

Abstract
Along with biocompatibility, chemical stability, and simplicity of structural prediction and modification, deoxyribozyme-based molecular sensors have the potential of an improved detection limit due to their ability to catalytically amplify signal. This study contributes to the understanding of the factors responsible for the limit of detection (LOD) of RNA-cleaving deoxyribozyme sensors. A new sensor that detects specific DNA/RNA sequences was designed from deoxyribozyme OA-II [Chiuman, W.; Li, Y. (2006) J. Mol. Biol. 357, 748–754]. The sensor architecture allows for a unique combination of high selectivity, low LOD and the convenience of fluorescent signal monitoring in homogeneous solution. The LOD of the sensor was found to be ~1.6 10×10 m after 3 h of incubation. An equation that allows estimation of the lowest theoretical LOD using characteristics of parent deoxyribozymes and their fluorogenic substrates was derived and experimentally verified. According to the equation, “catalytically perfect” enzymes can serve as scaffolds for the design of sensors with the LOD not lower than ~2×10−15 m after 3 h of incubation. A new value termed the detection efficiency (DE) is suggested as a time-independent characteristic of a sensor’s sensitivity. The expressions for the theoretical LOD and DE can be used to evaluate nucleic acid and protein enzymes for their application as biosensing platforms.
Keywords: deoxyribozyme sensor, DNA enzyme, DNA, fluorescence, limit of detection
Introduction
Deoxyribozymes (DNA enzymes, catalytic DNAs, or DNAzymes), are catalytic oligodeoxyribonucleotides derived by in vitro selection.[1] This relatively new class of catalytic molecules has been considered as a promising biochemical tool for a variety of applications including nucleic acid quantification,[2] site-specific RNA cleavage,[3] homogeneous assays for proteins,[4] detection of metal ions,[5] and molecular Boolean computation,[6] amongst others.[7] The advantages offered by catalytic DNAs include high chemical stability, low cost for synthesis, biocompatibility, and ease of structural prediction and modification. DNA enzymes that catalyze RNA cleavage are one of the first catalytic DNA molecules obtained[1a] and by far the largest and most extensively studied class of DNAzymes.
Deoxyribozymes represent an attractive tool for the analysis of specific DNA or RNA sequences because they can be easily tailored to recognize specific analytes by forming Watson–Crick base pairs. Importantly, DNAzyme-based sensors can potentially detect low concentrations of analyzed nucleic acids; this represents an alternative to other homogeneous assays (such as molecular beacon probes, for example)[8] that require PCR amplification of genetic material prior to its analysis. Although PCR represents the ultimate in terms of detection limit, it has significant drawbacks including complexity, sensitivity to contamination, cost, and lack of portability.[9] In this study we analyze a possibility of engineering a PCR-free deoxyribozyme sensor for nucleic acid analysis.
Based on the principle of complementarity, allosterically regulated RNA-cleaving ribozyme and deoxyribozyme sensors have been suggested for DNA/RNA analysis.[10–13] The design of such constructs implied their intrasteric inhibition by hybridization with specific internal fragments. The suppressed enzymes underwent conformational switching to their active forms upon hybridization with the cognate analytes. Some examples of intrasterically regulated deoxyribozyme sensors for nucleic acids include “catalytic molecular beacons” derived from E6[11] or 10–23[12] deoxyribozymes, as well as target-assisted self-cleaving (TASC) sensors[13] based on 8–17 deoxyribozyme. In “catalytic molecular beacons” the activated sensor cleaved a reporter oligonucleotide substrate that was double-labeled with a fluorophore and a quencher.[11,14] The deoxyribozyme-mediated substrate cleavage separated the fluorophore from the quencher, and this was accompanied by fluorescent signaling. The separation of the sensor and the enzyme in two different molecules enabled multiple substrate cleavage by a single activated sensor, thus potentially improving assay sensitivity. In this study, we took advantage of this approach for fluorescent detection of a sensor’s activity.
Selectivity and the detection limit are two major characteristics of any sensor that determine its efficiency. The selectivity of the intrasterically regulated deoxyribozyme sensors has to be further improved to enable single mismatch discrimination at ambient temperatures. For instance, “catalytic molecular beacon” could discriminate double-base-, but not single-base-substituted oligonucleotides from fully complementary targets;[11] A TASC probe distinguished single base substitution in a 16-mer, but not in 20-mer analytes.[13] We have recently addressed the selectivity issue of nucleic acid detection by developing a series of binary (split or two-component) probes.[15] In this approach hybridization of two short oligonucleotides to the adjacent positions of RNA/DNA analyte is required for signal generation. Binary probes have improved selectivity because each of the two short probe-analyte hybrids could be easily destabilized even by minor hybrid imperfection, such as a single base mispairing. The combination of the binary approach with amplification power of DNA enzymes is extremely attractive, because it potentially leads to the development of both specific and sensitive assays for nucleic acids. As proof of this concept, a binary probe based on E6 deoxyribozyme[16] was developed and studied.[15c] The probe demonstrated excellent selectivity by distinguishing single base substitutions in 20-mer DNA analyte at room temperature. The relatively high limit of detection (LOD) of this sensor (~1nm) was attributed to the low catalytic efficiency of the parent E6 deoxyribozyme. The achieved LOD is not low enough to be of significant practical use in PCR-free diagnostics. For example, the typical concentration of hepatitis C virus (HCV) varies between 104–106 copies per mL or ~6×10−14−6×1016 m.[17] Such low abundance of viral nucleic acids in clinical samples raises the following questions. Is it possible to obtain a deoxyribozyme sensor with such low LODs as to detect specific nucleic acids without using PCR amplification? What characteristics of DNAzyme-based sensors should be improved to achieve the lowest possible LOD?
