

Abstract
Rationale
Hepatic lipase (HL) and endothelial lipase (EL) are extracellular lipases that both hydrolyze triglycerides and phospholipids and display potentially overlapping or complementary roles in lipoprotein metabolism.
Objective
We sought to dissect the overlapping roles of HL and EL by generating mice deficient in both HL and EL (HL/EL-dko) for comparison with single HL-knockout (ko) and EL-ko mice, as well as wild-type mice.
Methods and Results
Reproduction and viability of the HL/EL-dko mice were impaired compared with the single-knockout mice. The plasma levels of total cholesterol, high-density lipoprotein (HDL) cholesterol, non–HDL cholesterol, and phospholipids in the HL/EL-dko mice were markedly higher than those in the single-knockout mice. Most notably, the HL/EL-dko mice exhibited an unexpected substantial increase in small low-density lipoproteins. Kinetic studies with [3H]cholesteryl ether–labeled very-low-density lipoproteins demonstrated that the HL/EL-dko mice accumulated counts in the smallest low-density lipoprotein–sized fractions, as assessed by size exclusion chromatography, suggesting that it arises from lipolysis of very-low-density lipoproteins. HDL from all 3 lipase knockout models had an increased cholesterol efflux capacity but reduced clearance of HDL cholesteryl esters versus control mice. Despite their higher HDL cholesterol levels, neither HL-ko, EL-ko, nor HL/EL-dko mice demonstrated an increased rate of macrophage reverse cholesterol transport in vivo.
Conclusions
These studies reveal an additive effect of HL and EL on HDL metabolism but not macrophage reverse cholesterol transport in mice and an unexpected redundant role of HL and EL in apolipoprotein B lipoprotein metabolism.
Keywords: lipase, lipoprotein, knockout mice, low-density lipoprotein metabolism, reverse cholesterol transport
Hepatic lipase (HL) and endothelial lipase (EL) are members of an extracellular lipase family that includes lipoprotein lipase (LPL).1–6 These 3 heparin-binding lipases are anchored to the endothelial surface and mediate the hydrolysis of triglycerides (TGs) and phospholipids (PLs) within circulating lipoproteins. Although all 3 lipases have both TG and PL hydrolytic activity, LPL is predominantly a TG lipase, whereas EL is predominantly a phospholipase; HL shares an intermediate TG lipase and phospholipase activity.7,8 In addition to their catalytic functions, these lipases are capable of “bridging” lipoproteins with cell surface proteoglycans independently of hydrolytic activity.9–12
Both HL and EL influence the metabolism of high-density lipoproteins (HDLs). Overexpression of HL in mice reduces plasma HDL levels13–16; in contrast, HL-ko mice have significantly elevated plasma levels of HDL.17 Likewise, the overexpression of EL in mice substantially reduces plasma HDL,5,18 whereas the loss of EL activity in mice significantly raises plasma HDL.18,19 Furthermore, numerous human studies have demonstrated that plasma HDL cholesterol (HDL-C) levels are strongly associated with variations in the genes encoding HL (LIPC)20–24 and EL (LIPG).23–26 However, the interaction between HL and EL on influencing HDL metabolism and its key function of reverse cholesterol transport (RCT) is unknown.
HL also modulates the metabolism of apolipoprotein (apo)B-containing lipoproteins (LpBs). Overexpression of HL in mice significantly reduces plasma levels of LpBs13,15,16; in contrast, HL-knockout (ko) mice fed a high-fat diet exhibit a modest increase in plasma LpB levels,17 which is exacerbated in the absence of apoE27 or the low-density lipoprotein (LDL) receptor.28 Genetic variation in LIPC is associated with TG and LpB levels,29–32 and human deficiency of HL causes elevated LpB levels.33 In contrast to HL, the role of EL in the metabolism of LpBs is uncertain. In vitro, EL is capable of hydrolyzing very-low-density lipoprotein (VLDL) TG and PL.8 Moderate transgenic overexpression of EL does not affect LpB levels,18 but adenoviral-mediated overexpression of EL in mice reduces LpB levels.34 Furthermore, very modest elevations of LpBs in the plasma were observed in EL-ko mice lacking either apoE35,36 or the LDL receptor.36 Human genetic studies to date have not associated LIPG with variation in LDL cholesterol or TG levels. Thus, the role of EL in LpB metabolism is uncertain, and its potential interaction with HL in modulating LpB metabolism is unknown.
Available data suggest that HL and EL may share a significant overlap in their roles in modulating HDL and LpB metabolism. It is plausible that EL and HL have a complementary relationship, but it is also possible that hidden functions exist for HL and EL that may be masked by redundancy or compensation. To investigate these possibilities, we generated mice deficient in both HL and EL (HL/EL-dko) and compared them with HL-ko, EL-ko, and wild-type (WT) mice in their effects on both HDL and LpB metabolism.
Methods
An expanded Methods section is available in the Online Data Supplement at http://circres.ahajournals.org.
HL/EL-double heterozygous mice were generated by crossing HL-ko and EL-ko mice. The double heterozygous mice were used to obtain the first generations of mice lacking both HL and EL (HL/EL-dko). The HL/EL-dko mice were subsequently obtained by breeding either the HL/EL-dko mice or the HL/EL-double heterozygous mice. Lipids from fasted plasma and fast performance liquid chromatography (FPLC) samples were quantified using commercially available kits. Apolipoproteins were detected by immunoblot analyses. Human and mouse lipoproteins were isolated by density gradient ultracentrifugation. Human VLDL was radiolabeled with [3H]cholesteryl ether (CEt), and human HDL3 was radiolabeled with [3H]CEt and [125I]tyramine cellobiose (TCB). Cholesterol efflux and in vivo RCT studies were performed using established methods. For metabolic studies, radiolabeled HDL3 was injected intravenously and plasma were collected at 2 minutes and at 1, 2, 4, 6, 9, and 24 hours postinjection for analyses. Radiolabeled VLDL was injected intravenously, and plasma were collected at 2 and 20 minutes and at 5 and 24 hours postinjection for analyses. All studies were approved by the University of Pennsylvania Institutional Animal Care and Use Committee.
