

Abstract
Tumor necrosis factor-like weak inducer of apoptosis (TWEAK) and its receptor fibroblast growth factor-inducible 14 (Fn14) are expressed in endothelial cells and perivascular astrocytes. Here, we show that TWEAK induces a dose-dependent increase in the expression of the chemokine monocyte chemoattractant protein-1 (MCP-1) in astrocytes, and that this effect is mediated by its interaction with Fn14 via nuclear factor-κB pathway activation. Exposure to oxygen-glucose deprivation (OGD) conditions increases TWEAK and Fn14 mRNA expression in wild-type (Wt) astrocytic cultures. Likewise, incubation under OGD conditions induces the expression of MCP-1 in Wt astrocytes but not in astrocytes deficient on either TWEAK (TWEAK−/−) or Fn14 (Fn14−/−). We also found that TWEAK induces the passage of neutrophils to the abluminal side of an in vitro model of the blood–brain barrier. Our earlier studies indicate that cerebral ischemia increases the expression of TWEAK and Fn14 in the endothelial cell-basement membrane-astrocyte interface. Here, we report that middle cerebral artery occlusion increases the expression of MCP-1 and the recruitment of neutrophils into the ischemic tissue in Wt but not in TWEAK−/− or Fn14−/− mice. These novel results indicate that during cerebral ischemia, the interaction between TWEAK and Fn14 leads to the recruitment of leukocytes into the ischemic tissue.
Keywords: cerebral ischemia, fibroblast growth factor-inducible 14 (Fn14), monocyte chemoattractant protein-1 (MCP-1), neutrophils, tumor necrosis factor-like weak inducer of apoptosis (TWEAK)
Introduction
Tumor necrosis factor-like weak inducer of apoptosis (TWEAK) is a member of the tumor necrosis factor superfamily of cytokines (Chicheportiche et al, 1997). The TWEAK is synthesized as a type II transmembrane protein that can be cleaved to generate a soluble factor with biological activity (Chicheportiche et al, 1997). Fibroblast growth factor-inducible 14 (Fn14) is the receptor for TWEAK (Wiley et al, 2001) and binding of TWEAK to Fn14 activates the nuclear factor (NF)-κB signal transduction pathway [Reviewed in Winkles (2008)], which has a central function in the generation of an inflammatory response in the central nervous system following a variety of stimuli including cerebral ischemia (Zheng and Yenari, 2004).
The TWEAK has been reported to stimulate cell proliferation (Lynch et al, 1999; Harada et al, 2002; Donohue et al, 2003), migration (Harada et al, 2002; Donohue et al, 2003; Tran et al, 2003), and differentiation (Polek et al, 2003), as well as the expression of proinflammatory molecules (Chicheportiche et al, 1997; Saas et al, 2000; Chicheportiche et al, 2002; Harada et al, 2002; Xu et al, 2004; Kim et al, 2004; Jin et al, 2004). The TWEAK induces the expression of MMP-9 in astrocytes (Polavarapu et al, 2005), MMP-1, IL-6, IL-8, RANTES and IP-10 in human synoviocytes and fibroblasts (Chicheportiche et al, 2002), monocyte chemoattractant protein-1 (MCP-1), MMP-9, IL-6 and IL-8 in macrophages (Kim et al, 2004), and MCP-1, E-selectin, IL-8 and ICAM-1 in HUVEC (Harada et al, 2002). Likewise, a growing body of evidence supports a role for TWEAK and Fn14 in the pathophysiology of inflammatory diseases. In addition, treatment with anti-TWEAK blocking antibodies improves clinical outcome and decreases the infiltration of inflammatory cells in animal models of collagen-induced arthritis (Kamata et al, 2006), experimental allergic encephalomyelitis (Desplat-Jego et al, 2005), and lupus nephritis (Zhao et al, 2007).
In the brain, TWEAK and Fn14 are expressed mainly in perivascular astrocytes and endothelial cells (Yepes et al, 2005). Moreover, experimental middle cerebral artery occlusion (MCAO) in mice (Yepes et al, 2005) and ischemic stroke in humans (Inta et al, 2008) increase the expression of this cytokine and its receptor in the ischemic tissue. Importantly, inhibition of TWEAK activity either with an Fn14-Fc decoy receptor (Yepes et al, 2005; Polavarapu et al, 2005) or anti-TWEAK neutralizing antibodies (Potrovita et al, 2004) or genetic deficiency of Fn14 (Zhang et al, 2007) decreases the volume of the ischemic lesion and preserves the barrier function of the blood–brain barrier (BBB) after experimental MCAO.
Ischemic stroke is the third cause of mortality and a leading cause of disability in the world (Lloyd-Jones et al, 2009). The onset of the ischemic insult is followed by the infiltration of inflammatory cells into the ischemic tissue, which has been directly associated with a poor outcome. In animal models of experimental cerebral ischemia, neutrophils are the first hematogenous cells that infiltrate the ischemic brain (peak at 24 h), followed by T cells and monocytes/macrophages (peak at 3 to 7 days) (Stoll et al, 1998; Stoll et al, 2002). The recruitment of inflammatory cells into the ischemic tissue is regulated by chemokines, which are a small group of cytokines (8 to 10 kDa in size) that directionally attract and activate leukocytes through chemokine gradients.
The MCP-1 (CCL2) is a member of the cysteine–cysteine chemokine gene family characterized by chemoattractant properties for monocytes, memory T-lymphocytes, and natural killer cells in vitro. In the brain, astrocytes are the main source for MCP-1 (Minami and Satoh, 2003). After MCAO, there is a progressive increase in the expression of MCP-1 in the ischemic tissue (Kim et al, 1995) associated with leukocyte migration into the ischemic area (Kim, 1996). Importantly, compared with wild-type (Wt) animals, mice lacking MCP-1 have smaller infarct volumes after MCAO as well as reduced infiltration of monocytes (Hughes et al, 2002) and polymorphonuclear neutrophils (Schilling et al, 2009).
In the work presented here, we show that during cerebral ischemia, the interaction between astrocytic-derived TWEAK and Fn14 induces the expression of astrocytic MCP-1 via activation of the NF-κB pathway, which leads to the recruitment of neutrophils into the ischemic brain. This is a novel pathway for the development of an inflammatory response after MCAO and a potential new target for the treatment of patient with ischemic stroke and other neurological pathologies characterized by the recruitment of blood-borne inflammatory cells.