In this study we aimed to answer these questions. We turned our attention to deoxyribozyme OA-II,[18] a more efficient catalyst than E6 DNA enzyme. In the first stage of the study we characterized the DNAzyme–substrate pair in terms of KM, kcat and the minimal amount of the substrate to be cleaved to generate a signal above the background. Then we designed a sensor based on OA-II, studied its selectivity and determined its detection limit. Using Michaelis–Menten kinetics, we aimed at predicting the LOD for enzyme-based sensors on the basis of the characteristics of parent enzymes and their substrates. Finally, we introduced the detection efficiency (DE), a time-independent parameter that can be conveniently used for the comparison of the detection limits for enzyme-based sensors.
Results and Discussion
Deoxyribozyme OA-II was obtained by Chuiman and Li by using standard in vitro selection strategy.[18] OA-II is a NiII-dependent DNAzyme capable of cleaving an RNA phosphodiester linkage positioned between fluorescein- and dabcyl-modified deoxyribonucleotides within a fluorogenic F-substrate (Figure 1 A).
Figure 1.

Design of biOA-II probe. A) Sequences of OA-II and the fluorogenic F-substrate. F and Q are fluorescein- and dabcyl-conjugated thymidines, respectively. B) The sequences of strands α and β of the biOA-II probe. The dithymidine linker is shown in lower case. C) Strands α and β hybridize to the abutting fragments of the A20 DNA analyte and reform the catalytic core. The active enzyme cleaves F-substrate and this generates fluorescent signal.
Michaelis–Menten parameters for deoxyribozyme OA-II
Deoxyribozyme OA-II was found to cleave the RNA phosphodiester bond with an observed catalytic rate constant of ~0.03 s−1.[18] The reported rate constant was measured under single-turnover conditions, that is, in the excess of the enzyme over substrate. In this work we determined the kinetic parameter for OA-II catalyzed cleavage of F-substrate under Michaelis–Menten conditions, that is, in the substrate excess. The analysis of double reciprocal slope of the initial velocity versus substrate concentration resulted in a kcat of (5.8±0.2)×10−3 s−1 and a KM of 560±170 nm with R2 = 0.98 (Figure S1, Supporting Information).
Design of binary deoxyribozyme probe
Binary probe for nucleic acid detection was designed from OA-II according to the earlier suggested Scheme for binary E6 probe.[15c] Specifically, OA-II was divided into two fragments, the stem-forming fragments of both strands were elongated to six nucleotides, forming inter-strand communication region, and the analyte binding arms were added to each strand (Figure 1 B). Strand α contained the analyte-binding arm connected to the stem-forming fragment through a dithymidine linker, because this modification was found to support the highest level of enzymatic activity restoration in the presence of an analyte (data not shown). This observation agreed with the earlier findings that bulged three-way junction structures have increased stability.[19] The sequence of substrate-binding arms of the binary deoxyribozyme was retained to allow F-substrate binding and cleavage. The concentrations of strands α and β were optimized to avoid substrate cleavage in the absence of the analyte. When the fully complementary A20 analyte was added, the two “subunits” of the enzyme cooperatively hybridized to the analyte and re-formed the deoxyribozyme catalytic core (Figure 1 C). The active enzyme cleaved the F-substrate; this was accompanied by separation of the fluorophore from the quencher.
To analyze the catalytic activity of the probe, the fluorescence enhancement of F-substrate in the presence of biOA-II and the analyte as a function of time was measured (Figure 2). The addition of A20 DNA analyte to the solution of biOA-II and F-substrate triggered the fluorescence increase (Figure 2, curve 3), and the rate of fluorescence enhancement was very close to that of OA-II catalyzed substrate cleavage (Figure 2, compare curves 3 and 4). No fluorescent increase was observed either for F-substrate alone or with biOA-II in the absence of DNA analyte (Figure 2, curves 1 and 2, respectively). Polyacrylamide gel analysis of the reaction mixtures revealed F-substrate cleavage product only when either OA-II deoxyribozyme or biOA-II together with the analyte were present in the reaction mixture (Figure 2 gel photograph inset, lanes 4 and 3, respectively). These data prove the model for the analyte-dependent binary deoxyribozyme activation and indicate that introduction of the analyte-binding arms does not disturb the catalytic action of the deoxyribozyme core.
Figure 2.

