that GAD65/67 expression levels are significantly decreased in the Fmr1 KOs, compared with WT in the BL. BL, Basolateral nucleus of the amygdala; Ce, central nucleus of the amygdala; ec, external capsule; EnD, endopiriform nucleus; L, lateral nucleus of the amygdala; Pir, piriform cortex. *p < 0.05. Scale bars: B, 100 μm; D, 10 μm.
Together with the above electrophysiological analyses, these results demonstrate that the absence of FMRP results in a dramatic and marked reduction in spontaneous and miniature IPSCs, tonic inhibition and GAD65/67 levels in the BL in Fmr1 KOs. These findings are indicative of potentially both presynaptic and postsynaptic alterations. At the presynaptic level, these observed changes can be explained by four broad nonmutually exclusive mechanisms: 1) a decrease in the number of GABAergic neurons; 2) a decrease in neurotransmitter availability; 3) a decrease in the number of functional inhibitory synapses; and 4) a decrease in the activity of presynaptic inhibitory neurons.
Numbers of inhibitory neurons are unaffected in Fmr1 KOs
To begin to distinguish between putative presynaptic mechanisms, we first examined Nissl-stained coronal brain sections to determine whether the total number of cells in the BL in Fmr1 KOs was altered at P21. At the gross morphological level, there were no alterations in the BL of Fmr1 KOs (supplemental Fig. 2A,B). Our cell counting analysis also revealed no differences in total cell numbers between Fmr1 KOs and WT (supplemental Fig. 2C, available at www.jneurosci.org as supplemental material). We also performed cell counting analyses of GABA-immunopositive cells in the BL, which similarly revealed no differences (supplemental Fig. 2D–F, available at www.jneurosci.org as supplemental material). These results suggest that generation and survival of inhibitory neurons in the BL up to P21 is unaffected by the absence of FMRP.
Reduced synaptic GABA in Fmr1 KOs
Given that decreased GAD65/67 expression in Fmr1 KOs is not accompanied by a reduction in the number of GABA-positive cells, we hypothesized that GABA production and/or availability might be decreased in the cytosol and/or terminals of GABAergic neurons. Additionally, since mIPSCs represent responses to single quanta of released GABA, the observed reduction in mIPSC amplitude (Fig. 2E) suggests the possibility of decreased vesicular, and thus synaptic GABA. To address this possibility more directly, we considered whether the GABA transient was decreased in Fmr1 KOs. For these experiments we examined the effect of TPMPA, a low-affinity competitive GABAA receptor antagonist. The block of postsynaptic GABAA receptor-mediated responses by TPMPA is dependent upon GABA concentration at the synapse (Barberis et al., 2005; Szabadics et al., 2007). When synaptic GABA levels are low, TPMPA is displaced from postsynaptic receptors to a lesser extent, and thus IPSCs are more effectively blocked. Bath application of TPMPA at its IC50 concentration for GABAA receptor inhibition (200 μm) (Ragozzino et al., 1996), resulted in a greater reduction in mIPSC amplitude in Fmr1 KOs (Fig. 4A–C; WT: 32.98 ± 4.52% reduction, n = 7; Fmr1 KO: 42.28 ± 1.80% reduction, n = 13; p = 0.034), with no apparent effect on mIPSC decay kinetics (Control vs TPMPA: WT: 14.66 ± 0.70 ms vs 14.93 ± 0.80 ms, n = 7; p = 0.62: Fmr1 KO: 15.77 ± 0.71 ms vs 16.39 ± 0.73 ms, n = 13; p = 0.14). This greater effect of TPMPA in Fmr1 KOs was also observed using lower concentrations (Fig. 4C; 100 μm: WT: 19.84 ± 4.32% reduction, n = 5; Fmr1 KO: 32.12 ± 2.98% reduction, n = 6; p = 0.040; 50 μm: WT: 15.91 ± 4.90% reduction, n = 5; Fmr1 KO: 27.87 ± 3.19% reduction, n = 6; p = 0.063). This finding illustrates reduced GABA concentration in the synaptic cleft in Fmr1 KOs.
Figure 4.
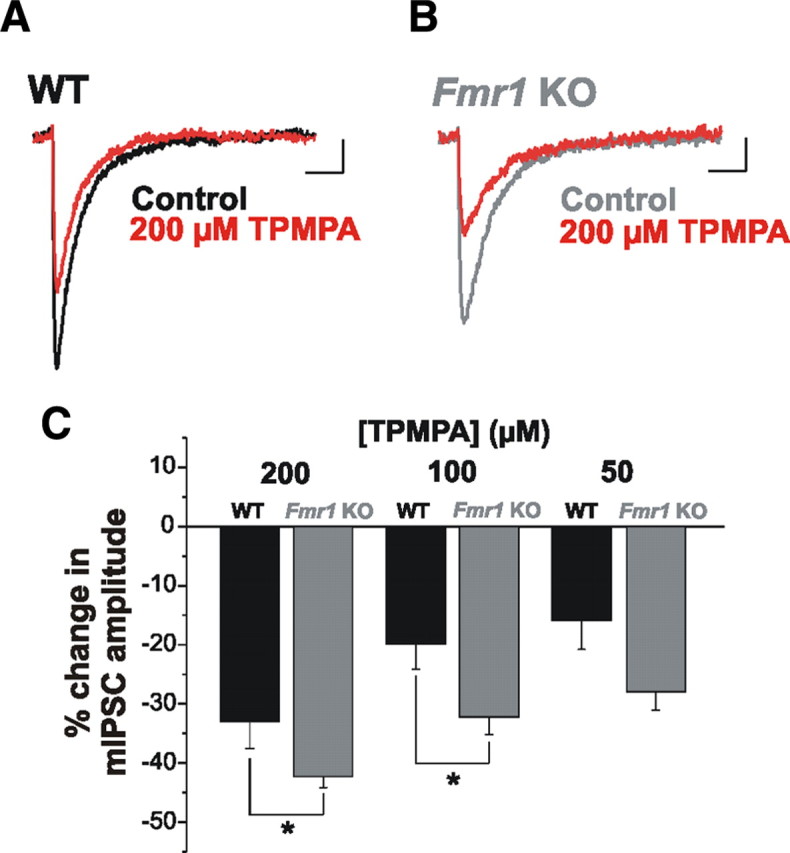
Synaptic GABA release is reduced in Fmr1 KOs. A–C, mIPSC amplitude is reduced to a greater extent by TPMPA in Fmr1 KO than in WT mice. Averaged mIPSCs from individual cells from WT (A) and Fmr1 KO (B) under control conditions and with TPMPA (200 μm) (red traces) illustrate the reduction in amplitude. C, Averaged group data indicating that TPMPA had a significantly greater effect on mIPSCs in Fmr1 KOs at doses of 200 and 100 μm. This trend was also observed using a 50 μm dose, but was not statistically significant. TPMPA did not exert a significant effect on mIPSC decay. *p < 0.05. Calibration: 2 pA, 20 ms.