Materials and methods
Animal Model of Cerebral Ischemia
Murine strains were TWEAK deficient (TWEAK−/−), and Fn14 deficient (Fn14−/−) mice, and their Wt littermate controls on a C57BL/6J genetic background, kindly provided by Dr Kyungmin Hahm (Biogen Idec Inc, Cambridge MA, USA). All procedures were approved by the Emory University Institutional Animal Care and Use Committee. Mice were anesthetized with 4% chloral hydrate (400 mg/kg IP). The rectal and masseter muscle temperatures were controlled at 37°C with a homeothermic blanket. The middle cerebral artery was exposed and occluded with a 10-to-0 suture as described elsewhere (Nagai et al, 1999). Cerebral perfusion in the distribution of the middle cerebral artery was monitored throughout the surgical procedure with a laser Doppler (Perimed Inc., North Royalton, OH, USA). Heart rate, systolic, diastolic, and mean arterial blood pressure were controlled with an IITC 229 System (IITC-Lice Science; Woodland Hills, CA, USA).
Cell Cultures, ELISA, and Exposure to Oxygen-Glucose Deprivation Conditions
Astrocytes were cultured from 1-day-old TWEAK−/−, Fn14−/−, and Wt C57BL/6J mice as described elsewhere (Yepes et al, 2003). The Wt cultures were treated 10 days later with serum-free media for 24 h, washed with PBS three times, and then incubated with progressive concentrations of recombinant TWEAK (0 to 1000 ng; R&D Systems, Minneapolis, MN, USA). A subset of Wt astrocytes was incubated with 10 μmol/L of the NF-κB inhibitor BAY 11-7085 (Alexis Biochemicals; Plymouth Meeting, PA, USA) followed 30 mins later by the addition of either vehicle (control) or TWEAK 100 ng. BAY 11-7085 has been shown earlier to successfully inhibit cytokine-induced IκBα phosphorylation (Pierce et al, 1997). A group of astrocytes was incubated with BAY 11-7085 alone. A subgroup of Wt and Fn14−/− astrocytes was incubated with rTWEAK 100 ng. A subset of Wt, TWEAK−/−, and Fn14−/− astrocytes were maintained under oxygen-glucose deprivation (OGD) conditions for 6 h in an anaerobic chamber (Billups-Rothenberg, Inc., Del Mar, CA, USA) as described earlier (Polavarapu et al, 2007) followed by 18 h of incubation under normoxic conditions and normal glucose concentration. A <5% oxygen concentration was confirmed with an oxymeter before starting the experiment. In preliminary studies, we used a colorimetric assay to quantify the release of lactate dehydrogenase in the media of cultured astrocytes (Oxford Biomedical research, Oxford, MI, USA). We found that exposure to OGD conditions for 6 h results in <10% release of lactate dehydrogenase compared with cultures treated with the cell lysing reagent provided by the manufacturer (data not shown). Twenty-four hours after incubation with rTWEAK under normoxic conditions or 6 h of OGD, we performed an ELISA for MCP-1 in the media with a kit purchased from eBiosciences (San Diego, CA, USA), following the manufacturer's instructions. All the samples were assayed by duplicate. The optical density was determined by a microplate reader with the wavelength set to 450 nm. Each observation was repeated six times. To determine the concentration of MCP-1 in the ischemic brain, 24 h after MCAO Wt, TWEAK−/−, and Fn14−/− mice were killed, and the brains were divided in ipsilateral and contralateral hemispheres. Tissue samples were weighted, homogenized in a lysis buffer, and centrifuged at 13,000g for 30 mins. The supernatants were collected and an ELISA for MCP-1 was performed as described above. Statistical analysis was performed with the Student's t and the Wilcoxon two-sample rank sum tests. A P<0.05 was considered as significant.
In Vitro Model of the Blood–Brain Barrier
To establish the in vitro model of the BBB, 1-μm pore size tissue culture inserts (Falcon Becton-Dikinson Labware; Franklin Lakes, NJ, USA) were coated with rat tail-collagen (Roche Diagnostics Corporation; Indianapolis IN, USA), followed by the addition of 1 × 105 Wt or Fn14−/− astrocytes to the underside and 1 × 105 bovine brain microvascular endothelial cells (Cell Applications, Inc.; San Diego, CA, USA) to the upper side of the insert. We also added 700 and 300 μl of the complete MEM culture medium to the wells and the inserts, respectively. Experiments were performed 72 h later.
Permeability Studies
For the permeability studies, cocultures were incubated with either vehicle (control) or rTWEAK 100 ng/mL for 24 h followed by the addition of 600 μl of FITC-albumin (400 μg/mL; Sigma-Aldrich, St Louis, MO, USA) to the upper chamber (endothelial cells) containing 0.5 mL of DMEM. The well (lower chamber) contained 1.2 mL of DMEM. After 4 h, 0.2 mL of this fluid was sampled and replaced with fresh DMEM and the permeability coefficient (PC; cm/min) was calculated with the following equation:
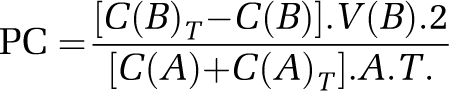 |
where C(B) and C(B)T are the concentrations (ng/mL) of FITC-albumin in the lower chamber at the start and at the end of the experiment, respectively, and V(B) is the volume of the basal chamber (in mL). C(A) and C(A)T are, respectively, the concentrations of FITC-albumin in the upper chamber at the beginning and at the end of the experiment. T is the duration of the time interval (minutes), whereas A is the area of the filter (in cm2). All samples were read on a fluorescent reader, emission 485 nm and excitation 540 nm, and the concentration of FITC-albumin was calculated from a standard curve derived from known concentrations of the tracer.