Results
Reproduction and Viability of HL/EL-dko Mice
To dissect the roles of HL and EL in lipoprotein metabolism, we generated HL/EL-dko mice for comparison and contrast with HL-ko, EL-ko, and WT mice. Like the HL-ko and EL-ko mouse models, the HL/EL-dko mice were viable. However, we observed significantly reduced litter sizes from the breeding of HL/EL-dko mice versus both HL-ko and EL-ko mice, which were similar to each other (Online Table I). We also noted from the HL/EL-dko litters that 2 pups were deceased at birth because of a lack of abdominal organs and 3 mice died before 12 weeks of age because of an infection that was secondary to poor healing of the umbilicus; no such observations were recorded with the HL-ko and EL-ko litters. These observations raise the possibility that HL or EL may have a novel function in development and vascular repair that warrants future exploration.
Plasma Lipids and Lipoproteins of HL/EL-dko Mice
This is the first time HL-ko and EL-ko mice have been examined side-by-side together with the HL/EL-dko lipid profiles. Using sex- and age-matched mice, our data show that both the HL-ko and EL-ko mice have elevated plasma levels of total cholesterol (TC), HDL-C, and PL compared to control mice (Figure 1). The deletion of EL has a more pronounced effect on plasma lipids versus the deletion of HL, likely because of the loss of the greater phospholipase activity of EL versus HL.8 The deletion of both HL and EL from mice raised plasma lipids beyond those observed with the individual knockouts. A gene dosage effect with the removal of HL or EL alleles leading to the double knockout was observed for the increase of plasma TC, HDL-C, and PL (Online Figure I). Plasma TG levels were comparable among all groups of mice (Figure 1D; Online Figure I, D).
Figure 1. The HL/EL-dko mice exhibit elevated plasma cholesterol and PL compared to WT and individual lipase knockout mice.

Fasted plasma from 2- to 3-month-old sex-matched WT, HL-ko, EL-ko, and HL/EL-dko (DKO) mice were assessed for total cholesterol (A), HDL-C (B), PL (C), and TG (D). The number of mice assessed (n) are indicated. *P<0.001 vs WT. All errors are ±SD.
We next assessed the plasma levels of apolipoproteins from the 4 groups of mice by immunoblot analyses (Figure 2) and densitometry of the immunoblots (Online Figure II). We observed a very striking 19-fold increase of apoB-100 in the HL/EL-dko plasma versus WT plasma. Plasma from both HL-ko and EL-ko mice displayed a less profound 7-fold increase of apoB-100 versus control mice. In contrast, only modest increases of apoB-48 were observed in the plasma from HL-ko, EL-ko, and HL/EL-dko mice. Plasma levels of apoE were 2-fold greater in the HL/EL-dko mice versus the control mice and 40% higher in both HL-ko and EL-ko mice. We observed no difference in the plasma levels of apoA-I between the HL-ko and WT mice and only a modest 13% and 18% increase in the plasma levels of apoA-I within the EL-ko and HL/EL-dko mice, respectively. The trends for plasma levels of apoB-100 and apoA-I correlate with the plasma particle concentrations of total LDL and HDL, respectively, as determined by nuclear magnetic resonance (Online Figure III). Notably, plasma very small LDL levels in the HL/EL-dko mice were substantially elevated compared to those within WT, HL-ko, and EL-ko mice.
Figure 2. Plasma levels of apoB, apoE, and apoA-I are elevated in the HL/EL-dko mice.
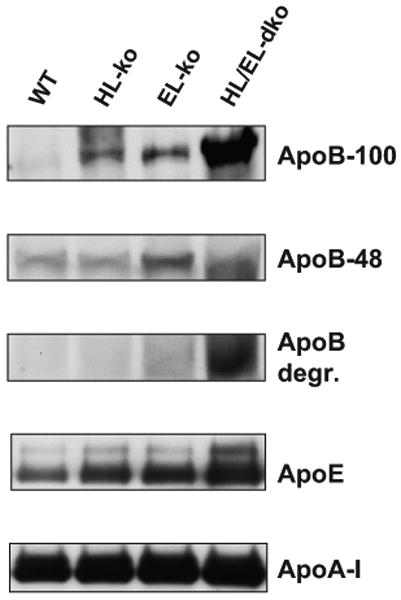
Fasted plasma proteins from 2-to 3-month-old sex-matched WT, HL-ko, EL-ko, and HL/EL-dko mice were separated by denaturing polyacrylamide gel electrophoresis, transferred to nitrocellulose, and subjected to chemiluminescent immunoblot analyses for apoB-100, apoB-48, apoE, and apoA-I. Apolipoproteins were quantified by scanning densitometry from 4 different sets of mice and data were graphed (see Online Figure II).
We separated lipoproteins within pooled plasma from our 4 groups of mice by FPLC (Figure 3). HDL-C was elevated in the 3 knockout groups versus control mice, in the order of control<HL-ko<EL-ko<HL/EL-dko. Unexpectedly, we observed a distinct peak from the HL/EL-dko plasma that was present in the smallest (s)LDL fractions; this peak was not observed from the plasma of either the HL-ko or EL-ko mice. A notable presence of apoB-100 (relative to apoB-48) and apoE was observed with this peak from the HL/EL-dko profile versus the WT profile (Online Figure IV). When the HL/EL-dko mice were placed on a Western diet for 6 months, this peak was greatly exacerbated (Online Figure V, A). In an attempt to determine whether the lipase bridging function alone would remove the sLDL from plasma, we expressed a catalytically inactive EL at supraphysiological levels in the HL/EL-dko mice by using adenoviruses. However, after 7 days of expression, catalytically inactive EL only modestly reduced cholesterol levels across all fractions relative to preinfection levels (Online Figure V, B); thus, it is likely that the activities of HL or EL are required for the clearance of sLDL.
Figure 3. Elevated cholesterol in the HDL and sLDL fractions of plasma from the HL/EL-dko mice.

Fasted plasma samples from six 2- to 3-month-old sex-matched WT (diamonds), HL-ko (squares), EL-ko (triangles), and HL/EL-dko mice (circles) were pooled (total of 100 μL) and fractionated (500 μL of each fraction) by FPLC. Total cholesterol from each fraction was quantified. IDL/LDL indicates intermediate-density lipoprotein and LDL. The sLDL cholesterol peak identified from the plasma of HL/EL-dko mice is indicated (arrow).