The observed reductions in the production and vesicular content of GABA suggest that AP-dependent release might also be reduced in Fmr1 KOs. We investigated this by evoking paired IPSCs in excitatory principal neurons and measuring the PPR. Under conditions in which GABA release is high, this form of synaptic plasticity results in the attenuation (depression) of the amplitude of the second IPSC. While inhibitory connections were strongly depressing in WT animals (Fig. 5A,C), this effect was significantly attenuated in Fmr1 KOs, such that the PPR was greater (Fig. 5B,C; WT: 0.73 ± 0.04, n = 16; Fmr1 KO: 0.95 ± 0.05, n = 8, p = 0.004). This difference does not appear to be related to an alteration in the capacity for auto-inhibition mediated by presynaptic GABAB receptors at inhibitory terminals. Under conditions that permitted the presynaptic effect of the GABAB receptor agonist Baclofen (1 μm) to be isolated, mIPSC frequency in excitatory principal neurons was reduced to the same degree in WT and Fmr1 KO mice (supplemental Fig. 3, available at www.jneurosci.org as supplemental material). Furthermore, the CV of the amplitude of the first response was also greater in Fmr1 KOs (Fig. 5D, WT: 0.28 ± 0.03, n = 16; Fmr1 KO: 0.41 ± 0.06, n = 8, p = 0.02). These findings are consistent with decreased GABA release in Fmr1 KO mice and suggest that inhibitory synapses are weakened.
Figure 5.

AP-dependent GABA release is impaired in Fmr1 KO mice. A–C, The PPR of evoked IPSCs is greater in excitatory principal neurons in Fmr1 KO than WT mice. Averaged traces from single cells from WT (A) and Fmr1 KO mice (B) illustrate that the strong depression observed in WT is attenuated in Fmr1 KOs. C, Averaged group data. D, Average amplitude CV of the first evoked response is also greater in Fmr1 KOs. *p < 0.05; ***p < 0.005. Calibration: 50 pA, 100 ms.
Reduction in number of inhibitory synapses in Fmr1 KOs
As mentioned above, another potential mechanism to explain the decrease in inhibitory function might be a decrease in the number of inhibitory synapses. To assess inhibitory synapse numbers in the BL in Fmr1 KOs, we performed transmission electron microscopy. We quantified symmetric (putative inhibitory) and asymmetric (putative excitatory) synapse density using electron micrographs from WT and Fmr1 KOs at P25 (Fig. 6A–C). Our results revealed a striking decrease in the number of inhibitory synapses in Fmr1 KOs (Fig. 6D; WT: n = 3; Fmr1 KO: n = 3; p = 0.0000002). Interestingly, the number of excitatory synapses was also decreased, although to a much lesser extent (Fig. 6D; WT: n = 3; Fmr1 KO: n = 3; p = 0.010). Interestingly, this finding contrasts with other studies which showed that developing hippocampal Fmr1 KO neurons in culture display increased synapse numbers (Pfeiffer and Huber, 2007). Surprisingly, despite a marked reduction in inhibitory synapses as measured by EM in the BL of Fmr1 KO, we did not observe reductions in the expression of VGAT (Fig. 6E–I), which specifically labels GABAergic inhibitory terminals (Chaudhry et al., 1998; Gibson et al., 2009). Thus, these results indicate that the absence of FMRP results in a dramatic preferential reduction in the number of inhibitory synapses in the BL in Fmr1 KOs, without changes in the amount of VGAT protein. The decrease in inhibitory synapse number is also consistent with our finding that mIPSC frequency is decreased in Fmr1 KOs (Fig. 2D).
Figure 6.