Neutrophil Isolation and Transmigration Assay
Neutrophils were isolated using Ficoll-gradient centrifugation method. Briefly, 20 mL of whole blood from a human donor were collected into 4 mL of acetate-citrate-dextrose-anticoagulated followed by the addition of 12 mL of a 6% Dextran/0.9% NaCl solution. The mixture was pipetted into 15 mL tubes and kept at room temperature for 1 h. The yellowish supernatant containing leukocytes and lymphocytes was separated into a new 50 mL tube and centrifuged at 238g for 12 mins at 4°C. The supernatant was discarded and the pellet was resuspended in 12 mL of ice-cold filter deionized water. After 20 secs, 4 mL of 0.6 M KCL was added to the mixture, and the solution was diluted to 50 mL with PBS and centrifuged at 304g for 6 mins at 4°C. The pellet was resuspended in 2.5 mL PBS and layered over 3 mL of Ficoll-Paque™ PLUS (GE Healthcare Bio-Sciences AB, Uppsala, Sweden) in a 15-mL tube and centrifuged at 405g for 30 mins at 4°C. For the transmigration assays, the complete medium was replaced with serum-free DMEM medium on both sides of the insert followed by the addition of 100 ng/mL of recombinant TWEAK to the lower chamber of the system. As controls, a subset of cocultures were left untreated. Twenty-four hours later, 5 × 105 neutrophils were added to the upper chamber and the number of neutrophils in the bottom chamber of the inserts was determined using a hemocytometer after 4 h. Each observation was repeated six times.
Quantitative Real-Time Polymerase Chain Reaction Analysis
Astrocytes cultured from TWEAK−/− and Fn14−/− mice were exposed to OGD conditions. Wild-type, TWEAK−/−, and Fn14−/− mice underwent MCAO. Sham-operated animals and astrocytes kept under normoxic conditions were included as controls for each experiment. Six hour after either exposure to OGD conditions or MCAO, cells and brains were harvested. Total RNA was isolated using the RNAeasy mini kit (Qiagen; Valencia, CA, USA) according to the manufacturer's instructions and equal amounts of RNA were taken for cDNA synthesis using high-capacity cDNA Kit (Applied Biosystems; Foster City, CA, USA). Real-time (RT) quantitative polymerase chain reaction (PCR) analysis for TWEAK and Fn14 (in astrocytes) or MCP-1 (in brains) was performed using TaqMan Gene Expression Assays (Applied Biosystems) with forward and reverse primers as well as an internal probe also purchased from Applied Biosystems. Polymerase chain reactions were performed using a 7500 Fast RT-PCR System (Applied Biosystems) under the following conditions: 50°C for 2 mins, 95°C for 10 mins, 40 cycles at 95°C for 15 secs, and 60°C for 1 min. Each observation was repeated eight times.
Immunohistochemistry and Definition of Areas of Interest
Twenty-four hours after MCAO, Wt, TWEAK−/−, and Fn14−/− mice were transcardially perfused for 10 mins. Brains were harvested and 20 frozen brain sections 10 μm each were stained with a monoclonal antibody that detects a polymorphic 40 kDa antigen expressed only by polymorphonuclear cells (MCA771GA; AbD Serotec, Raleigh, NC, USA). Sections were costained with 4′-6-Diamidino-2-phenylindole (DAPI, Sigma-Aldrich). Each coronal section was then divided into 16 square areas (150 mm2 each one) that involved the necrotic core and the area of ischemic penumbra, and comparable areas in the nonischemic hemisphere. Two areas of interest (AOI) were chosen in the boundaries between the ischemic penumbra and necrotic core (AOI-1 and AOI-3), whereas a third zone was located in the necrotic core (AOI-2). To determine the number of neutrophils, images were digitized in a Zeiss Axioplan 2 microscope (20-fold objective) with a Zeiss AxioCam and imported into AxioVision. Images were then viewed at 150% of the original × 20 images with an Image MetaMorph Software. The number of neutrophils counted in the described AOIs was organized in a two-way comparison table for the ischemic and the nonischemic hemispheres. Four animals were included in each experimental group. Each experiment was repeated eight times.
Myeloperoxidase Activity Assay
Tissue myeloperoxidase (MPO) activity was quantified with a commercially available kit purchased from Cytostore (Calgary; Alberta, Canada). The brains of Wt, TWEAK−/−, and Fn14−/− mice were harvested 24 h after MCAO, homogenized and suspended in 0.5% hexadecyltrimethylammonium bromide in 50 mmol/L potassium phosphate buffer, pH 6.0 (HTAB buffer), cooled on ice and homogenized and sonicated for 30 secs, followed by centrifugation at 40,000g for 20 mins at 4°C. A measure of 30 μL of supernatant were assayed for MPO activity in a microtiter plate in HTAB buffer at room temperature by monitoring its OD at 450 nm. The reaction was stopped with 0.1% sodium azide. Murine polymorphonuclear MPO standards were included in each assay to quantitate and standardize MPO content. Each observation was repeated eight times.
Results
Tumor Necrosis Factor-like Weak Inducer of Apoptosis Induces the Passage of Neutrophils Through the Endothelial Cell-Basement Membrane-Astrocyte Interface
Earlier studies indicate that TWEAK induces the recruitment of inflammatory cells in animal models of collagen-induced arthritis (Kamata et al, 2006), experimental allergic encephalomyelitis (Desplat-Jego et al, 2005), and lupus nephritis (Zhao et al, 2007). Thus, we decided to investigate, whether TWEAK also induces the passage of inflammatory cells through the BBB. To study this possibility, the lower compartment of an in vitro model of the BBB seeded with either Wt or Fn14−/− astrocytes was incubated with either TWEAK 100 ng/mL or vehicle control followed by the addition of 5 × 105 neutrophils to the upper chamber containing bovine brain microvascular endothelial cells. The number of neutrophils in the lower chamber was determined 4 h later. We found that compared with vehicle-control-treated cocultures, incubation with TWEAK induced a significant increase in the number of neutrophils infiltrating the lower chamber of the coculture system (5.8±104 neutrophils/mL. Figure 1; n=6, P<0.0001). In contrast, rTWEAK failed to induce the passage of neutrophils in a model assembled with Fn14−/− astrocytes. Importantly, our permeability studies showed that incubation with comparable doses of rTWEAK does not induce a significant increase in the PC to FITC-albumin, indicating that the passage of neutrophils through the endothelial cell-astrocyte interface induced by TWEAK is mediated by transcellular transport and not by degradation of structural components of the BBB.
Figure 1.