Analyte-dependent activation of biOA-II probe. F-substrate (1 μm) and biOA-II (50 nm) were incubated in the absence (curve 2) or presence (curve 3) of 1 nm A20 DNA analyte in 50 mm HEPES, pH 7.4, 50 mm MgCl2, 1mm NiCl2. Curve 1: F-substrate was incubated in the same buffer without the enzyme (negative control). Curve 4: F-substrate was incubated with deoxyribozyme OA-II (1 nm; positive control). The fluorescent intensities at 517 nm are plotted against the incubation time. The data are average values of three independent measurements. Inset: Electrophoretic analysis of reaction mixtures 1–4 in a 20 % polyacrylamide gel containing 7 m urea.
Selectivity of biOA-II probe
Binary probes were reported to recognize single-base substitutions in nucleic acid analytes with exceptional selectivity.[15] Herein, we verified that biOA-II, like other binary probes, discriminated single-base substitutions in a 20 nucleotide analyte at room temperature. The probe was incubated with the fully matched A20 or an oligonucleotide containing a single base substitution (A20-6, A20-12 and A20-16, Figure 3 A). Discrimination factors (DFs), calculated as ratios of the fluorescence intensity of the probe in the presence of the true target to that in the presence of the mismatched analytes after subtracting the background, were found to be 3.6±0.3, 2.3±0.4, and 3.8±0.2 for the mismatched oligonucleotides A20-6, A20-12, and A20-16, respectively. These data demonstrate that the probe reliably discriminated the true target from the single base mismatched targets at ambient temperatures without the need for precise temperature control.
Figure 3.

A) Sequences of the mismatched analytes. B) Primary and suggested secondary structure for biOA-II (stem-loop) probe. C) Discrimination against mismatched oligonucleotides by biOA-II (stem-loop). Fully complementary A20 analyte or one of the single base mismatched targets A20-6, A20-12 or A20-16 were incubated with biOAII (stem-loop) mixed with F-substrate. The fluorescent intensities at 517 nm were measured after 1, 3, and 20 hours of incubation.
The discrimination power of binary probes can be further improved by introducing conformational constraints in the analyte recognition fragments of the probes.[15b,c,20] Indeed, when a biOA-II probe with stem-loop structures in the analyte binding arms was used (Figure 3 B), the fluorescence increase was observed only in the presence of the fully matched oligonucleotide indicating extremely high DFs (Figure 3 C). Remarkably, this selectivity was achieved at room temperature. The selectivity of this probe can be further improved by elongating the stem region or by shortening the analyte binding arm that is complementary to the mismatch-containing site. Therefore, the sensor designed in this work is one of the most selective, structurally flexible, and inexpensive probe for nucleic acid analysis. It might be especially useful for single nucleotide polymorphism genotyping.
Limit of detection for biOA-II probe
To determine the LOD, we carried out seven independent measurements of the F-substrate fluorescence (blank sample).[21,22] The LOD is considered equal to the signal corresponding to the average blank plus three standard deviations of the blank.[22] The average fluorescence intensity of the blank was determined to be 8.8±0.8 fluorescence arbitrary units (a.u.; data not shown). Therefore, the fluorescence corresponding to the LOD was calculated as 8.8 + 3×0.8=11.2 a.u. In practice, however, it is easier to operate with relative rather than absolute values. Based on the data, the criterion of signal-to-background ratio (F/F0) being equal to 1.3 (calculated as 11.2/8.8) was used to distinguish signals statistically above the background in the data analysis presented below.
To determine the detection limit for biOA-II, the probe was incubated in the presence of various concentrations of A20 analyte. Figure 4 A shows F/F0 plotted against the analyte concentration. From this graph F/F0 threshold of 1.3 was found to be reached at 0.63 nm, 0.16 nm and 0.04 nm analyte after 1, 3 and 20 h of incubation, respectively. These values are among the lowest achieved by deoxyribozyme sensors[10–13] or by other state-of-the-art homogeneous PCR-free fluorescent assays.[8,23]
Figure 4.
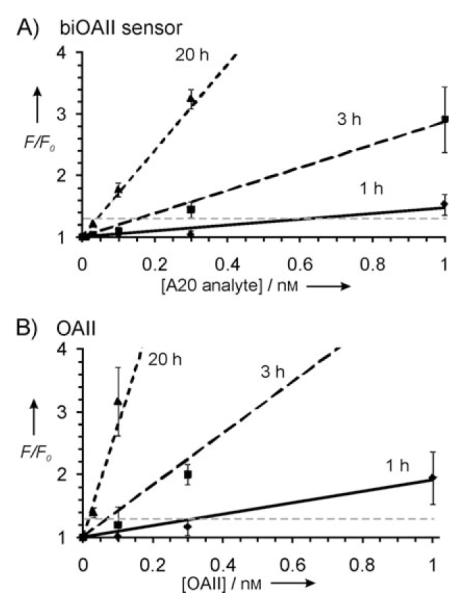
Fluorescent signal-to-background ratios (F/F0) for biOA-II probe and OA-II deoxyribozyme after different incubation periods. A) F/F0 for biOA-II probe in the presence of various concentrations of A20 analyte after 1 (●), 3 (■), and 20 (▲) hours of incubation. B) F/F0 for F-substrate in the presence of different OA-II concentrations after 1 (●), 3 (■), and 20 (▲) hours of incubation. The F/F0 threshold 1.3 is indicated by the horizontal dashed line. The data are average values of three independent measurements. The solid lines are the least squares fit of the experimental data for low analyte or enzyme concentrations.