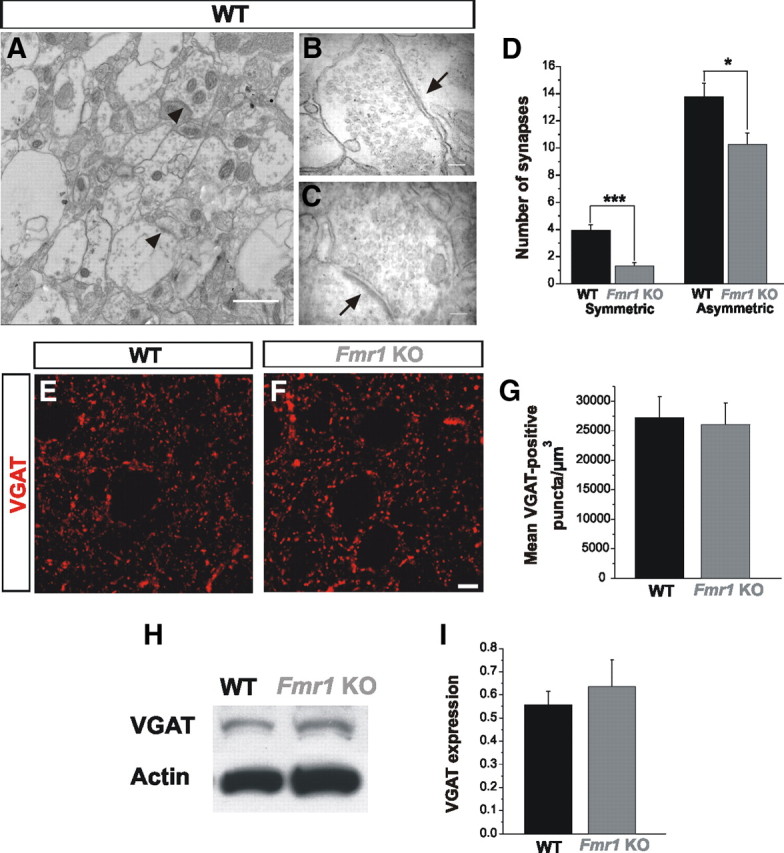
Reduced number of inhibitory synapses but no changes in VGAT expression in Fmr1 KOs. A–D, Quantitative analyses of electron micrographs of the BL show a remarkable decrease in inhibitory synapses in Fmr1 KOs with respect to WT. Excitatory synapses are decreased as well in Fmr1 KOs, although to a lesser extent (D). Arrowheads in A identify synaptic contacts. Detail of a symmetric (B) and asymmetric (C) synaptic contacts in the BL (arrows). E, F, Qualitative images of VGAT-immunostaining in the BL show no difference between WT (E) and Fmr1 KOs (F). G, Quantification of VGAT puncta (5 sections from 4 different mice) show no differences between WT and the Fmr1 KOs. H, I, Western blot analyses demonstrate that VGAT expression levels are similar in Fmr1 KOs and WT mice in the BL (WT, n = 4; Fmr1 KO, n = 4). *p < 0.05; ***p < 0.005. Scale bars, 20 μm.
THIP rescues neuronal hyperexcitability in the BL in Fmr1 KOs
In addition to a dampened inhibitory tone (Fig. 2), we also observed increased excitability in principal neurons of the BL amygdala in Fmr1 KO mice. Excitatory neurons from Fmr1 KOs consistently fired a greater number of APs in response to a series of depolarizing current steps (Fig. 7A–C; WT: n = 29; Fmr1 KO: n = 44). The threshold for AP generation was also significantly lower in Fmr1 KO mice (Fig. 7D; WT Control ACSF: 39.0 ± 3.5 pA, n = 10; Fmr1 KO Control ACSF: 23.3 ± 4.2 pA, n = 15; p = 0.009). Given the decreased GABA availability and weakened tonic currents in Fmr1 KOs, we hypothesized that augmentation of tonic inhibitory tone might alleviate this hyperexcitability. Activation of tonic inhibitory currents is highly dependent upon the availability of ambient GABA, and represents a means to decrease membrane excitability by decreasing input resistance and increasing conductance (Semyanov et al., 2004). In an attempt to rescue the hyperexcitability phenotype in principal neurons from Fmr1 KOs, we performed a depolarizing current step protocol in control ACSF, and then during bath application of THIP. THIP is a superagonist at δ subunit-containing perisynaptic and extrasynaptic GABAA receptors known to mediate a strong tonic inhibitory conductance in the CNS (Brown et al., 2002; Glykys et al., 2007). As expected, significant increases in the AP threshold were observed in both WT and Fmr1 KOs with THIP application. Strikingly, the AP threshold in Fmr1 KOs was restored to WT levels by THIP application (Fig. 7B,D; Fmr1 KO THIP: 40.7 ± 4.6 pA, n = 15; p = 0.78). Thus, the neuronal hyperexcitability in the Fmr1 KO amygdala can be dramatically rescued by augmenting tonic inhibitory tone.
Figure 7.

THIP rescues neuronal excitability in Fmr1 KOs. A, B, Concatenated traces from single cells in response to depolarizing current steps of increasing amplitude (10 pA steps). In control ACSF (A, B1), cells from Fmr1 KO mice exhibit higher action potential firing rates for a given depolarizing current step, as well as a lower threshold for action potential generation than cells from WT mice. C, Group F–I plot illustrating hyperexcitability of cells in Fmr1 KOs. No differences in passive membrane properties were observed between cells in the two groups (Vm: WT, −61.5 ± 0.5 mV; Fmr1 KO, −61.2 ± 0.7 mV; p = 0.80; Rm: WT, 231 ± 21 MΩ; Fmr1 KO, 242 ± 12 MΩ; p = 0.19). B2, D, Bath application of THIP increases action potential threshold in Fmr1 KOs to WT levels. *p < 0.05; **p < 0.01; n.s. not statistically significant. Calibration: 40 mV, 1 s.
Discussion
Recent studies of several developmental brain disorders, including autism spectrum disorders and their animal models, have led to the hypothesis that the primary dysfunction lies at the level of the synapse (Rubenstein and Merzenich, 2003; Zoghbi, 2003; Belmonte and Bourgeon, 2006; Johnston, 2006; Moy and Nadler, 2008). Since FMRP plays an important role in regulating protein synthesis at the synapse, one prediction of this hypothesis is that the absence of FMRP leads to synaptic alterations that in turn causes circuit-specific changes in neuronal excitation and inhibition in FXS. While there is strong evidence that excitatory transmission and synaptic plasticity are abnormal in various brain regions of the FXS mouse model (Bassell and Warren, 2008), underlying synaptic disturbances in relevant brain structures such as the amygdala remain largely unexplored.