The TWEAK induces the passage of neutrophils through the endothelial cell-basement membrane-astrocyte interface. Average number of neutrophils in the lower compartment of an in vitro model of the BBB assembled with either Wt or Fn14−/− astrocytes after 24 h of incubation with either vehicle (−, white bars) or rTWEAK 100 ng (+, black bars) and the addition of 5 × 105 neutrophils to the upper chamber of the system containing bovine brain microvascular endothelial cells. n=6. Lines denote s.d.
The Interaction Between Tumor Necrosis Factor-like Weak Inducer of Apoptosis and Fibroblast Growth Factor-Inducible 14 Induces the Expression of Monocyte Chemoattractant Protein-1 in Astrocytes via Nuclear Factor-κB Pathway Activation
It has been shown that MCP-1 has a pivotal function in the infiltration of neutrophils through the endothelial cell-basement membrane-astrocyte interface (Schilling et al, 2009). As astrocytes are the main source of MCP-1 in the central nervous system, we decided to study the effect of the interaction between TWEAK and Fn14 on the expression of astrocytic MCP-1. Astrocytes were cultured from Wt mice and incubated with TWEAK 0 to 1000 ng followed 24 h later by quantification of MCP-1 protein in the media by ELISA. Our results show that TWEAK induces a dose-dependent increase in the expression of MCP-1 in the media of Wt astrocytes from 847±55.6 to 1200±59.39 and 1462±52.13 pg/mL after incubation with 10,100 and 1,000 ng of TWEAK, respectively. Each determination was repeated six times (Figure 2A). As in earlier studies we showed that TWEAK induces NF-κB pathway activation in astrocytes (Polavarapu et al, 2005; Zhang et al, 2007), we decided to see whether the effect of TWEAK on MCP-1 expression was mediated by activation of the NF-κB pathway. The Wt astrocytes were incubated with either vehicle (control) or the NF-κB inhibitor BAY 11-7085 30 mins before the addition of TWEAK 100 ng, followed 24 h later by determination of MCP-1 protein by ELISA. A subset of astrocytes was incubated with BAY 11-7085 alone. We found that inhibition of the NF-κB pathway attenuated the effect of TWEAK on MCP-1 expression in astrocytes from 1272.86±192.04 pg/mL in TWEAK-treated astrocytes to 328.28±119.88 pg/mL in astrocytes pretreated with the NF-κB inhibitor BAY 11-7085 (n=6; P<0.0001) (Figure 2B). Importantly, incubation with BAY 11-7085 alone did not have any effect on MCP-1 expression (data not shown). It has been recently reported that TWEAK also interacts with CD163 (Bover et al, 2007). Thus, we decided to investigate whether the effect of TWEAK on MCP-1 expression was mediated by its interaction with Fn14 or any other receptor. To study this possibility, we quantified the expression of MCP-1 in the media of astrocytes cultured from Wt and Fn14−/− mice and incubated with TWEAK 100 ng for 24 h. Our results indicate that incubation with TWEAK induces a sharp increase in MCP-1 expression in Wt astrocytes from 404.25±28.85 pg/mL to 1464.2±197 pg/mL (n=6, P<0.0001), and that this effect is abrogated by genetic deficiency of Fn14 (Figure 2C).
Figure 2.

The binding of TWEAK to Fn14 induces the expression of MCP-1 in astrocytes via NF-κB pathway activation. (A) Mean MCP-1 protein concentration in Wt astrocytes incubated for 24 h with rTWEAK 0 to 1000 ng. Lines denote s.d. n=6 observations per time-point. (B) Mean MCP-1 protein concentration in Wt astrocytes pretreated with either TWEAK alone (black bar,−) or with the NF-κB inhibitor BAY 11-7085 and TWEAK 100 ng (white bar, +). Lines denote s.d. n=6. (C) Mean MCP-1 protein concentration in wild-type (Wt) and Fn14−/− astrocytes incubated with either vehicle control (black bars, −) or rTWEAK 100 ng (white bars, +) for 24 h. Lines denote s.d. n=6.
Endogenous Tumor Necrosis Factor-like Weak Inducer of Apoptosis-Fibroblast Growth Factor-Inducible 14 Mediate Hypoxia-Induced Astrocytic Monocyte Chemoattractant Protein-1 Expression
Then we decided to study whether TWEAK and Fn14 mediate the effect of hypoxia on the expression of astrocytic MCP-1. First, we studied the expression of astrocytic TWEAK and Fn14 mRNA after 6 h of exposure to OGD conditions. We found that compared with cultures maintained under normoxic conditions, exposure of Wt astrocytes to OGD conditions induces a 4.3±0.9-fold increase in TWEAK mRNA and 6.3±1.2-fold in Fn14 mRNA expression (Figure 3A). To study the effect of hypoxia on astrocytic MCP-1 expression, we performed an ELISA for MCP-1 in the media of Wt astrocytes maintained under either normoxic (N in Figure 3B) or hypoxic (H in Figure 3B) conditions for 6 h as described above in the Materials and methods section. We found that OGD induces a sharp increase in MCP-1 expression in Wt astrocytes from 658.8±128.2 to 1427.5±248 pg/mL (n=6, P<0.005). To see whether the effect of hypoxia on MCP-1 expression is mediated by endogenous TWEAK and Fn14, we performed similar experiments with TWEAK−/− and Fn14−/− astrocytes. Our data indicate that in contrast to Wt astrocytes, OGD failed to induce a significant increase in MCP-1 expression in TWEAK−/− or Fn14−/− astrocyes. Importantly, incubation of TWEAK−/− astrocytes with rTWEAK increased the expression of MCP-1 to 1138±121 pg/mL (P<0.005 compared with TWEAK−/− astrocytes maintained under hypoxic OGD conditions in absence of rTWEAK.
Figure 3.

The interaction between endogenous TWEAK and Fn14 mediates hypoxia-induced astrocytic MCP-1 expression. (A) Real-time quantitative PCR analysis of TWEAK and Fn14 mRNA expression in Wt astrocytes exposed to OGD conditions for 6 h. n=6; *P<0.005 compared with controls kept under normoxic conditions. (B) Mean MCP-1 protein concentration in Wt, TWEAK−/−, and Fn14−/− astrocytes exposed to normoxic (N, black bars) or hypoxic (H, white bars) conditions for 6 h followed by 18 h of incubation under normoxic conditions. A subset of TWEAK−/− astrocytes was incubated under hypoxic conditions with rTWEAK (gray bar; **P<0.005 compared with TWEAK−/− astrocytes maintained under OGD conditions in the absence of rTWEAK). Lines denote s.d. n=6 observations per time-point.