We also isolated LDL and HDL within plasma from the 4 groups of mice by ultracentrifugation and determined their TC, PL, TG, and protein compositions as a mass percentage (Online Figure VI). Compared to LDL from control mice, the LDL from HL/EL-dko mice had no appreciable change of TC content but had notable differences for TG (81% lower), PL (37% higher), and protein (29% lower) (Online Figure VI, A). The marked reduction for LDL-TG and increased LDL-PL from the HL/EL-dko mice likely point to the hydrolysis of TG (likely by LPL) to generate a smaller LDL particle that is enriched with PL because of the lack of phospholipase activity from either HL or EL. Interestingly, the HL/EL-dko mice have a 45% elevation of postheparin plasma TG lipase activity (Online Figure VII). The composition of LDL isolated from HL-ko and EL-ko mice also differed versus LDL from control mice. Compared to HDL from control mice, the HDL from HL/EL-dko mice also had no appreciable change of TC and TG content but had differences for PL (33% higher) and protein (16% lower) (Online Figure VI, B). The composition of HDL isolated from HL-ko and EL-ko mice also differed versus HDL from control mice.
Identifying the Precursor of the sLDL Within HL/EL-dko Mice
The combination of markedly elevated plasma apoB-100 levels, LDL particle concentrations, FPLC profiles, and LDL composition are all consistent with the accumulation of sLDL that may be derived from VLDL within the plasma of HL/EL-dko mice. To determine whether the sLDL is derived from abnormal VLDL catabolism, we injected [3H]CEt-labeled VLDL into HL/EL-dko and control mice and followed the progression of the [3H]CEt in the plasma over 24 hours by FPLC analyses (Figure 4). As expected, the association of [3H]CEt progressed from VLDL to LDL throughout the study. Some [3H]CEt was associated with HDL, but this may be attributable, in part, to de novo lipoprotein formation driven by the association of endogenous apoA-I with lipids shed from VLDL. At 5 hours after injection, [3H]CEt was associated with the LDL fractions in both WT and HL/EL-dko mice (Figure 4C). By 24 hours, most of the 3H label had cleared, but there was a clear difference in the LDL fractions between the 2 groups of mice: the control animals accumulated [3H]CEt in the normal-sized LDL range, whereas this peak was shifted toward sLDL in the HL/EL-dko mice (Figure 4D). These data confirm that this lipoprotein is derived from VLDL and it accumulates when LDL cannot be removed from the circulation in the HL/EL-dko mice.
Figure 4. VLDL is the precursor for the formation of the sLDL particles in the HL/EL-dko mice.

Pooled plasma from 4- to 6-month-old sex-matched WT mice and HL/EL-dko mice (used as described in Methods) were diluted, and lipoproteins were separated by FPLC to identify the location of the sLDL peak (arrows) (A). [3H]CEt-labeled human VLDL was injected into these mice and plasma lipoproteins were collected and separated by FPLC at 20 minutes after injection (B), 5 hours after injection (C), and 24 hours after injection (D). Each fraction was assessed for [3H]CEt content and is presented as a percentage of initial [3H]CEt injected. VLDL, fractions 3 to 8. IDL/LDL, fractions 10 to 24. sLDL, fractions 25 to 30. HDL, fractions 31 to 43.
Cholesterol Efflux Ex Vivo and RCT In Vivo
The differences observed in HDL composition among the control, HL-ko, EL-ko, and HL/EL-dko mice suggested that the abilities of HDL to efflux cholesterol and affect RCT may also vary. We tested the ability of both plasma and isolated HDL from the 4 groups of mice to efflux [3H]cholesterol from J774 macrophages ex vivo (Online Figure VIII). The efflux capacities of equal quantities of plasma from HL-ko and EL-ko mice were comparable but significantly greater than control plasma, whereas the HL/EL-dko plasma had a significantly greater capacity versus the control and individual knockout plasma (Online Figure VIII, A). The efflux capacities of equal quantities of HDL from HL-ko and EL-ko mouse plasma were comparable but significantly greater than HDL from control plasma, whereas the HDL from HL/EL-dko plasma had a significantly greater capacity versus the control and individual knockout HDL (Online Figure VIII, B).
The ex vivo efflux data combined with the HDL composition data suggest that a higher HDL-PL content attributable to the loss of phospholipase activity within the circulation would allow for greater efflux of peripheral cholesterol to HDL. To determine whether this increased efflux capacity would be beneficial in vivo, we measured macrophage-to-feces RCT in the lipase knockout mice. To accomplish this, [3H]cholesterol-labeled J774 macrophages were injected into the intraperitoneal cavity, and the appearance of macrophage-derived [3H]cholesterol in the plasma and feces was monitored for 48 hours (Figure 5). Over 48 hours, the amount of [3H]sterol in the plasma of HL-ko mice was higher than that observed in the plasma of control mice (Figure 5A). The [3H]sterol levels in the plasma from the EL-ko mice were significantly greater than those in the HL-ko mice (Figure 5A). The plasma [3H]sterol levels in the HL/EL-dko mice trended higher than those for the EL-ko plasma (Figure 5A) but were not statistically significant. Despite higher HDL-C levels and increased [3H]sterol in the plasma of knockout mice, the fecal excretion of [3H]sterol among control, HL-ko, EL-ko, and HL/EL-dko mice was not significantly different (Figure 5B).
Figure 5. RCT is unaffected in lipase-deficient mice, despite markedly elevated [3H]cholesterol in the plasma.

Age-matched 4- to 6-month-old male WT, HL-ko, EL-ko, and HL/EL-dko (DKO) mice (n=6 for each group) were injected with [3H]cholesterol-labeled macrophages intraperitoneally and [3H]sterol levels (presented as percentage of total [3H] injected) were assessed postinjection. A, Plasma [3H]sterol levels were measured from WT (diamonds), HL-ko (squares), EL-ko (triangles), and HL/EL-dko mice (circles) at 6, 24, and 48 hours postinjection. *P<0.05 vs WT; **P<0.001 vs WT. B, Total fecal [3H]sterol levels collected over 48 hours. All errors are ±SD.