In the present study we identified a number of major inhibitory defects in the amygdala of the Fmr1 KO mouse model of FXS. We reveal dampened inhibitory neurotransmission, reductions in inhibitory synapse number, and decreased cellular and synaptic levels of GABA. Together, these findings uncover a striking reduction in inhibitory tone that likely stems, at least in part, from presynaptic defects. This finding is also consistent with recent studies in a mouse model of Rett syndrome indicating that reduced expression of presynaptic components of inhibitory synapses contribute to decreases in inhibitory neurotransmission (Medrihan et al., 2008). Thus, combined with our findings, it appears that alterations in inhibitory neurotransmission may be a common mechanism in a number of neurodevelopmental disorders. In addition to uncovering novel defects in GABA neurotransmission in the FXS brain, we were able to strikingly rescue cellular hyperexcitability by enhancing neurotransmission at high-affinity extrasynaptic GABAA receptors sensitive to ambient GABA. Therefore, considering the central role of the amygdala in behaviors that are altered in FXS, our study provides basic findings that have implications for the pathogenic mechanisms and putative correction of underlying amygdala-based phenotypes in FXS.
Global reductions in inhibitory transmission: evidence for cumulative presynaptic defects
In the present study, both synaptic and tonic inhibitory transmission in the BL of Fmr1 KO mice are shown to be remarkably deficient. These findings are consistent with previous studies which have shown alterations in the expression of several important presynaptic and postsynaptic components of the inhibitory transmission machinery, including GABAA receptor subunits, GABA transporters, GAD, and gephyrin in other brain regions of Fmr1 KOs (Brown et al., 2001; El Idrissi et al., 2005; D'Hulst et al., 2006, 2009; Curia et al., 2009). Interestingly, the absence of FMRP has distinct effects on GABAergic circuit function in different brain regions. For example, reductions in the numbers and altered activity of parvalbumin (PV)-positive inhibitory neurons have been demonstrated in somatosensory cortex but not in hippocampus of Fmr1 KOs (Selby et al., 2007; Gibson et al., 2008), suggesting a region-dependent defect in inhibitory neuronal genesis, migration and/or survival. In addition, in stark contrast to our findings in the amygdala, studies in the striatum have revealed increased GABAergic neurotransmission (Centonze et al., 2008). Therefore, the putative mechanisms that result in alterations in inhibitory drive in the amygdala may involve, at least in part, different processes than in the cerebral cortex and striatum.
Our results reveal normal numbers of inhibitory neurons indicating, although indirectly, that interneuron subtype specification and migration to the BL in Fmr1 KOs are unaffected. While it remains to be determined whether the numbers of specific inhibitory neuron subtypes are altered in Fmr1 KOs, we propose that overall global decreases in GABA production and release, reduced synaptic connectivity and interneuron activity are primary mechanisms for decreased inhibitory tone. In support of these mechanisms we identify deficits in both basal and AP-dependent inhibitory transmission. Interestingly, the values for sIPSC frequency in Fmr1 KOs compared with mIPSC frequency in WTs are similar. Moreover, in Fmr1 KOs sIPSC frequency is reduced to a greater extent than mIPSC frequency. Together these data are indicative of reduced AP-dependent release from interneurons. Consistent with this finding, tonic inhibitory current in Fmr1 KOs, which relies heavily on AP-dependent GABA release in both WT and Fmr1 KOs is also reduced. Thus reduced inhibitory neuron activity—in addition to reduced neurotransmitter availability—may also contribute significantly to the inhibitory deficits observed in the BL of Fmr1 KO mice.
Additionally, we report decreased GAD65/67 expression that coincides with a reduced GABA transient and decreased release probability. Interestingly, decreased GABA production can alter not only vesicular neurotransmitter content, as indicated by a weaker GABA transient, but AP-dependent release as well. A reduction in GABA release probability in response to sustained stimulation has been reported in GAD65 KO mice, reflecting decreased numbers of vesicles at the synaptic terminal or a mechanism by which only filled vesicles are released (Tian et al., 1999). Indeed, our finding that PPR is increased in Fmr1 KO mice suggests that AP-dependent GABA release is reduced, and this effect could result at least in part from a reduction in GABA production.
With respect to connectivity, our study reveals a marked reduction in the number of inhibitory synapses at the ultrastructural level, which correlates functionally with a reduction in mIPSC frequency. Surprisingly, however, we did not observe a correlative reduction in VGAT expression. Discrepancies remain regarding VGAT expression in animal models of autism spectrum disorders; for example, recent findings in Neuroligin-2 knock-out mice showed reduced synaptic function and without a reduction of VGAT puncta (Gibson et al., 2009). Studies in Neuroligin3 knock-in mice showed changes in VGAT expression that were not accompanied by changes in the numbers of inhibitory synapses (Tabuchi et al., 2007). These apparent discrepancies in the VGAT levels compared with inhibitory synapse numbers suggests the possibility of compensatory mechanisms, similar to those found after induced cholinergic neuron loss or in Alzheimer's disease (Kish et al., 1990; Wenk and Mobley, 1996) or in Neuroligin-2 knock-out mice (Gibson et al., 2009). An alternative interpretation is that numbers of GABAergic synapses in the BL of Fmr1 KO mice are actually normal, and that the reduction of inhibitory symmetric synaptic contacts observed in our study is due to solely to the decrease in large cholinergic projections from the basal forebrain to the BL, which also mainly form inhibitory symmetric synaptic contacts (Li et al., 2001). While this is a possibility, the dramatic reduction in the numbers of symmetric synaptic contacts in combination with the decrease in mIPSC frequency and tonic inhibition is more readily explained by a major loss of GABAergic synapses in BL of Fmr1 KO mice.