Tumor Necrosis Factor-like Weak Inducer of Apoptosis/Fibroblast Growth Factor-Inducible 14 Mediates Cerebral Ischemia-Induced Monocyte Chemoattractant Protein-1 Expression
As it has been reported that MCAO induces a progressive increase in MCP-1 expression in the ischemic brain, thus we decided to investigate whether endogenous TWEAK and Fn14 mediate this effect. To test this hypothesis, we performed quantitative RT-PCR analysis for TWEAK and Fn14 mRNA in brain extracts of Wt, TWEAK−/− and Fn14−/− mice 6 h after MCAO. Our results indicate that compared with nonischemic brains, cerebral ischemia induces an 11.3±2.5-fold increase in MCP-1 mRNA expression in the ischemic tissue of Wt mice, and that this effect is significantly decreased in TWEAK−/− (5.5±1.4-fold increase) and Fn14−/− (4.4±1.0-fold increase) mice (Figure 4A). To further characterize these results, we determined the concentration of MCP-1 protein in brain extracts prepared from the ipsilateral (ischemic) and contralateral (nonischemic) hemispheres of Wt, TWEAK−/− and Fn14−/− mice 24 h after MCAO. We found that cerebral ischemia induces a sharp increase in MCP-1 protein expression in the ischemic hemisphere of Wt mice (2168±37.8 pg/mL) that is significantly attenuated by genetic deficiency of either TWEAK (1007±84 pg/mL) or Fn14 (1106±95 pg/mL; Figure 4B).
Figure 4.

Effect of genetic deficiency of TWEAK and Fn14 on cerebral ischemia-induced MCP-1 expression. (A) Real-time quantitative PCR analysis of MCP-1 in brain extracts from wild-type (Wt, white bar), TWEAK−/− (black bar), and Fn14−/− (gray bar) mice 6 h after MCAO. Lines denote s.d. n=6 observations per time-point. *P<0.001 compared with the other experimental groups. (B) Mean MCP-1 protein concentration in the ischemic (Ipsi., black bars) and nonischemic (Con., white bars) hemispheres of Wt, TWEAK−/−, and Fn14−/− mice 24 h after MCAO. Lines denote s.d. n=6 observations per time-point. NS, not significant.
Effect of Genetic Deficiency of Tumor Necrosis Factor-like Weak Inducer of Apoptosis and Fibroblast Growth Factor-Inducible 14 on Cerebral Ischemia-Induced Neutrophil Infiltration
As a growing body of evidence indicates that MCP-1 has a pivotal function in the infiltration of neutrophils into the ischemic brain (Schilling et al, 2009), we decided to investigate the effect of genetic deficiency of TWEAK or Fn14 on the infiltration of neutrophils into the ischemic tissue after MCAO. First, we determined the average number of cells immunopositive for a polymorphic 40 kDa antigen present only in neutrophils in each of the AOI of Wt, TWEAK−/−, and Fn14−/− mice 24 h after MCAO. We found that the average number of neutrophils in each AOI in Wt mice was 78±32, in contrast to 42.6±6 in TWEAK−/−, and 32±9 in Fn14−/− mice (Figures 5A and 5B; n=6, P<0.001 when Wt brains are compared with the other experimental groups). To further characterize these results, we performed an MPO activity assay in the ischemic brain of Wt, TWEAK−/− and Fn14−/− 24 h after MCAO. Our results indicate that cerebral ischemia induces a sharp increase in MPO activity in the ischemic tissue of Wt mice (0.44±0.05 MPO units/g tissue), and that this effect is attenuated in TWEAK−/− (0.18±0.047 MPO units/g tissue) and Fn14−/− (0.1±0.05 MPO units/g tissue) mice (Figure 5C).
Figure 5.

Effect of genetic deficiency of TWEAK and Fn14 on the recruitment of neutrophils into the ischemic tissue after MCAO. (A) Representative immunohistochemical staining for neutrophils in each AOI of wild-type (A), TWEAK−/− (B), and Fn14−/− mice (C) mice 24 h after MCAO. Green is a 40-kDa antigen expressed only by polymorphonuclear cells and blue is DAPI. Magnification × 40. (B) Mean number of infiltrating neutrophils in AOI 1-3 in wild-type (Wt), TWEAK−/−, and Fn14−/− mice 24 h after MCAO. Lines depict s.e.m. n=6. *P<0.001 compared with TWEAK−/− and Fn14−/− mice. (C) Myeloperoxidase activity in the ischemic brain tissue of Wt, TWEAK−/−, and Fn14−/− mice 24 h after MCAO. Lines depict s.e.m. n=6. *P<0.001 compared with TWEAK−/− and Fn14−/− mice.
Discussion
The TWEAK is a member of the tumor necrosis factor ligand family originally identified as a protein with proapoptotic properties on interferon-γ-treated human HT-29 colon carcinoma cells. The Fn14 is a member of the tumor necrosis factor receptor superfamily and the cognate receptor for TWEAK (Winkles, 2008). It was initially shown that TWEAK binds to Fn14 but not to any other tumor necrosis factor receptor superfamily member (Bossen et al, 2006). However, recent evidence indicates that TWEAK binds to CD163 as well (Bover et al, 2007). Our results show that the effect of TWEAK on the recruitment of blood-borne inflammatory cells in the ischemic brain is mediated by its interaction with Fn14.