Figure 4 B demonstrates the fluorescence increase of F-substrate in the presence of different concentrations of parent OA-II. Remarkably, these curves correlate well with the data obtained for biOA-II probe (Figure 4, compare panels A and B). The OA-II concentrations that triggered substrate fluorescence equal to the F/F0 threshold were 0.34 nm, 0.07 nm and 0.02 nm after 1, 3 and 20 h of incubation, respectively. These values are about 2-fold lower than the actual LOD for biOA-II sensor. This difference can be attributed to the incomplete formation of the probe-analyte complex, when some analyte molecules are not in the complex with biOA-II probe and, therefore, fail to induce the assembling of active deoxyribozyme catalytic core.
The correlation of the data shown in Figure 4 A and B led us to the two following conclusions. First, the design of the biOA-II probe was nearly optimal: the hybridization of the probe analyte binding arms to the analyte did not significantly disturb the catalytic core and the substrate binding activity of the formed binary deoxyribozyme, as compared with parent OA-II. The LOD for the probe is, therefore, confined by the efficiency of the parent deoxyribozyme rather than by the probe design. Second, it is possible to estimate the LOD for a binary deoxyribozyme sensor by finding the lowest concentration of the original deoxyribozyme that generates a detectable signal. In other words, the minimal concentration of the parent deoxyribozyme that is sufficient to catalyze detectable substrate cleavage can mimic the same concentrations of the activated binary deoxyribozyme sensor, which is equal to or lower than the actual analyte concentration corresponding to the LOD. This finding opens the possibility to predict the LOD for a potential deoxyribozyme sensor by determining the minimal concentration of a parent DNAzyme that generates a fluorescence signal equal to the LOD threshold. This simple assay is of practical value when, for instance, screening of many enzymatic platforms for sensor design is required. Taking into consideration the above mentioned observations, we further derived the dependence of the lowest possible LOD for a deoxyribozyme sensor from the kinetic characteristics of the parent deoxyribozyme.
Theoretical prediction of the lowest possible LOD for deoxyribozyme sensors
Herein, we define the theoretical lowest LOD as the least DNA-zyme concentration that generates the fluorescence signal reliably above the threshold. Because such signal is usually achieved earlier in the reaction, it is possible to use the Michaelis–Menten equation (1) for the initial reaction velocity:
| (1) |
in which E is a deoxyribozyme, S is a fluorogenic substrate and P is the substrate cleavage products. Since the initial rate of the reaction equals product concentration over time (V0 = [P]/t), the enzyme concentration can be determined from Equation (2):
| (2) |
Equation (2) contains the cleavage degree α, which is a ratio of the product concentration to the initial concentration of the substrate (α = [P]/[S]0). For the LOD Equation (3), the cleavage degree sufficient to generate a signal with F/F0 = 1.3 is referred to as α1.3.
| (3) |
We determined that 0.53 % of F-substrate should be cleaved to generate a F/F0 of 1.3, which corresponds to α1.3 = 0.0053 (Figure S2 in the Supporting Information). The theoretical detection limit for OA-II calculated from Equation (3) (kcat = 5.8×10−3 s−1, KM = 560 nm, and [S0] = 1000 nm) is 0.4, 0.13, and 0.02 nm after 1, 3, and 20 h. These data correlate with the values 0.34, 0.07, and 0.02 nm experimentally found for OA-II deoxyribozyme and about two times lower the detection limits found for biOA-II sensor. This correlation speaks in favor of the suggestion that Equation (3) can be used to estimate lower LOD border for a sensor designed from a chosen deoxyribozyme.
The theoretical LOD depends on time. This time-dependence might be inconvenient for the comparison of sensors’ efficiencies, since different incubation periods are used for the LOD determination in different studies. Here we introduce a time-independent parameter termed “detection efficiency” (DE).
| (4) |
DE is inversely proportional to the active enzyme (analyte) concentration by time [Eq. (4)]. In the case of OA-II deoxyribozyme, DE was calculated to be ~7×105 m−1 s−1. A molecular sensor with such efficiency generates a signal reliably above the background in the presence of 1.4×10−6m analyte in one second. Such sensor would be capable of detecting ~1.3×10−10m analyte after 3 h of incubation, which correlates with the observed LOD of 1.6×10−10m.