Catabolism and Selective Uptake of HDL3
Although HL-ko, EL-ko, and HL/EL-dko mice all had higher HDL-C levels and increased plasma levels of macrophage-derived [3H]sterol in our RCT studies, the overall rate of macrophage-to-feces RCT, as assessed by fecal excretion of macrophage-derived [3H]sterol, was surprisingly unaffected. We hypothesized that this might be attributable to impaired clearance of HDL from the plasma. To address this possibility, we injected [125I]TCB/[3H]CEt-labeled HDL3 into the 4 groups of mice and compared their plasma clearance. The fractional catabolic rates (FCRs) for the clearance of plasma [3H]CEt were significantly slower for both the EL-ko and HL/EL-dko mice versus control mice but not significantly different from each other (Table). The FCRs for the clearance of plasma [125I]TCB-labeled proteins in the HL-ko and EL-ko mice were significantly slower than control mice but were not significantly different from each other (Table). The whole-body selective uptake (calculated as the difference between the FCR for [3H]CEt and [125I]TCB) was comparable between HL-ko and control mice but was significantly lower in the EL-ko and HL/EL-dko mice versus the control mice (Table). These data clearly show the importance of EL in the selective uptake of cholesteryl ester from HDL3 and it solidifies the notion that EL is the preferential lipase associated with whole-body HDL3 catabolism compared with HL.
Table. FCRs of [3H]CEt- and [125I]TCB-Labeled Protein From Human HDL3 by Lipase Knockout Mice.
| Strain | [3H]CEt FCR (d−1) |
[125I]TCB FCR (d−1) |
CEt Selective Uptake ([3H]CEt FCR–[125I]TCB FCR) |
|---|---|---|---|
| WT (C57BL/6) | 5.36±0.22 | 1.87±0.23 | 3.49±0.32 |
| HL-ko | 5.07±0.28 | 1.41 ±0.09* | 3.66±0.25 |
| EL-ko | 4.29±0.36‡ | 1.35±0.34† | 2.94±0.25* |
| HL/EL-dko | 4.47±0.13‡ | 1.54±0.21 | 2.93±0.34* |
FCRs for CEt and TCB were calculated as described in Methods (n=6 for each group).
P<0.05,
P<0.01,
P<0.001 vs WT.
Discussion
Although HL and EL display overlapping catalytic functions and lipoprotein substrate specificities, important physiological distinctions exist between these lipases. The HL/EL-dko mice share some similarities with the individual knockout mice, but they also have some remarkable differences. The greatly increased HDL in these mice improves cholesterol efflux capacity but does not increase RCT because of reduced whole-body uptake of HDL-derived cholesterol. Our study also uncovered unexpected redundant roles for both HL and EL in the metabolism of LpBs, such that a combined deficiency of these lipases results in an accumulation of sLDL.
The HL/EL-dko mice are viable, which is in contrast to the LPL-ko mice, which die as neonates.37,38 However, our data suggest that both HL and EL share overlapping roles in reproduction and survival. Wade et al39 previously addressed the impaired reproduction ability of HL-ko mice. Litter sizes for HL-ko mice in our study exactly match those of previous reported data39; litter sizes of HL-ko mice and EL-ko mice are comparable, which suggests that a reproductive impairment may exist in the absence of EL. The HL/EL-dko mice exhibit a significant impairment compared to both HL-ko and EL-ko mice, which is manifested by markedly reduced litter sizes from dko-dko breeding, as well as early neonatal lethality. The placenta was identified to have one of the highest levels of EL expression,5 yet its function in this tissue remains to be investigated. Although the causes for reduced litter sizes in EL-ko and HL/EL-dko mice remain to be determined, it may be similar to the reduced progesterone levels that lead to impaired ovulation in HL-ko mice,39 or it may be attributable to the loss of the undefined role of placental EL, which is augmented by reduced ovulation in the absence of HL. The roles of HL and EL in these processes are poorly understood and require further future studies.
The unexpected presence of a PL-rich sLDL in the HL/EL-dko mice clearly shows that HL and EL play a redundant role in the normal catabolism of LDL, which is otherwise masked in the HL-ko and EL-ko mice. Hepatic HL and EL mRNA from the HL-ko and EL-ko mice showed that EL and HL, respectively, were not elevated versus control mice (data not shown); thus, it is unlikely that a compensatory mechanism of transcriptional upregulation of the other lipase in the single-knockout models prevented the observation of sLDL. Our data point to a mechanism whereby the phospholipase activities of either HL or EL are necessary for the effective processing of LpBs and prevention of the accumulation of sLDL. Although LPL has a low phospholipase activity relative to HL and EL,8 the activity appears to be insufficient in the HL/EL-dko mice to properly hydrolyze LDL lipids to effectively promote catabolism.
We postulate that sLDL accumulates in the HL/EL-dko mice as a result of aberrant LDL catabolism mediated by LPL. Previous studies have demonstrated that elevated activities of LPL or HL contribute to the formation of sLDL.40 This may be attributable to the increased TG lipase activity within the plasma relative to the combined phospholipase activities by HL and EL. The HL/EL-dko mouse model eliminates the major phospholipase activities toward plasma lipoproteins, thus dramatically shifting the balance of plasma lipase activity toward TG lipase because of the remaining activity by LPL. Effective hydrolysis of PL by HL and EL in LpBs undergoing TG lipolysis by LPL may be required for the efficient metabolism of LDL, and in the absence of both HL and EL, a PL-rich and TG-depleted sLDL particle is the result. Another potential possibility to account for the presence of sLDL is an upregulation of LPL activity to compensate for the loss of TG lipase activities from both HL and EL. Although epidemiological studies support a proatherogenic role for sLDL,41–43 the causality of this relationship has not been directly and cleanly examined in an animal model. The HL/EL-dko mice may be a useful model to study the effects of sLDL on atherogenesis, particularly given that when these mice are fed a Western diet, the levels of sLDL are further increased.