In the central nervous system, TWEAK and Fn14 are found mainly in perivascular astrocytes and endothelial cells. Likewise, the expression of TWEAK and Fn14 increases in the ischemic tissue of stroke patients (Inta et al, 2008) and in two different animal models of MCAO (Potrovita et al, 2004; Yepes et al, 2005). Importantly, it has been recently described that TWEAK is expressed in human atherosclerotic plaques and that the interaction between TWEAK and CD163 is a biomarker of clinical and subclinical atherosclerosis. Together, these findings indicate that changes in TWEAK expression may constitute a risk factor for ischemic stroke (Moreno et al, 2009). Our data show that exposure to OGD conditions increases astrocytic TWEAK and Fn14 expression. As astrocytes are important regulators of the permeability of the BBB, thus we postulated that the interaction between TWEAK and Fn14 in the endothelial cell-basement membrane-astrocyte interface regulates the permeability of the BBB under hypoxic/ischemic conditions. This hypothesis is supported by earlier observations, indicating that inhibition of TWEAK-Fn14 binding preserves the architecture and barrier function of the BBB in an animal model of cerebral ischemia (Potrovita et al, 2004; Yepes et al, 2005; Zhang et al, 2007). Here, we propose that the binding of astrocytic-derived TWEAK to Fn14 promotes the recruitment of blood-borne leukocytes into the ischemic tissue via NF-κB pathway activation.
In earlier studies, we showed that the intracerebral injection of TWEAK induces the expression and activity of MMP-9 in the brain (Polavarapu et al, 2005). As neutrophils are an important source of MMP-9 in the ischemic brain, based on the results presented here, it is plausible to postulate that the interaction between TWEAK and Fn14 in the endothelial cell-basement membrane-astrocyte interface induces the infiltration of neutrophils into the ischemic tissue leading to an augmentation in neutrophil-derived MMP-9 activity, which has been recognized as an important cause of increase in the permeability of the BBB during cerebral ischemia.
The onset of ischemic stroke leads to the induction of a robust inflammatory reaction characterized not only by the influx of blood-borne leukocytes into the cerebral parenchyma (Stoll et al, 1998), but also by the expression of astrocytic-derived proinflammatory cytokines. A growing body of evidence indicates that TWEAK is a cytokine with proinflammatory activity in the CNS. Indeed, earlier studies showed that incubation of human astrocytes with TWEAK induces the expression of ICAM-1 (Saas et al, 2000). Subsequently, we showed that TWEAK induces the expression of MMP-9 in Wt astrocytes (Polavarapu et al, 2005) and that this effect was linked to TWEAK's ability to increase the permeability of the BBB. Here, we show that the interaction between TWEAK and Fn14 in astrocytes induces a dose-dependent increase in the expression of the chemokine MCP-1, which has an important function in the infiltration of inflammatory cells into the ischemic brain. Together, our data suggest that the interaction between TWEAK and Fn14 in astrocytes is an important regulator of the inflammatory response of the ischemic brain. Nevertheless, we acknowledge that our in vivo studies cannot rule out the possibility of a ‘cross-talk' between astrocytic-derived TWEAK and endothelial cell Fn14, or between astrocytic-derived Fn14 and endothelial cell TWEAK.
Chemokines are small proinflammatory cytokines that act as chemmoatractants of neutrophils, macrophages, and other inflammatory cells. Indeed, in several neurological diseases such as cerebral ischemia, high-chemokine levels in the perivascular space result in the formation of a chemoattractant gradient that leads to the influx of leukocytes. The MCP-1 is a member of the cysteine–cysteine chemokine gene family that attracts monocytes, natural killer cells, and memory T lymphocytes in vitro and induces monocyte and neutrophil infiltration in vivo (Schilling et al, 2009). It has been shown that TWEAK induces the expression of MCP-1 in several cell lines (Winkles, 2008). Our results indicate that the interaction between TWEAK and Fn14 induces the expression of MCP-1 in astrocytes and Wt brains, and that this effect is decreased but not completely abrogated by genetic deficiency of either TWEAK or Fn14. This suggests the existence of other inductors of MCP-1 under hypoxic/ischemic conditions.
Activation of the NF-κB pathway is an important regulator of MCP-1 expression (Carlos and Harlan, 1994). Likewise, our earlier studies indicate that the interaction between TWEAK and Fn14 induces NF-κB pathway activation in astrocytes (Polavarapu et al, 2005). It is known that TWEAK may also activate the MAP kinase pathway. However, the data presented here show that the effect of TWEAK on astrocytic MCP-1 expression is mediated by activation of the NF-κB pathway. Thus, together our results suggest a model where after the onset of the ischemic/hypoxic insult the interaction between astrocytic-derived TWEAK and Fn14 induces an NF-κB-dependent increase in MCP-1 expression.
Cerebral ischemia is accompanied by a progressive increase in the expression of MCP-1 expression in the ischemic tissue. Importantly, this effect is seen in animal models of MCAO with (Kim et al, 1995) and without reperfusion (Che et al, 2001), and in both cases, the presence of MCP-1 is associated with the migration of monocytes and neutrophils into the ischemic tissue. Hence, mice genetic deficiency on MCP-1 (CCL2) or its receptor CCR2 have a significant decrease in the infiltration of inflammatory cells into the ischemic tissue after MCAO (Hughes et al, 2002). Our results indicate that the interaction between TWEAK and Fn14 mediate cerebral ischemia/hypoxia induced increase in MCP-1 expression. Accordingly, genetic deficiency of either TWEAK or Fn14 not only attenuates the expression of MCP-1 but also the influx of neutrophils into the ischemic tissue.
In summary, in this study we propose a model where in response to the ischemic insult, the interaction between TWEAK and Fn14 in perivascular astrocytes leads to the activation of the NF-κB pathway, with NF-κB-dependent induction of MCP-1 expression and MCP-1 regulated recruitment of neutrophils into the ischemic tissue. This is a novel pathway for cerebral ischemia/hypoxia-induced neuroinflammatory response and a potential target for the treatment of patients with ischemic stroke.
Acknowledgments
This work was supported in part by National Institutes of Health Grants NS-062073 and HL-095063 (to MY).
The authors declare no conflict of interest.