Importantly, there is a number of characterized RNA-cleaving deoxyribozymes and ribozymes with the determined catalytic characteristics. The expressions for LOD and DE can help to evaluate these nucleic acid enzymes as a starting point for a sensor design without carrying out actual experiments. For example, relatively inefficient deoxyribozyme E6 (KM = 13 000 nm kcat = 0.002 s−1)[16] in the presence of a fluorogenic substrate with α1.3 = 0.0053 has DE of ~3×104 m−1 s−1, which is high enough to detect 3×10−9m analyte after 3 h of incubation. These parameters are sufficient for E6 to be a good sensor platform for a number of environmentally important analytes. For example, U.S. Environmental Protection Agency (EPA) defined the toxic HgII, and UO22+ levels in drinking water to be 10 and 130 nm, respectively, which are within the dynamic range of a putative E6-based sensor.
In general, the LOD of deoxyribozyme sensors can be limited by the low affinity of the sensors to the analytes. This important interaction has not been taken into consideration by the above analysis. However, in assumption that sensor LOD is limited by the efficiency of deoxyribozyme core and the substrate signal enhancement power, the approach provides important insight into the design of deoxyribozyme-based sensors. DE is proportional to the catalytic rate constant (kcat) and inversely proportional to both α1.3 and KM. Thus, the efficient sensor should use substrates with the highest fluorescence enhancement upon cleavage (corresponds to the lowest αLOD) and be designed from a deoxyribozyme with the highest kcat/KM values. Moreover, the substrate should be used in concentrations below KM to be neglected in the sum. In the later case, DE can be estimated ~kcat/(αLOD*KM). For the “catalytically perfect” enzymes kcat/KM is ~108m−1 s−1. It is important to note that not only the protein enzymes, but also some deoxyribozymes demonstrate catalytic efficiency close to the 108 m−1 s−1 diffusional limit.[24] The best double labeled fluorogenic substrates can increase fluorescent 180 times upon complete cleavage,[25] which makes α1.3 to be ~0.002. The DE for such an “ideal” deoxyribozyme sensor is limited by the value of 5×1010m−1 s−1. Such a sensor would be capable of detecting analyte at concentrations not lower than 2×10−15m after 3 h of incubation.[26] This DE is high enough to analyze clinical samples with high content of viral nucleic acids without PCR amplifications, but insufficient to detect 10−15–10−16m analytes without additional amplification steps. Importantly, the equations for DE and LOD and the following conclusions are equally applicable to the assays based on both nucleic acid and protein enzymes.
Conclusions
The LOD for a binary deoxyribozyme probe can be predicted by using kcat, KM and the characteristic of substrate signal enhancement. This hypothesis was validated in this study using the probe designed from DNA enzyme OA-II. The lowest possible LOD for a “perfect” deoxyribozyme sensor was estimated to be ~2×10−15m. This detection limit is satisfactory for the analysis of many practically significant targets including metal ions and a number of biological molecules including proteins, but might be insufficient for the detection of viral nucleic acids in clinical samples without using additional amplification procedures.
Sensors that allow nonlinear (exponential) signal amplification are required for the design of more sensitive assays. Deoxyribozyme-based cascades for signal amplification is an alternative to PCR amplification. Deoxyribozymes represent a versatile platform for such cascades as was demonstrated by several prove-of-concept studies.[27]
Experimental Section
Materials
DNAse-/RNAse-free water was purchased from Fisher Scientific, Inc. (Pittsburgh, PA) and used for all buffers and for the stock solutions of oligonucleotides. Fluorogenic F-substrate was custom-made by TriLink BioTechnologies, Inc. (San Diego, CA). All other oligonucleotides were obtained from Integrated DNA Technologies, Inc. (Coralville, IA).
Instrumentation
Fluorescent spectra were recorded on a Perkin–Elmer (San Jose, CA) LS-55 Luminescence Spectrometer equipped with a Hamamatsu Xenon lamp. Experiments were performed at an excitation wavelength of 485 nm and emission monitored from 500–550 nm. Excitation and emission slits were 5 and 20 nm, respectively. Fluorescence intensities at 517 nm were taken for all calculations. The data were processed using Microsoft Excel.
Statistical LOD determination
A blank solution (240 μL) of F-substrate (1 μm) in HEPES (50 mm, pH 7.4), MgCl2 (50 mm), and NiCl2 (1 mm) was incubated at RT (20 °C), and the fluorescence intensities at 517 nm were measured after an hour of incubation. Seven independent measurements were performed to calculate the average and the standard deviation of the blank. The fluorescence intensity corresponding to the LOD was calculated as the blank average plus three blank standard deviations, as was recommended by Kirchmer.[20]
Michaelis–Menten parameters for deoxyribozyme OA-II
To provide a way of calibrating the fluorescence enhancement upon the fluorogenic substrate cleavage, the completely cleaved (0.2m NaOH at 37 °C, overnight) fluorogenic substrate was mixed with the initial F-substrate to obtain mixtures with the cleavage degree (α) of 0, 0.01, 0.02, 0.05, 0.10, 0.20, 0.50 and 1.00 at each substrate concentration used in the experiments. The fluorescence intensities of the mixtures at 517 nm were taken to calculate F/F0 ratio (F0 and F are fluorescence intensities in the absence and in the presence of deoxyribozyme OA-II, respectively), and F/F0 values were plotted against α. Linear correlation of these values was observed (Figure S2 in the Supporting Information).