The additive effect of HL and EL deficiency on plasma levels of HDL-C demonstrates that these enzymes work in concert to modulate HDL remodeling and metabolism. We performed additional studies to assess the effects on HDL function with regard to cholesterol efflux and RCT. The deficiency of HL, EL, or both increased the capacity of HDL to accept cholesterol from macrophages, likely because of the increased HDL-PL content and a larger HDL particle number. However, the increased efflux capacity is offset by a slower clearance of HDL-C from the circulation; thus, macrophage RCT is ultimately unaffected. Pharmacological inhibition of EL, with or without additional HL inhibition, might be expected to raise HDL-C levels and improve cholesterol efflux capacity, but the effect of this approach on RCT and atherogenesis remains uncertain.
Unlike humans, mice do not have cholesteryl ester transfer protein (CETP), which exchanges TG from LpBs with cholesteryl esters from HDL.44 Hydrolysis of TG-enriched HDL by lipases enhances HDL clearance44 while shedding apoA-I from the particle.45 ApoA-I could accept cholesterol from macrophages and thus contribute to enhanced RCT in mice expressing CETP.46 We speculate that overexpressing CETP in HL-ko or EL-ko mice might improve RCT; however, no enhancement may be observed in the HL/EL-dko mice. We further hypothesize that the expression of CETP in HL/EL-dko mice would result in a significant accumulation of sLDL, which might amplify any atherogenic phenotype. Thus, in humans, EL inhibition might have an adverse effect on the metabolism of LpBs, particularly if HL is also partially inhibited.
In summary, deletion of both HL and EL in mice yielded a novel phenotype of extremely high HDL-C without an overall increase in macrophage RCT, and an unexpected substantial increase in plasma apoB-100 and sLDL. This demonstrates the complementary effects of HL and EL on the metabolism of both HDL and LpBs. It also raises important questions about the advisability of pharmacological inhibition of EL, particularly if HL were also partially inhibited.
Novelty and Significance.
What Is Known?
Hepatic lipase (HL) and endothelial lipase (EL) are extracellular heparin-binding lipases that hydrolyze the triglycerides and phospholipids associated with circulating lipoproteins.
HL and EL appear to have overlapping roles in modulating the metabolism of apolipoprotein B-containing lipoproteins (LpBs) and high-density lipoproteins (HDLs).
Targeted deletion of either HL or EL in animal models raises HDL levels, which might be predicted to reduce atherosclerosis by promoting reverse cholesterol transport.
What New Information Does This Article Contribute?
HL and EL share a redundant role in LpB metabolism; in the absence of both enzymes in mice, a very small low-density lipoprotein particle accumulates in plasma, likely because of impaired phospholipid hydrolysis coupled with increased triglyceride lipase activity by lipoprotein lipase.
HL and EL share a redundant role in HDL metabolism; a combined deficiency of both enzymes results in a substantial increase in HDL cholesterol (HDL-C) levels, but does not improve in vivo macrophage reverse cholesterol transport likely because of a slow clearance of HDL from the plasma.
Both HL and EL are members of a family of extracellular lipases that also includes lipoprotein lipase. Each of these lipases can modulate the levels of plasma lipoproteins by hydrolyzing their triglycerides and phospholipids. Previously published in vitro and in vivo studies suggest that HL and EL share a significant overlap in the metabolism of HDL, thus making either of the 2 lipases desirable candidates for pharmacological inhibition to raise plasma HDL levels and, thereby, reducing atherosclerotic burden. We attempted to dissect the physiological roles of HL and EL by generating mice deficient in both lipases (HL/EL-dko) for comparison with single HL-knockout (ko) and EL-ko mice, as well as wild-type control mice. We observed elevated HDL-C in the plasma all knockout models. The elevated HDL-C in the HL/EL-dko mice resulted in increased cholesterol efflux from macrophages, but this did not translate into improved reverse cholesterol transport in vivo. Unexpectedly, we also observed a significant accumulation of potentially proatherogenic sLDL particles in the plasma of HL/EL-dko mice but not in the plasma of single-knockout mice. Overall, our study questions the advisability of EL inhibition as a therapeutic target, particularly if HL inhibition cannot be excluded.
Supplementary Material
Acknowledgments
We thank our animal care staff (Edwige Edouard, Aisha Faruqi, and Mao-Sen Sun), Debra Cromley, Anna DiFlioro, Michelle Joshi, Linda Morrell, and Amrith Rodrigues. We also appreciate the assistance from some of our students with genotyping. We thank Dr John Millar (University of Pennsylvania) for advice with modeling our fractional catabolic studies and Dr Jeff Billheimer (University of Pennsylvania) for advice with some of our animal studies.
Sources of Funding: This work was supported by NIH grants HL022633 and HL55323 from the National Heart, Lung, and Blood Institute; an Established Investigator Award from the American Heart Association; and a Burroughs Wellcome Fund Clinical Scientist Award in Translational Research (to D.J.R.). R.J.B. was supported by a Research Fellowship of the Heart & Stroke Foundation of Canada.
Non-standard Abbreviations and Acronyms
- apo
apolipoprotein
- CEt
cholesteryl ether
- CETP
cholesteryl ester transfer protein
- EL
endothelial lipase
- FCR
fractional catabolic rate
- FPLC
fast-performance liquid chromatography
- HDL
high-density lipoprotein
- HDL-C
high-density lipoprotein cholesterol
- HL
hepatic lipase
- HL/EL-dko
hepatic lipase/endothelial lipase double-knockout
- ko
knockout
- LDL
low-density lipoprotein
- LPL
lipoprotein lipase
- PL
phospholipid
- RCT
reverse cholesterol transport
- sLDL
small low density lipoprotein
- TC
total cholesterol
- TCB
tyramine cellobiose
- TG
triglyceride
- VLDL
very-low-density lipoprotein
- WT
wild type
Footnotes
Disclosures: D.J.R. is a coinvestigator of a patent related to EL as a target for pharmacological inhibition and is a consultant to several companies with an interest in novel HDL-targeted therapies.
Contributor Information
Robert J. Brown, Department of Medicine and Institute for Translational Medicine and Therapeutics, University of Pennsylvania School of Medicine, Philadelphia
William R. Lagor, Department of Medicine and Institute for Translational Medicine and Therapeutics, University of Pennsylvania School of Medicine, Philadelphia
Sandhya Sankaranaravanan, Division of Gastroenterology and Nutrition, The Children's Hospital of Philadelphia, Pa.