References
- Bossen C, Ingold K, Tardivel A, Bodmer JL, Gaide O, Hertig S, Ambrose C, Tschopp J, Schneider P. Interactions of tumor necrosis factor (TNF) and TNF receptor family members in the mouse and human. J Biol Chem. 2006;281:13964–13971. doi: 10.1074/jbc.M601553200. [DOI] [PubMed] [Google Scholar]
- Bover LC, Cardo-Vila M, Kuniyasu A, Sun J, Rangel R, Takeya M, Aggarwal BB, Arap W, Pasqualini R. A previously unrecognized protein-protein interaction between TWEAK and CD163: potential biological implications. J Immunol. 2007;178:8183–8194. doi: 10.4049/jimmunol.178.12.8183. [DOI] [PubMed] [Google Scholar]
- Carlos TM, Harlan JM. Leukocyte-endothelial adhesion molecules. Blood. 1994;84:2068–2101. [PubMed] [Google Scholar]
- Che X, Ye W, Panga L, Wu DC, Yang GY. Monocyte chemoattractant protein-1 expressed in neurons and astrocytes during focal ischemia in mice. Brain Res. 2001;902:171–177. doi: 10.1016/s0006-8993(01)02328-9. [DOI] [PubMed] [Google Scholar]
- Chicheportiche Y, Bourdon PR, Xu H, Hsu YM, Scott H, Hession C, Garcia I, Browning JL. TWEAK, a new secreted ligand in the tumor necrosis factor family that weakly induces apoptosis. J Biol Chem. 1997;272:32401–32410. doi: 10.1074/jbc.272.51.32401. [DOI] [PubMed] [Google Scholar]
- Chicheportiche Y, Chicheportiche R, Sizing I, Thompson J, Benjamin CB, Ambrose C, Dayer JM. Proinflammatory activity of TWEAK on human dermal fibroblasts and synoviocytes: blocking and enhancing effects of anti-TWEAK monoclonal antibodies. Arthritis Res. 2002;4:126–133. doi: 10.1186/ar388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desplat-Jego S, Creidy R, Varriale S, Allaire N, Luo Y, Bernard D, Hahm K, Burkly L, Boucraut J. Anti-TWEAK monoclonal antibodies reduce immune cell infiltration in the central nervous system and severity of experimental autoimmune encephalomyelitis. Clin Immunol. 2005;117:15–23. doi: 10.1016/j.clim.2005.06.005. [DOI] [PubMed] [Google Scholar]
- Donohue PJ, Richards CM, Brown SA, Hanscom HN, Buschman J, Thangada S, Hla T, Williams MS, Winkles JA. TWEAK is an endothelial cell growth and chemotactic factor that also potentiates FGF-2 and VEGF-A mitogenic activity. Arterioscler Thromb Vasc Biol. 2003;23:594–600. doi: 10.1161/01.ATV.0000062883.93715.37. [DOI] [PubMed] [Google Scholar]
- Harada N, Nakayama M, Nakano H, Fukuchi Y, Yagita H, Okumura K. Pro-inflammatory effect of TWEAK/Fn14 interaction on human umbilical vein endothelial cells. Biochem Biophys Res Commun. 2002;299:488–493. doi: 10.1016/s0006-291x(02)02670-0. [DOI] [PubMed] [Google Scholar]
- Hughes PM, Allegrini PR, Rudin M, Perry VH, Mir AK, Wiessner C. Monocyte chemoattractant protein-1 deficiency is protective in a murine stroke model. J Cereb Blood Flow Metab. 2002;22:308–317. doi: 10.1097/00004647-200203000-00008. [DOI] [PubMed] [Google Scholar]
- Inta I, Frauenknecht K, Dorr H, Kohlhof P, Rabsilber T, Auffarth GU, Burkly L, Mittelbronn M, Hahm K, Sommer C, Schwaninger M. Induction of the cytokine TWEAK and its receptor Fn14 in ischemic stroke. J Neurol Sci. 2008;275:117–120. doi: 10.1016/j.jns.2008.08.005. [DOI] [PubMed] [Google Scholar]
- Jin L, Nakao A, Nakayama M, Yamaguchi N, Kojima Y, Nakano N, Tsuboi R, Okumura K, Yagita H, Ogawa H. Induction of RANTES by TWEAK/Fn14 interaction in human keratinocytes. J Invest Dermatol. 2004;122:1175–1179. doi: 10.1111/j.0022-202X.2004.22419.x. [DOI] [PubMed] [Google Scholar]
- Kamata K, Kamijo S, Nakajima A, Koyanagi A, Kurosawa H, Yagita H, Okumura K. Involvement of TNF-like weak inducer of apoptosis in the pathogenesis of collagen-induced arthritis. J Immunol. 2006;177:6433–6439. doi: 10.4049/jimmunol.177.9.6433. [DOI] [PubMed] [Google Scholar]
- Kim JS. Cytokines and adhesion molecules in stroke and related diseases. J Neurol Sci. 1996;137:69–78. doi: 10.1016/0022-510x(95)00338-3. [DOI] [PubMed] [Google Scholar]
- Kim JS, Gautam SC, Chopp M, Zaloga C, Jones ML, Ward PA, Welch KM. Expression of monocyte chemoattractant protein-1 and macrophage inflammatory protein-1 after focal cerebral ischemia in the rat. J Neuroimmunol. 1995;56:127–134. doi: 10.1016/0165-5728(94)00138-e. [DOI] [PubMed] [Google Scholar]
- Kim SH, Kang YJ, Kim WJ, Woo DK, Lee Y, Kim DI, Park YB, Kwon BS, Park JE, Lee WH. TWEAK can induce pro-inflammatory cytokines and matrix metalloproteinase-9 in macrophages. Circ J. 2004;68:396–399. doi: 10.1253/circj.68.396. [DOI] [PubMed] [Google Scholar]
- Lloyd-Jones D, Adams R, Carnethon M, De SG, Ferguson TB, Flegal K, Ford E, Furie K, Go A, Greenlund K, Haase N, Hailpern S, Ho M, Howard V, Kissela B, Kittner S, Lackland D, Lisabeth L, Marelli A, McDermott M, Meigs J, Mozaffarian D, Nichol G, O'Donnell C, Roger V, Rosamond W, Sacco R, Sorlie P, Stafford R, Steinberger J, Thom T, Wasserthiel-Smoller S, Wong N, Wylie-Rosett J, Hong Y. Heart disease and stroke statistics--2009 update: a report from the American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Circulation. 2009;119:480–486. doi: 10.1161/CIRCULATIONAHA.108.191259. [DOI] [PubMed] [Google Scholar]
- Lynch CN, Wang YC, Lund JK, Chen YW, Leal JA, Wiley SR. TWEAK induces angiogenesis and proliferation of endothelial cells. J Biol Chem. 1999;274:8455–8459. doi: 10.1074/jbc.274.13.8455. [DOI] [PubMed] [Google Scholar]