While the present study addresses the multiple presynaptic components of inhibitory dysfunction in the amygdala, it is likely that postsynaptic alterations in the GABAergic system also contribute to circuit abnormalities in FXS. Consistent with this, we observed a prolongation of sIPSC decay (plus a trend toward mIPSC decay prolongation) in Fmr1 KOs, which suggests possible differences in GABAA receptor subunit expression (Huntsman and Huguenard, 2000). Additionally, reduced tonic inhibitory tone may be a cumulative effect arising from reduced expression of δ and α5 receptor subunits, as previously reported in the subiculum (Curia et al., 2009) combined with lower ambient GABA.
Results in the present study suggest the hypothesis that an intrinsic deficit in FMRP expression in inhibitory neurons leads to cellular and synaptic defects. However, it should be noted that these defects may also in part arise secondarily from alterations in excitatory transmission. Consistent with this possibility, our EM data reveal a decrease of asymmetric (i.e., excitatory) synapses in Fmr1 KOs. In addition, transient alterations in cortical excitatory transmission during the critical period have been described in Fmr1 KOs (Bureau et al., 2008; Harlow et al., 2010).
Tonic inhibition as a therapeutic target in FXS
Complementing our findings of alterations in inhibitory neuronal efficacy in Fmr1 KOs, we restored principal neuron excitability to WT levels by pharmacological enhancement of tonic inhibitory transmission. This form of inhibition generates a powerful inhibitory shunt, thus decreasing membrane excitability (Semyanov et al., 2004). Accumulating evidence implicates tonic inhibition as a potential therapeutic target in various disorders, such as epilepsy and premenstrual dysphoric disorder, involving alterations in the balance of excitation/inhibition (Maguire et al., 2005; Glykys and Mody, 2007). Our data suggest that a lack of ambient GABA, due to deficient AP-dependent release from inhibitory interneurons, is a contributor to hyperexcitability in the BL amygdala of Fmr1 KOs. This neuronal hyperexcitability was corrected in vitro using THIP, a GABAA receptor agonist. Increasing GABA exposure during development has recently been shown to rescue the biochemical, morphological, and behavioral phenotypes of the Drosophila model of FXS (Chang et al., 2008), an intriguing finding given the proposed role of GABA as a regulator of synaptic maturation during critical periods of development (Hensch, 2005). The therapeutic value of increasing GABAergic transmission therefore appears to lie in its ability to either compensate for increased excitatory glutamatergic signaling and/or to boost intrinsically deficient inhibitory signaling, the latter of which is suggested by our findings. Regardless of the mechanism, our findings support the notion, as suggested by others (El Idrissi et al., 2005; D'Hulst et al., 2006; D'Hulst and Kooy, 2007), that inhibitory GABAergic transmission is an intriguing target for the treatment of FXS.
Footnotes
This work was supported by the US National Institute on Drug Abuse (J.G.C., Grant DA020140), the US National Institute of Neurological Disorders and Stroke (M.M.H., Grant NS053719), the Hazeltine Foundation (J.G.C.), the Neurodevelopmental Disorders Fund (J.G.C.), the Cellular Imaging Core of Intellectual and Developmental Disabilities Research Center (P30HD40677), the FRAXA Research Foundation (W.E.K.), Autism Speaks (JG.C., M.M.H.), the Epilepsy Foundation (B.S.M.), and a Canadian Institutes of Health Research Doctoral Research Award (S.M.P., #181393). We thank Vittorio Gallo and Tarik Haydar for helpful comments and the critical reading of this manuscript, Anastas Popratiloff from the Center for Microscopy and Image Analysis, The George Washington University, for assistance with electron microscopy, Cristina Pineda Delgado for technical assistance, and members of the Corbin, Haydar, and Huntsman laboratories for valuable input during the course of this study. All authors were involved in the conception and design and interpretation of experiments. J.L.O.-S. conducted histological procedures, electron microscopy, Western blot, immunohistochemistry, and nonelectrophysiological data analysis, and organized and led in writing of the manuscript. S.M.P. performed the majority of electrophysiological experiments, data analysis, and writing of electrophysiological aspects. B.S.M. and M.M.H. conducted a subset of electrophysiological experiments. W.E.K. also provided intellectual insight and data analysis. J.G.C. and M.M.H. oversaw the project and writing of the manuscript.