To obtain Michaelis–Menten parameters, F-substrate (150, 250, 500, 750, 1000, 1500, 2000, 3000 or 4000 nm) and deoxyribozyme OA-II (3 nm) were incubated in the reaction buffer (50 mm HEPES, pH 7.4, 50 mm MgCl2, 1mm NiCl2) at RT (20 °C) during 70 min. The fluorescent emission spectra were recorded every 7 min, and the fluorescence intensities at 517 nm were used to calculate product concentration at each time point using the calibration curve. The initial velocities (nm min−1) obtained from the kinetic curves for two independent measurements were plotted against substrate concentrations. The data fit well to the Michaelis–Menten equation (Figure S1 in the Supporting Information). Reported kcat and KM values were determined from the double reciprocal plot.
Deoxyribozyme OA-II fluorescent assay
To determine minimal detectable concentration of deoxyribozyme OA-II, a solution (960 μL) of F-substrate (1 μm) in HEPES (50 mm, pH 7.4), MgCl2 (50 mm), and NiCl2 (1 mm) was split into eight tubes (120 μL each). Deoxyribozyme OA-II was added to seven tubes at final concentrations of 0.01, 0.03, 0.1, 0.3, 1, 3, or 10 nm, while an equal volume of water was added to the eighth tube (control sample). All solutions were incubated at room temperature (20 °C), and the fluorescent emission spectra were recorded after 1, 3 and 20 h of incubation. The fluorescence intensities at 517 nm were plotted against analyte concentration.
LOD assay for binary deoxyribozyme OA-II (biOA-II) probe
A solution (1080 μL) of F-substrate (1 μm) in HEPES (50 mm, pH 7.4), MgCl2 (50 mm), and NiCl2 (1 mm) was split into two tubes (960 μL and 120 μL). Both strands of biOAII probe were added to the first tube at final concentration of 50 nm, while an equal volume of water was added to the control sample 1 (second tube). The content of the first tube was further divided into eight tubes (120 μL each) and A20 analyte (ATG GAG AGA GTG GGT GCG AG) was added to seven tubes at final concentration of 0.01, 0.03, 0.1, 0.3, 1, 3 an 10 nm, while an equal volume of water was added to the eighth tube (control sample 2). All solutions were incubated at room temperature (20 °C), and the fluorescent emission spectra were recorded after 1, 3 and 20 h of incubation.
Kinetics of F-substrate cleavage in the presence of either deoxyribozyme OA-II or biOA-II probe
A solution (480 μL) of F-substrate (1 μm) in HEPES (50 mm, pH 7.4), MgCl2 (50 mm), and NiCl2 (1 mm) was split into three tubes (120 μL, 240 μL and 120 μL). A solution of deoxyribozyme OA-II to a final concentration of 1 nm was added to the first tube to obtain positive control. Both strands of biOA-II probe were added to the second tube at the final concentration of 50 nm followed by a further division of the solution into two tubes. A20 was added to one of them to a final concentration of 1 nm, while an equal volume of water was added to the control sample. The third tube represented negative control. The fluorescent emission spectra were recorded after 5, 15, 30 and 60 min of incubation. The fluorescence intensities at 517 nm were plotted against time.
Discrimination factors (DFs) for biOA-II
A solution (720 mL) of F-substrate (1 μm) in HEPES (50 mm, pH 7.4), MgCl2 (50 mm), and NiCl2 (1 mm) was split into two tubes (600 μL and 120 μL) Both strands of biOAII probe were added to the first tube at final concentration of 50 nm, while an equal volume of water was added to the control sample 1 (second tube). The content of the first tube was further divided into five tubes (120 μL each) and either fully matched A20 analyte (5′-ATG GAG AGA GTG GGT GCG AG) or any of mismatched oligonucleotides A20–6 (5′-ATG GAT AGA GTG GGT GCG AG), A20–12 (5′-ATG GAG AGA GTT GGT GCG AG) and A20–16 (5′-ATG GAG AGA GTG GGT ACG AG) were added to each of the four tubes at final concentration of 3 nm, while an equal volume of water was added to the fifth tube (control sample 2). All solutions were incubated at room temperature (20 °C), and the fluorescent emission spectra were recorded after 1 hour of incubation. To calculate the DFs the fluorescence intensity at 517 nm of the probe in the presence of A20 was divided by that of the probe in the presence of a mismatched oligonucleotide after subtracting the background fluorescence, which is the fluorescence of biOA-II probe incubated in the absence of analyte.
Supplementary Material
Acknowledgements
The authors are grateful to Dr. Diego Diaz for helpful discussion. This study was funded by UCF Office of Research and Commercialization, College of Science and Chemistry Department and NHGRI R21
Footnotes
Supporting information for this article is available on the WWW under http://dx.doi.org/10.1002/cbic.201000006.