Tomoyuki Yasuda, Department of Medicine and Institute for Translational Medicine and Therapeutics, University of Pennsylvania School of Medicine, Philadelphia.
Thomas Quertermous, Donald W. Reynolds Cardiovascular Clinical Research Center and Divisions of Cardiovascular Medicine and Gastroenterology, Stanford University School of Medicine, Stanford, Calif.
George H. Rothblat, Division of Gastroenterology and Nutrition, The Children's Hospital of Philadelphia, Pa
Daniel J. Rader, Department of Medicine and Institute for Translational Medicine and Therapeutics, University of Pennsylvania School of Medicine, Philadelphia
References
- 1.Stahnke G, Sprengel R, Augustin J, Will H. Human hepatic triglyceride lipase: cDNA cloning, amino acid sequence and expression in a cultured cell line. Differentiation. 1987;35:45–52. doi: 10.1111/j.1432-0436.1987.tb00150.x. [DOI] [PubMed] [Google Scholar]
- 2.Datta S, Luo CC, Li WH, VanTuinen P, Ledbetter DH, Brown MA, Chen SH, Liu SW, Chan L. Human hepatic lipase. Cloned cDNA sequence, restriction fragment length polymorphisms, chromosomal localization, and evolutionary relationships with lipoprotein lipase and pancreatic lipase. J Biol Chem. 1988;263:1107–1110. [PubMed] [Google Scholar]
- 3.Martin GA, Busch SJ, Meredith GD, Cardin AD, Blankenship DT, Mao SJT, Rechtin AE, Woods CW, Racke MM, Schafer MP, Fitzgerald MC, Burke DM, Flanagan MA, Jackson RL. Isolation and cDNA sequence of human postheparin plasma hepatic triglyceride lipase. J Biol Chem. 1988;263:10907–10914. [PubMed] [Google Scholar]
- 4.Hide WA, Chan L, Li WH. Structure and evolution of the lipase super-family. J Lipid Res. 1992;33:167–178. [PubMed] [Google Scholar]
- 5.Jaye M, Lynch KJ, Krawiec J, Marchadier D, Maugeais C, Doan K, South V, Amin D, Perrone M, Rader DJ. A novel endothelial-derived lipase that modulates HDL metabolism. Nat Genet. 1999;21:424–428. doi: 10.1038/7766. [DOI] [PubMed] [Google Scholar]
- 6.Hirata K, Dichek HL, Cioffi JA, Choi SY, Leeper NJ, Quintana L, Kronmal GS, Cooper AD, Quertermous T. Cloning of a unique lipase from endothelial cells extends the lipase gene family. J Biol Chem. 1999;274:14170–14175. doi: 10.1074/jbc.274.20.14170. [DOI] [PubMed] [Google Scholar]
- 7.Ehnholm C, Shaw W, Greten H, Brown WV. Purification from human plasma of a heparin-released lipase with activity against triglyceride and phospholipids. J Biol Chem. 1975;250:6756–6761. [PubMed] [Google Scholar]
- 8.McCoy MG, Sun GS, Marchadier D, Maugeais C, Glick JM, Rader DJ. Characterization of the lipolytic activity of endothelial lipase. J Lipid Res. 2002;43:921–929. [PubMed] [Google Scholar]
- 9.Ji ZS, Lauer SJ, Fazio S, Bensadoun A, Taylor JM, Mahley RW. Enhanced binding and uptake of remnant lipoproteins by hepatic lipase-secreting hepatoma cells in culture. J Biol Chem. 1994;269:13429–13436. [PubMed] [Google Scholar]
- 10.Krapp A, Ahle S, Kersting S, Hua Y, Kneser K, Nielsen M, Gliemann J, Beisiegel U. Hepatic lipase mediates the uptake of chylomicrons and beta-VLDL into cells via the LDL receptor-related protein (LRP) J Lipid Res. 1996;37:926–936. [PubMed] [Google Scholar]
- 11.Ji ZS, Dichek HL, Miranda RD, Mahley RW. Heparan sulfate proteoglycans participate in hepatic lipase and apolipoprotein E-mediated binding and uptake of plasma lipoproteins, including high density lipoproteins. J Biol Chem. 1997;272:31285–31292. doi: 10.1074/jbc.272.50.31285. [DOI] [PubMed] [Google Scholar]
- 12.Fuki IV, Blanchard N, Jin W, Marchadier DH, Millar JS, Glick JM, Rader DJ. Endogenously produced endothelial lipase enhances binding and cellular processing of plasma lipoproteins via heparan sulfate proteoglycan-mediated pathway. J Biol Chem. 2003;278:34331–34338. doi: 10.1074/jbc.M302181200. [DOI] [PubMed] [Google Scholar]
- 13.Busch SJ, Barnhart RL, Martin GA, Fitzgerald MC, Yates MT, Mao SJ, Thomas CE, Jackson RL. Human hepatic triglyceride lipase expression reduces high density lipoprotein and aortic cholesterol in cholesterol-fed transgenic mice. J Biol Chem. 1994;269:16376–16382. [PubMed] [Google Scholar]
- 14.Applebaum-Bowden D, Kobayashi J, Kashyap VS, Brown DR, Berard A, Meyn S, Parrott C, Maeda N, Shamburek R, Brewer HB, Jr, Santamarina-Fojo S. Hepatic lipase gene therapy in hepatic lipase-deficient mice. Adenovirus-mediated replacement of a lipolytic enzyme to the vascular endothelium. J Clin Invest. 1996;97:799–805. doi: 10.1172/JCI118479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dichek HL, Brecht W, Fan J, Ji ZS, McCormick SP, Akeefe H, Conzo L, Sanan DA, Weisgraber KH, Young SG, Taylor JM, Mahley RW. Overexpression of hepatic lipase in transgenic mice decreases apolipoprotein B-containing and high density lipoproteins. Evidence that hepatic lipase acts as a ligand for lipoprotein uptake. J Biol Chem. 1998;273:1896–1903. doi: 10.1074/jbc.273.4.1896. [DOI] [PubMed] [Google Scholar]