References
- Abitbol M, Menini C, Delezoide AL, Rhyner T, Vekemans M, Mallet J. Nucleus basalis magnocellularis and hippocampus are the major sites of FMR-1 expression in the human fetal brain. Nat Genet. 1993;4:147–153. doi: 10.1038/ng0693-147. [DOI] [PubMed] [Google Scholar]
- Antar LN, Afroz R, Dictenberg JB, Carroll RC, Bassell GJ. Metabotropic glutamate receptor activation regulates fragile x mental retardation protein and FMR1mRNA localization differentially in dendrite and at synapses. J Neurosci. 2004;24:2648–2655. doi: 10.1523/JNEUROSCI.0099-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakker CE, de Diego Otero Y, Bontekoe C, Raghoe P, Luteijn T, Hoogeveen AT, Oostra BA, Willemsen R. Immunocytochemical and biochemical characterization of FMRP, FXR1P, and FXR2P in the mouse. Exp Cell Res. 2000;258:162–170. doi: 10.1006/excr.2000.4932. [DOI] [PubMed] [Google Scholar]
- Barberis A, Lu C, Vicini S, Mozrzymas JW. Developmental changes of GABA synaptic transient in cerebellar granule cells. Mol Pharmacol. 2005;67:1221–1228. doi: 10.1124/mol.104.006437. [DOI] [PubMed] [Google Scholar]
- Bassell GJ, Warren ST. Fragile X syndrome: loss of local mRNA regulation alters synaptic development and function. Neuron. 2008;60:201–214. doi: 10.1016/j.neuron.2008.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belmonte MK, Bourgeron T. Fragile X syndrome and autism at the intersection of genetic and neural networks. Nat Neurosci. 2006;9:1221–1225. doi: 10.1038/nn1765. [DOI] [PubMed] [Google Scholar]
- Brickley SG, Cull-Candy SG, Farrant M. Development of a tonic form of synaptic inhibition in rat cerebellar granule cells resulting from persistent activation of GABAA receptors. J Physiol. 1996;15:497, 753–759. doi: 10.1113/jphysiol.1996.sp021806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bright DP, Aller MI, Brickley SG. Synaptic release generates a tonic GABA(A) receptor-mediated conductance that modulates burst precision in thalamic relay neurons. J Neurosci. 2007;27:2560–2569. doi: 10.1523/JNEUROSCI.5100-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown N, Kerby J, Bonnert TP, Whiting PJ, Wafford KA. Pharmacological characterization of a novel cell line expressing human alpha(4)beta(3)delta GABA(A) receptors. Br J Pharmacol. 2002;136:965–974. doi: 10.1038/sj.bjp.0704795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown V, Jin P, Ceman S, Darnell JC, O'Donnell WT, Tenenbaum SA, Jin X, Feng Y, Wilkinson KD, Keene JD, Darnell RB, Warren ST. Microarray identification of FMRP-associated brain mRNAs and altered mRNA translational profiles in fragile X syndrome. Cell. 2001;107:477–487. doi: 10.1016/s0092-8674(01)00568-2. [DOI] [PubMed] [Google Scholar]
- Budimirovic DB, Bukelis I, Cox C, Gray RM, Tierney E, Kaufmann WE. Autism spectrum disorder in fragile X syndrome: differential contribution of adaptive socialization and social withdrawal. Am J Med Genet. 2006;140A:1814–1826. doi: 10.1002/ajmg.a.31405. [DOI] [PubMed] [Google Scholar]
- Bureau I, Shepherd GM, Svoboda K. Circuit and plasticity defects in the developing somatosensory cortex of FMR1 knock-out mice. J Neurosci. 2008;28:5178–5188. doi: 10.1523/JNEUROSCI.1076-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Centonze D, Rossi S, Mercaldo V, Napoli I, Ciotti MT, De Chiara V, Musella A, Prosperetti C, Calabresi P, Bernardi G, Bagni C. Abnormal striatal GABA transmission in the mouse model for the fragile X syndrome. Biol Psychiatry. 2008;63:963–973. doi: 10.1016/j.biopsych.2007.09.008. [DOI] [PubMed] [Google Scholar]
- Chang S, Bray SM, Li Z, Zarnescu DC, He C, Jin P, Warren ST. Identification of small molecules rescuing fragile X syndrome phenotypes in Drosophila. Nat Chem Biol. 2008;4:256–263. doi: 10.1038/nchembio.78. [DOI] [PubMed] [Google Scholar]
- Chaudhry FA, Reimer RJ, Bellocchio EE, Danbolt NC, Osen KK, Edwards RH, Storm-Mathisen J. The vesicular GABA transporter, VGAT, localizes to synaptic vesicles in sets of glycinergic as well as GABAergic neurons. J Neurosci. 1998;18:9733–9750. doi: 10.1523/JNEUROSCI.18-23-09733.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curia G, Papouin T, Séguéla P, Avoli M. Downregulation of tonic GABAergic inhibition in a mouse model of fragile X syndrome. Cereb Cortex. 2009;19:1515–1520. doi: 10.1093/cercor/bhn159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devys D, Lutz Y, Rouyer N, Bellocq JP, Mandel JL. The FMR-1 protein is cytoplasmic most abundant in neurons and appears normal in carriers of a fragile x premutation. Nat Genet. 1993;4:335–340. doi: 10.1038/ng0893-335. [DOI] [PubMed] [Google Scholar]
- D'Hulst C, Kooy RF. The GABAA receptor: a novel target for treatment of fragile X? Trends Neurosci. 2007;30:425–431. doi: 10.1016/j.tins.2007.06.003. [DOI] [PubMed] [Google Scholar]
- D'Hulst C, De Geest N, Reeve SP, Van Dam D, De Deyn PP, Hassan BA, Kooy RF. Decreased expression of the GABAa receptor in fragile X syndrome. Brain Res. 2006;1121:238–245. doi: 10.1016/j.brainres.2006.08.115. [DOI] [PubMed] [Google Scholar]