References
- [1]a).Breaker RR, Joyce GF. Chem. Biol. 1994;1:223–229. doi: 10.1016/1074-5521(94)90014-0. [DOI] [PubMed] [Google Scholar]; b) Joyce GF. Annu. Rev. Biochem. 2004;73:791–836. doi: 10.1146/annurev.biochem.73.011303.073717. [DOI] [PubMed] [Google Scholar]
- [2].Todd AV, Fuery CJ, Impey HL, Applegate TL, Haughton MA. Clin. Chem. 2000;46:625–630. [PubMed] [Google Scholar]
- [3]a).Peracchi A. Rev. Med. Virol. 2004;14:47–64. doi: 10.1002/rmv.415. [DOI] [PubMed] [Google Scholar]; b) Schubert S, Kurreck J. Curr. Drug Targets. 2004;5:667–681. doi: 10.2174/1389450043345092. [DOI] [PubMed] [Google Scholar]
- [4]a).Stojanovic MN, de Prada P, Landry DW. Nucleic Acids Res. 2000;28:2915–2918. doi: 10.1093/nar/28.15.2915. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Li D, Shlyahovsky B, Elbaz J, Willner I. J. Am. Chem. Soc. 2007;129:5804–5805. doi: 10.1021/ja070180d. [DOI] [PubMed] [Google Scholar]
- [5]a).Brown AK, Liu J, He Y, Lu Y. ChemBioChem. 2009;10:486–492. doi: 10.1002/cbic.200800632. [DOI] [PubMed] [Google Scholar]; b) Liu J, Lu Y. Angew. Chem. 2007;119:7731–7734. [Google Scholar]; Angew. Chem. Int. Ed. Angew. Chem. Int. Ed. Engl. 2007;46:7587–7590. [Google Scholar]; c) Liu J, Lu Y. J. Am. Chem. Soc. 2007;129:9838–9839. doi: 10.1021/ja0717358. [DOI] [PubMed] [Google Scholar]; d) Liu J, Lu Y. Methods Mol. Biol. 2006;335:275–288. doi: 10.1385/1-59745-069-3:275. [DOI] [PubMed] [Google Scholar]
- [6]a).Stojanovic MN. Prog. Nucleic Acid Res. Mol. Biol. 2008;82:199–217. doi: 10.1016/S0079-6603(08)00006-8. [DOI] [PubMed] [Google Scholar]; b) Chen X, Wang Y, Liu Q, Zhang Z, Fan C, He L. Angew. Chem. 2006;118:1791–1794. [Google Scholar]; Angew. Chem. Int. Ed. 2006;45:1759–1762. doi: 10.1002/anie.200502511. [DOI] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2006;45:1759–1762. doi: 10.1002/anie.200502511. [DOI] [PubMed] [Google Scholar]; c) Moshe M, Elbaz J, Willner I. Nano Lett. 2009;9:1196–1200. doi: 10.1021/nl803887y. [DOI] [PubMed] [Google Scholar]
- [7]a).Silverman SK. Nucleic Acids Res. 2005;33:6151–6163. doi: 10.1093/nar/gki930. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Baum DA, Silverman SK. Cell. Mol. Life Sci. 2008;65:2156–2174. doi: 10.1007/s00018-008-8029-y. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Schlosser K, Li Y. Chem. Biol. 2009;16:311–322. doi: 10.1016/j.chembiol.2009.01.008. [DOI] [PubMed] [Google Scholar]
- [8]a).Tyagi S, Kramer FR. Nat. Biotechnol. 1996;14:303–308. doi: 10.1038/nbt0396-303. [DOI] [PubMed] [Google Scholar]; b) Marras SA, Tyagi S, Kramer FR. Clin. Chim. Acta. 2006;363:48–60. doi: 10.1016/j.cccn.2005.04.037. [DOI] [PubMed] [Google Scholar]; c) Wang K, Tang Z, Yang CJ, Kim Y, Fang X, Li W, Wu Y, Medley CD, Cao Z, Li J, Colon P, Lin H, Tan W. Angew. Chem. 2009;121:870–885. doi: 10.1002/anie.200800370. [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2009;48:856–870. doi: 10.1002/anie.200800370. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Tsourkas A, Behlke MA, Rose SD, Bao G. Nucleic Acid Res. 2003;31:1319–1330. doi: 10.1093/nar/gkg212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Rosi NL, Mirkin CA. Chem. Rev. 2005;105:1547–1562. doi: 10.1021/cr030067f. [DOI] [PubMed] [Google Scholar]
- [10].Hartig JS, Grune I, Najafi-Shoushtari SH, Famulok M. J. Am. Chem. Soc. 2004;126:722–723. doi: 10.1021/ja038822u. [DOI] [PubMed] [Google Scholar]
- [11].Stojanovic MN, de Prada P, Landry DW. ChemBioChem. 2001;2:411–415. doi: 10.1002/1439-7633(20010601)2:6<411::AID-CBIC411>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- [12].Tian Y, Mao C. Talanta. 2005;67:532–537. doi: 10.1016/j.talanta.2005.06.044. [DOI] [PubMed] [Google Scholar]
- [13]a).Sando S, Sasaki T, Kanatani K, Aoyama Y. J. Am. Chem. Soc. 2003;125:15720–15721. doi: 10.1021/ja0386492. [DOI] [PubMed] [Google Scholar]; b) Sando S, Narita A, Sasaki T, Aoyama Y. Org. Biomol. Chem. 2005;3:1002–1007. doi: 10.1039/b418078j. [DOI] [PubMed] [Google Scholar]