- 16.Braschi S, Couture N, Gambarotta A, Gauthier BR, Coffill CR, Sparks DL, Maeda N, Schultz JR. Hepatic lipase affects both HDL and apoB-containing lipoprotein levels in the mouse. Biochim Biophys Acta. 1998;1392:276–290. doi: 10.1016/s0005-2760(98)00046-0. [DOI] [PubMed] [Google Scholar]
- 17.Homanics GE, de Silva HV, Osada J, Zhang SH, Wong H, Borensztajn J, Maeda N. Mild dyslipidemia in mice following targeted inactivation of the hepatic lipase gene. J Biol Chem. 1995;270:2974–2980. doi: 10.1074/jbc.270.7.2974. [DOI] [PubMed] [Google Scholar]
- 18.Ishida T, Choi S, Kundu RK, Hirata K, Rubin EM, Cooper AD, Quertermous T. Endothelial lipase is a major determinant of HDL level. J Clin Invest. 2003;111:347–355. doi: 10.1172/JCI16306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ma K, Cilingiroglu M, Otvos JD, Ballantyne CM, Marian AJ, Chan L. Endothelial lipase is a major genetic determinant for high-density lipoprotein concentration, structure, and metabolism. Proc Natl Acad Sci USA. 2003;100:2748–2753. doi: 10.1073/pnas.0438039100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Guerra R, Wang J, Grundy SM, Cohen JC. A hepatic lipase (LIPC) allele associated with high plasma concentrations of high density lipoprotein cholesterol. Proc Natl Acad Sci U S A. 1997;94:4532–4537. doi: 10.1073/pnas.94.9.4532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Murtomäki S, Tahvanainen E, Antikainen M, Tiret L, Nicaud V, Jansen H, Ehnholm C. Hepatic lipase gene polymorphisms influence plasma HDL levels. Results from the Finnish EARS participants. European Atherosclerosis Research Study. Arterioscler Thromb Vasc Biol. 1997;17:1879–1884. doi: 10.1161/01.atv.17.10.1879. [DOI] [PubMed] [Google Scholar]
- 22.Zambon A, Deeb SS, Hokanson JE, Brown BG, Brunzell JD. Common variants in the promoter of the hepatic lipase gene are associated with lower levels of hepatic lipase activity, buoyant LDL, and higher HDL2 cholesterol. Arterioscler Thromb Vasc Biol. 1998;18:1723–1729. doi: 10.1161/01.atv.18.11.1723. [DOI] [PubMed] [Google Scholar]
- 23.Willer CJ, Sanna S, Jackson AU, Scuteri A, Bonnycastle LL, Clarke R, Heath SC, Timpson NJ, Najjar SS, Stringham HM, Strait J, Duren WL, Maschio A, Busonero F, Mulas A, Albai G, Swift AJ, Morken MA, Narisu N, Bennett D, Parish S, Shen H, Galan P, Meneton P, Hercberg S, Zelenika D, Chen WM, Li Y, Scott LJ, Scheet PA, Sundvall J, Watanabe RM, Nagaraja R, Ebrahim S, Lawlor DA, Ben-Shlomo Y, Davey-Smith G, Shuldiner AR, Collins R, Bergman RN, Uda M, Tuomilehto J, Cao A, Collins FS, Lakatta E, Lathrop GM, Boehnke M, Schlessinger D, Mohlke KL, Abecasis GR. Newly identified loci that influence lipid concentrations and risk of coronary artery disease. Nat Genet. 2008;40:161–169. doi: 10.1038/ng.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kathiresan S, Melander O, Guiducci C, Surti A, Burtt NP, Rieder MJ, Cooper GM, Roos C, Voight BF, Havulinna AS, Wahlstrand B, Hedner T, Corella D, Tai ES, Ordovas JM, Berglund G, Vartiainen E, Jousilahti P, Hedblad B, Taskinen MR, Newton-Cheh C, Salomaa V, Peltonen L, Groop L, Altshuler DM, Orho-Melander M. Six new loci associated with blood low-density lipoprotein cholesterol, high-density lipoprotein cholesterol or triglycerides in humans. Nat Genet. 2008;40:189–197. doi: 10.1038/ng.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Edmondson AC, Brown RJ, Kathiresan S, Cupples LA, Demissie S, Manning AK, Jensen MK, Rimm EB, Wang J, Rodrigues A, Bamba V, Khetarpal SA, Wolfe ML, Derohannessian S, Li M, Reilly MP, Aberle J, Evans D, Hegele RA, Rader DJ. Loss-of-function variants in endothelial lipase are a cause of elevated HDL cholesterol in humans. J Clin Invest. 2009;119:1042–1050. doi: 10.1172/JCI37176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brown RJ, Edmondson AC, Griffon N, Hill TB, Fuki IV, Badellino KO, Li M, Wolfe ML, Reilly MP, Rader DJ. A naturally occurring variant of endothelial lipase associated with elevated HDL exhibits impaired synthesis. J Lipid Res. 2009;50:1910–1916. doi: 10.1194/jlr.P900020-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mezdour H, Jones R, Dengremont C, Castro G, Maeda N. Hepatic lipase deficiency increases plasma cholesterol but reduces susceptibility to atherosclerosis in apolipoprotein E-deficient mice. J Biol Chem. 1997;272:13570–13575. doi: 10.1074/jbc.272.21.13570. [DOI] [PubMed] [Google Scholar]
- 28.Barcat D, Amadio A, Palos-Pinto A, Daret D, Benlian P, Darmon M, Bérard AM. Combined hyperlipidemia/hyperalphalipoproteinemia associated with premature spontaneous atherosclerosis in mice lacking hepatic lipase and low density lipoprotein receptor. Atherosclerosis. 2006:347–355. doi: 10.1016/j.atherosclerosis.2005.11.022. [DOI] [PubMed] [Google Scholar]
- 29.Jansen H, Chu G, Ehnholm C, Dallongeville J, Nicaud V, Talmud PJ. The T allele of the hepatic lipase promoter variant C-480T is associated with increased fasting lipids and HDL and increased preprandial and postprandial LpCIII:B: European Atherosclerosis Research Study (EARS) II. Arterioscler Thromb Vasc Biol. 1999;19:303–308. doi: 10.1161/01.atv.19.2.303. [DOI] [PubMed] [Google Scholar]