- D'Hulst C, Heulens I, Brouwer JR, Willemsen R, De Geest N, Reeve SP, De Deyn PP, Hassan BA, Kooy RF. Expression of the GABAergic system in animal models for fragile X syndrome and fragile X associated tremor/ataxia syndrome (FXTAS) Brain Res. 2009;1253:176–183. doi: 10.1016/j.brainres.2008.11.075. [DOI] [PubMed] [Google Scholar]
- Dolen G, Bear MF. Role for metabotropic glutamate receptor 5 (mGluR5) in the pathogenesis of fragile X syndrome. J Neurophysiol. 2007;586:1503–1508. doi: 10.1113/jphysiol.2008.150722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrlich I, Humeau Y, Grenier F, Ciocchi S, Herry C, Lüthi A. Amygdala inhibitory circuits and the control of fear memory. Neuron. 2009;62:757–771. doi: 10.1016/j.neuron.2009.05.026. [DOI] [PubMed] [Google Scholar]
- El Idrissi A, Ding XH, Scalia J, Trenkner E, Brown WT, Dobkin C. Decreased GABA(A) receptor expression in the seizure-prone fragile X mouse. Neurosci Lett. 2005;377:141–146. doi: 10.1016/j.neulet.2004.11.087. [DOI] [PubMed] [Google Scholar]
- Farrant M, Nusser Z. Variations on an inhibitory theme: phasic and tonic activation of GABA(A) receptors. Nat Rev Neurosci. 2005;6:215–229. doi: 10.1038/nrn1625. [DOI] [PubMed] [Google Scholar]
- Gibson JR, Bartley AF, Hays SA, Huber KM. Imbalance of neocortical excitation and inhibition and altered UP states reflect network hyperexcitability in the mouse model of fragile X syndrome. J Neurophysiol. 2008;100:2615–2626. doi: 10.1152/jn.90752.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson JR, Huber KM, Südhof TC. Neuroligin-2 deletion selectively decreases inhibitory synaptic transmission originating from fast-spiking but not from somatostatin-positive interneuron's. J Neurosci. 2009;29:13883–13897. doi: 10.1523/JNEUROSCI.2457-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glykys J, Mody I. Activation of GABAA receptors: views from outside the synaptic cleft. Neuron. 2007;56:763–770. doi: 10.1016/j.neuron.2007.11.002. [DOI] [PubMed] [Google Scholar]
- Glykys J, Peng Z, Chandra D, Homanics GE, Houser CR, Mody I. A new naturally occurring GABA(A) receptor subunit partnership with high sensitivity to ethanol. Nat Neurosci. 2007;10:40–48. doi: 10.1038/nn1813. [DOI] [PubMed] [Google Scholar]
- Gothelf D, Furfaro JA, Hoeft F, Eckert MA, Hall SS, O'Hara R, Erba HW, Ringel J, Hayashi KM, Patnaik S, Golianu B, Kraemer HC, Thompson PM, Piven J, Reiss AL. Neuroanatomy of fragile X syndrome is associated with aberrant behavior and the fragile X mental retardation protein (FMRP) Ann Neurol. 2008;63:40–51. doi: 10.1002/ana.21243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagerman RJ, Berry-Kravis E, Kaufmann WE, Ono MY, Tartaglia N, Lachiewicz A, Kronk R, Delahunty C, Hessl D, Visootsak J, Picker J, Gane L, Tranfaglia M. Advances in the treatment of fragile X syndrome. Pediatrics. 2009;123:378–390. doi: 10.1542/peds.2008-0317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harlow EG, Till SM, Russell TA, Wijetunge LS, Kind P, Contractor A. Critical period plasticity is disrupted in the barrel cortex of Fmr1 knockout mice. Neuron. 2010;65:385–398. doi: 10.1016/j.neuron.2010.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hensch TK. Critical period mechanisms in developing visual cortex. Curr Top Dev Biol. 2005;69:215–237. doi: 10.1016/S0070-2153(05)69008-4. [DOI] [PubMed] [Google Scholar]
- Hessl D, Rivera SM, Reiss AL. The neuroanatomy and neuroendocrinology of fragile X syndrome. Ment Retard Dev Disabil Res Rev. 2004;10:17–24. doi: 10.1002/mrdd.20004. [DOI] [PubMed] [Google Scholar]
- Huber KM, Gallagher SM, Warren ST, Bear MF. Altered synaptic plasticity in a mouse model of fragile X mental retardation. Proc Natl Acad Sci U S A. 2002;99:7746–7750. doi: 10.1073/pnas.122205699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huntsman MM, Huguenard JR. Nucleus-specific differences in GABA(A)-receptor-mediated inhibition are enhanced during thalamic development. J Neurophysiol. 2000;83:350–358. doi: 10.1152/jn.2000.83.1.350. [DOI] [PubMed] [Google Scholar]
- Huntsman MM, Porcello DM, Homanics GE, DeLorey TM, Huguenard JR. Reciprocal inhibitory connections and network synchrony in mammalian thalamus. Science. 1999;283:541–543. doi: 10.1126/science.283.5401.541. [DOI] [PubMed] [Google Scholar]
- Johnston MV. Fresh ideas for treating developmental cognitive disorders. Curr Opin Neurol. 2006;19:115–118. doi: 10.1097/01.wco.0000218224.43184.b0. [DOI] [PubMed] [Google Scholar]
- Kish SJ, Distefano LM, Dozic S, Robitaille Y, Rajput A, Deck JH, Hornykiewicz O. [3H]Vesamicol binding in human cholinergic deficiency disorders. Neurosci Lett. 1990;117:347–352. doi: 10.1016/0304-3940(90)90689-7. [DOI] [PubMed] [Google Scholar]
- Krook-Magnuson EI, Li P, Paluszkiewicz SM, Huntsman MM. Tonically active inhibition selectively controls feedforward circuits in mouse barrel cortex. J Neurophys. 2008;100:932–944. doi: 10.1152/jn.01360.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeDoux J. The emotional brain, fear, and the amygdala. Cell Mol Neurobiol. 2003;23:727–738. doi: 10.1023/A:1025048802629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li R, Nishijo H, Wang Q, Uwano T, Tamura R, Ohtani O, Ono T. Light and electron microscopic study of cholinergic and noradrenergic elements in the basolateral nucleus of the rat amygdala: evidence for interactions between the two systems. J Comp Neurol. 2001;439:411–425. doi: 10.1002/cne.1359. [DOI] [PubMed] [Google Scholar]