- [14].Singh KK, Parwaresch R, Krupp G. RNA. 1999;5:1348–1356. doi: 10.1017/s1355838299991185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15]a).Kolpashchikov DM. J. Am. Chem. Soc. 2005;127:12442–12443. doi: 10.1021/ja0529788. [DOI] [PubMed] [Google Scholar]; b) Kolpashchikov DM. J. Am. Chem. Soc. 2006;128:10625–10628. doi: 10.1021/ja0628093. [DOI] [PubMed] [Google Scholar]; c) Kolpashchikov DM. ChemBioChem. 2007;8:2039–2042. doi: 10.1002/cbic.200700384. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Kolpashchikov DM. J. Am. Chem. Soc. 2008;130:2934–2935. doi: 10.1021/ja711192e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Breaker RR, Joyce GF. Chem. Biol. 1995;2:655–660. doi: 10.1016/1074-5521(95)90028-4. [DOI] [PubMed] [Google Scholar]
- [17].Steininger C, Kundi M, Jatzko G, Kiss H, Lischka A, Holzmann H. J. Infect. Dis. 2003;187:345–351. doi: 10.1086/367704. [DOI] [PubMed] [Google Scholar]
- [18].Chiuman W, Li Y. J. Mol. Biol. 2006;357:748–754. doi: 10.1016/j.jmb.2006.01.036. [DOI] [PubMed] [Google Scholar]
- [19].Leontis NB, Kwok W, Newman JS. Nucleic Acids Res. 1991;19:759–766. doi: 10.1093/nar/19.4.759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20]a).Roberts RW, Crothers DM. Proc. Natl. Acad. Sci. USA. 1991;88:9397–9401. doi: 10.1073/pnas.88.21.9397. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Bonnet G, Tyagi S, Libchaber A, Kramer FR. Proc. Natl. Acad. Sci. USA. 1999;96:6171–6176. doi: 10.1073/pnas.96.11.6171. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Kolpashchikov DM. ChemBioChem. 2009;10:1443–1445. doi: 10.1002/cbic.200900264. [DOI] [PubMed] [Google Scholar]
- [21].Kirchmer CJ. In: Detection in Analytical Chemistry: Importance, Theory, and Practice. Currie LA, editor. American Chemical Society; Washington, DC: 1988. pp. 78–93. [Google Scholar]
- [22].MacDougall D, Crummett WB. Anal. Chem. 1980;52:2242–2249. [Google Scholar]
- [23]a).Grossmann TN, Seitz O. Chem. Eur. J. 2009;15:6723–6730. doi: 10.1002/chem.200900025. [DOI] [PubMed] [Google Scholar]; b) Sueda S, Yuan J, Matsumoto K. Bioconjugate Chem. 2002;13:200–205. doi: 10.1021/bc010049+. [DOI] [PubMed] [Google Scholar]; c) Franzini RM, Kool ET. J. Am. Chem. Soc. 2009;131:16021–16023. doi: 10.1021/ja904138v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Santoro SW, Joyce GF. Biochemistry. 1998;37:13330–13342. doi: 10.1021/bi9812221. [DOI] [PubMed] [Google Scholar]
- [25].Kelemen BR, Klink TA, Behlke MA, Eubanks SR, Leland PA, Raines RT. Nucleic Acid Res. 1999;27:3696–3701. doi: 10.1093/nar/27.18.3696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Note added in proof: After our initial submission, a binary probe based on catalytically the most efficient deoxyribozyme 10–23 was reported: Mokany E, Bone SM, Young PE, Doan TB, Todd AV. J. Am. Chem. Soc. 2010;132:1051–1059. doi: 10.1021/ja9076777. The probe can detect as little as 5 pm ana- lyte after 1 hour of incubation. Taking into account the kinetic parameters of 10–23 and the equation derived in the present work, we estimated the theoretical LOD for 10–23-based sensors to be in the range of 4 to 17 pm, depending on the substrate fluorescence-enhancement parameter α. This range correlates well with the experimentally determined LOD.
- [27]a).Levy M, Ellington AD. Proc. Natl. Acad. Sci. USA. 2003;100:6416–6421. doi: 10.1073/pnas.1130145100. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Elbaz J, Moshe M, Shlyahovsky B, Willner I. Chem. Eur. J. 2009;15:3411–3418. doi: 10.1002/chem.200802004. [DOI] [PubMed] [Google Scholar]; c) Willner I, Shlyahovsky B, Zayats M, Willner B. Chem. Soc. Rev. 2008;37:1153–1165. doi: 10.1039/b718428j. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.