- 30.Isaacs A, Sayed-Tabatabaei FA, Njajou OT, Witteman JC, van Duijn CM. The -514 C->T hepatic lipase promoter region polymorphism and plasma lipids: a meta-analysis. J Clin Endocrinol Metab. 2004;89:3858–3863. doi: 10.1210/jc.2004-0188. [DOI] [PubMed] [Google Scholar]
- 31.Lindi V, Schwab U, Louheranta A, Vessby B, Hermansen K, Tapsell L, Riccardi G, Rivellese AA, Laakso M, Uusitupa MI, KANWU Study Group The G-250A polymorphism in the hepatic lipase gene promoter is associated with changes in hepatic lipase activity and LDL cholesterol: the KANWU Study. Nutr Metab Cardiovasc Dis. 2008;18:88–95. doi: 10.1016/j.numecd.2006.09.008. [DOI] [PubMed] [Google Scholar]
- 32.Fan YM, Raitakari OT, Kähönen M, Hutri-Kähönen N, Juonala M, Marniemi J, Viikari J, Lehtimäki T. Hepatic lipase promoter C-480T polymorphism is associated with serum lipids levels, but not subclinical atherosclerosis: the Cardiovascular Risk in Young Finns Study. Clin Genet. 2009;76:46–53. doi: 10.1111/j.1399-0004.2009.01180.x. [DOI] [PubMed] [Google Scholar]
- 33.Connelly PW, Hegele RA. Hepatic lipase deficiency. Crit Rev Clin Lab Sci. 1998;35:547–572. doi: 10.1080/10408369891234273. [DOI] [PubMed] [Google Scholar]
- 34.Broedl UC, Maugeais C, Millar JS, Jin W, Moore RE, Fuki IV, Marchadier D, Glick JM, Rader DJ. Endothelial lipase promotes the catabolism of apoB-containing lipoproteins. Circ Res. 2004;94:1554–1561. doi: 10.1161/01.RES.0000130657.00222.39. [DOI] [PubMed] [Google Scholar]
- 35.Ishida T, Choi SY, Kundu RK, Spin J, Yamashita T, Hirata K, Kojima Y, Yokoyama M, Cooper AD, Quertermous T. Endothelial lipase modulates susceptibility to atherosclerosis in apolipoprotein-E-deficient mice. J Biol Chem. 2004;279:45085–45092. doi: 10.1074/jbc.M406360200. [DOI] [PubMed] [Google Scholar]
- 36.Ko KW, Paul A, Ma K, Li L, Chan L. Endothelial lipase modulates HDL but has no effect on atherosclerosis development in apoE−/− and LDLR−/− mice. J Lipid Res. 2005;46:2586–2594. doi: 10.1194/jlr.M500366-JLR200. [DOI] [PubMed] [Google Scholar]
- 37.Weinstock PH, Bisgaier CL, Aalto-Setälä K, Radner H, Ramakrishnan R, Levak-Frank S, Essenburg AD, Zechner R, Breslow JL. Severe hypertriglyceridemia, reduced high density lipoprotein, and neonatal death in lipoprotein lipase knockout mice. Mild hypertriglyceridemia with impaired very low density lipoprotein clearance in heterozygotes. J Clin Invest. 1995;96:2555–2568. doi: 10.1172/JCI118319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Coleman T, Seip RL, Gimble JM, Lee D, Maeda N, Semenkovich CF. COOH-terminal disruption of lipoprotein lipase in mice is lethal in homozygotes, but heterozygotes have elevated triglycerides and impaired enzyme activity. J Biol Chem. 1995;270:12518–12525. doi: 10.1074/jbc.270.21.12518. [DOI] [PubMed] [Google Scholar]
- 39.Wade RL, Van Andel RA, Rice SG, Banka CL, Dyer CA. Hepatic lipase deficiency attenuates mouse ovarian progesterone production leading to decreased ovulation and reduced litter size. Biol Reprod. 2002;66:1076–1082. doi: 10.1095/biolreprod66.4.1076. [DOI] [PubMed] [Google Scholar]
- 40.Aviram M, Bierman EL, Chait A. Modification of low density lipoprotein by lipoprotein lipase or hepatic lipase induces enhanced uptake and cholesterol accumulation in cells. J Biol Chem. 1988;263:15416–15422. [PubMed] [Google Scholar]
- 41.Lamarche B, Lemieux I, Després JP. The small, dense LDL phenotype and the risk of coronary heart disease: epidemiology, patho-physiology and therapeutic aspects. Diabetes Metab. 1999;25:199–211. [PubMed] [Google Scholar]
- 42.Bossé Y, Pérusse L, Vohl MC. Genetics of LDL particle heterogeneity: from genetic epidemiology to DNA-based variations. J Lipid Res. 2004;45:1008–1026. doi: 10.1194/jlr.R400002-JLR200. [DOI] [PubMed] [Google Scholar]
- 43.Packard CJ. Small dense low-density lipoprotein and its role as an independent predictor of cardiovascular disease. Curr Opin Lipidol. 2006;17:412–417. doi: 10.1097/01.mol.0000236367.42755.c1. [DOI] [PubMed] [Google Scholar]
- 44.de Grooth GJ, Klerkx AH, Stroes ES, Stalenhoef AF, Kastelein JJ, Kuivenhoven JA. A review of CETP and its relation to atherosclerosis. J Lipid Res. 2004;45:1967–1974. doi: 10.1194/jlr.R400007-JLR200. [DOI] [PubMed] [Google Scholar]
- 45.Clay MA, Newnham HH, Forte TM, Barter PJ. Cholesteryl ester transfer protein and hepatic lipase activity promote shedding of apo A-I from HDL and subsequent formation of discoidal HDL. Biochim Biophys Acta. 1992;1124:52–58. doi: 10.1016/0005-2760(92)90125-f. [DOI] [PubMed] [Google Scholar]
- 46.Tanigawa H, Billheimer JT, Tohyama J, Zhang Y, Rothblat G, Rader DJ. Expression of cholesteryl ester transfer protein in mice promotes macrophage reverse cholesterol transport. Circulation. 2007;116:1267–1273. doi: 10.1161/CIRCULATIONAHA.107.704254. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.