- Maguire JL, Stell BM, Rafizadeh M, Mody I. Ovarian cycle-linked changes in GABA(A) receptors mediating tonic inhibition alter seizure susceptibility and anxiety. Nat Neurosci. 2005;8:797–804. doi: 10.1038/nn1469. [DOI] [PubMed] [Google Scholar]
- McNaughton CH, Moon J, Strawderman MS, Maclean KN, Evans J, Strupp BJ. Evidence for social anxiety and impaired social cognition in a mouse model of fragile X syndrome. Behav Neurosci. 2008;122:293–300. doi: 10.1037/0735-7044.122.2.293. [DOI] [PubMed] [Google Scholar]
- Medrihan L, Tantalaki E, Aramuni G, Sargsyan V, Dudanova I, Missler M, Zhang W. Early defects of GABAergic synapses in the brain stem of a MeCP2 mouse model of Rett syndrome. J Neurophysiol. 2008;99:112–121. doi: 10.1152/jn.00826.2007. [DOI] [PubMed] [Google Scholar]
- Moy SS, Nadler JJ. Advances in behavioral genetics: mouse models of autism. Mol Psychiatry. 2008;13:4–26. doi: 10.1038/sj.mp.4002082. [DOI] [PubMed] [Google Scholar]
- Pacey LK, Doering LC. Developmental expression of FMRP in the astrocyte lineage: implication for fragile X syndrome. Glia. 2007;55:1601–1609. doi: 10.1002/glia.20573. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Franklin KBJ. The mouse brain in stereotaxic coordinates. Ed 2. San Diego: Academic; 2001. [Google Scholar]
- Pfeiffer BE, Huber KM. Fragile X mental retardation protein induces synapse loss through acute postsynaptic translational regulation. J Neurosci. 2007;27:3120–3130. doi: 10.1523/JNEUROSCI.0054-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ragozzino D, Woodward RM, Murata Y, Eusebi F, Overman LE, Miledi R. Design and in vitro pharmacology of a selective gamma-aminobutyric acid C receptor antagonist. Mol Pharmacol. 1996;50:1024–1030. [PubMed] [Google Scholar]
- Rubenstein JL, Merzenich MM. Model of autism: increase ratio of excitation/inhibition in key neural systems. Genes Brain Behav. 2003;2:255–267. doi: 10.1034/j.1601-183x.2003.00037.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sah P, Faber ES, Lopez De Armentia M, Power J. The amygdaloid complex: anatomy and physiology. Physiol Rev. 2003;83:803–834. doi: 10.1152/physrev.00002.2003. [DOI] [PubMed] [Google Scholar]
- Selby L, Zhang C, Sun QQ. Major defects in neocortical GABAergic circuits in mice lacking the fragile X mental retardation protein. Neurosci Lett. 2007;412:227–232. doi: 10.1016/j.neulet.2006.11.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semyanov A, Walker MC, Kullmann DM, Silver RA. Tonically active GABA A receptors: modulating gain and maintaining the tone. Trends Neurosci. 2004;27:262–269. doi: 10.1016/j.tins.2004.03.005. [DOI] [PubMed] [Google Scholar]
- Suvrathan A, Hoeffer CA, Wong H, Klann E, Chattarji S. Characterization and reversal of synaptic defects in the amygdala in a mouse model of fragile X syndrome. Proc Natl Acad Sci U S A. 2010;107:11591–11596. doi: 10.1073/pnas.1002262107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabadics J, Tamás G, Soltesz I. Different transmitter transients underlie presynaptic cell type specificity of GABAASlow and GABAAFast. Proc Natl Acad Sci U S A. 2007;104:14831–14836. doi: 10.1073/pnas.0707204104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabuchi K, Blundell J, Etherton MR, Hammer RE, Liu X, Powell CM, Südhof TC. A neuroligin-3 mutation implicated in autism increases inhibitory synaptic transmission in mice. Science. 2007;318:71–76. doi: 10.1126/science.1146221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tervonen TA, Louhivuori V, Sun X, Hokkanen ME, Kratochwil CF, Zebryk P, Castrén E, Castrén ML. Aberrant differentiation of glutamatergic cells in neocortex of mouse model for fragile X syndrome. Neurobiol Dis. 2009;33:250–259. doi: 10.1016/j.nbd.2008.10.010. [DOI] [PubMed] [Google Scholar]
- Tian N, Petersen C, Kash S, Baekkeskov S, Copenhagen D, Nicoll R. The role of the synthetic enzyme GAD65 in the control of neuronal gamma-aminobutyric acid release. Proc Natl Acad Sci U S A. 1999;96:12911–12916. doi: 10.1073/pnas.96.22.12911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson C, Hoeft F, Garrett AS, Hall SS, Reiss AL. Aberrant brain activation during gaze processing in boys with fragile X syndrome. Arch Gen Psychiatry. 2008;65:1315–1323. doi: 10.1001/archpsyc.65.11.1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenk GL, Mobley SL. Choline acetyltransferase activity and vesamicol binding in Rett Syndrome and in rats with nucleus basalis lesions. Neuroscience. 1996;73:79–84. doi: 10.1016/0306-4522(96)00019-x. [DOI] [PubMed] [Google Scholar]
- Zhao MG, Toyoda H, Ko SW, Ding HK, Wu LJ, Zhuo M. Deficits in trace fear memory and long-term potentiation in a mouse model of fragile X syndrome. J Neurosci. 2005;25:7385–7392. doi: 10.1523/JNEUROSCI.1520-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoghbi HY. Postnatal neurodevelopmental disorders: meeting at the synapse? Science. 2003;302:826–830. doi: 10.1126/science.1089071. [DOI] [PubMed] [Google Scholar]