- Minami M, Satoh M. Chemokines and their receptors in the brain: pathophysiological roles in ischemic brain injury. Life Sci. 2003;74:321–327. doi: 10.1016/j.lfs.2003.09.019. [DOI] [PubMed] [Google Scholar]
- Moreno JA, Munoz-Garcia B, Martin-Ventura JL, Madrigal-Matute J, Orbe J, Paramo JA, Ortega L, Egido J, Blanco-Colio LM. The CD163-expressing macrophages recognize and internalize TWEAK: potential consequences in atherosclerosis. Atherosclerosis. 2009;207:103–110. doi: 10.1016/j.atherosclerosis.2009.04.033. [DOI] [PubMed] [Google Scholar]
- Nagai N, De Mol M, Lijnen HR, Carmeliet P, Collen D. Role of plasminogen system components in focal cerebral ischemic infarction: a gene targeting and gene transfer study in mice. Circulation. 1999;99:2440–2444. doi: 10.1161/01.cir.99.18.2440. [DOI] [PubMed] [Google Scholar]
- Pierce JW, Schoenleber R, Jesmok G, Best J, Moore SA, Collins T, Gerritsen ME. Novel inhibitors of cytokine-induced IkappaBalpha phosphorylation and endothelial cell adhesion molecule expression show anti-inflammatory effects in vivo. J Biol Chem. 1997;272:21096–21103. doi: 10.1074/jbc.272.34.21096. [DOI] [PubMed] [Google Scholar]
- Polavarapu R, Gongora MC, Winkles JA, Yepes M. Tumor necrosis factor-like weak inducer of apoptosis increases the permeability of the neurovascular unit through nuclear factor-kappaB pathway activation. J Neurosci. 2005;25:10094–10100. doi: 10.1523/JNEUROSCI.3382-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polavarapu R, Gongora MC, Yi H, Ranganthan S, Lawrence DA, Strickland D, Yepes M. Tissue-type plasminogen activator-mediated shedding of astrocytic low-density lipoprotein receptor-related protein increases the permeability of the neurovascular unit. Blood. 2007;109:3270–3278. doi: 10.1182/blood-2006-08-043125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polek TC, Talpaz M, Darnay BG, Spivak-Kroizman T. TWEAK mediates signal transduction and differentiation of RAW264.7 cells in the absence of Fn14/TweakR. Evidence for a second TWEAK receptor. J Biol Chem. 2003;278:32317–32323. doi: 10.1074/jbc.M302518200. [DOI] [PubMed] [Google Scholar]
- Potrovita I, Zhang W, Burkly L, Hahm K, Lincecum J, Wang MZ, Maurer MH, Rossner M, Schneider A, Schwaninger M. Tumor necrosis factor-like weak inducer of apoptosis-induced neurodegeneration. J Neurosci. 2004;24:8237–8244. doi: 10.1523/JNEUROSCI.1089-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saas P, Boucraut J, Walker PR, Quiquerez AL, Billot M, Desplat-Jego S, Chicheportiche Y, Dietrich PY. TWEAK stimulation of astrocytes and the proinflammatory consequences. Glia. 2000;32:102–107. [PubMed] [Google Scholar]
- Schilling M, Strecker JK, Schabitz WR, Ringelstein EB, Kiefer R. Effects of monocyte chemoattractant protein 1 on blood-borne cell recruitment after transient focal cerebral ischemia in mice. Neuroscience. 2009;161:806–812. doi: 10.1016/j.neuroscience.2009.04.025. [DOI] [PubMed] [Google Scholar]
- Stoll G, Jander S, Schroeter M. Detrimental and beneficial effects of injury-induced inflammation and cytokine expression in the nervous system. Adv Exp Med Biol. 2002;513:87–113. doi: 10.1007/978-1-4615-0123-7_3. [DOI] [PubMed] [Google Scholar]
- Stoll G, Jander S, Schroeter M. Inflammation and glial responses in ischemic brain lesions. Prog Neurobiol. 1998;56:149–171. doi: 10.1016/s0301-0082(98)00034-3. [DOI] [PubMed] [Google Scholar]
- Tran NL, McDonough WS, Donohue PJ, Winkles JA, Berens TJ, Ross KR, Hoelzinger DB, Beaudry C, Coons SW, Berens ME. The human Fn14 receptor gene is up-regulated in migrating glioma cells in vitro and overexpressed in advanced glial tumors. Am J Pathol. 2003;162:1313–1321. doi: 10.1016/S0002-9440(10)63927-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiley SR, Cassiano L, Lofton T, Davis-Smith T, Winkles JA, Lindner V, Liu H, Daniel TO, Smith CA, Fanslow WC. A novel TNF receptor family member binds TWEAK and is implicated in angiogenesis. Immunity. 2001;15:837–846. doi: 10.1016/s1074-7613(01)00232-1. [DOI] [PubMed] [Google Scholar]
- Winkles JA. The TWEAK-Fn14 cytokine-receptor axis: discovery, biology and therapeutic targeting. Nat Rev Drug Discov. 2008;7:411–425. doi: 10.1038/nrd2488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H, Okamoto A, Ichikawa J, Ando T, Tasaka K, Masuyama K, Ogawa H, Yagita H, Okumura K, Nakao A. TWEAK/Fn14 interaction stimulates human bronchial epithelial cells to produce IL-8 and GM-CSF. Biochem Biophys Res Commun. 2004;318:422–427. doi: 10.1016/j.bbrc.2004.04.036. [DOI] [PubMed] [Google Scholar]
- Yepes M, Brown SA, Moore EG, Smith EP, Lawrence DA, Winkles JA. A soluble Fn14-Fc decoy receptor reduces infarct volume in a murine model of cerebral ischemia. Am J Pathol. 2005;166:511–520. doi: 10.1016/S0002-9440(10)62273-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yepes M, Moore E, Brown SA, Hanscom HN, Smith EP, Lawrence DA, Winkles JA. Progressive ankylosis (Ank) protein is expressed by neurons and Ank immunohistochemical reactivity is increased by limbic seizures. Lab Invest. 2003;83:1025–1032. doi: 10.1097/01.lab.0000075640.49586.e6. [DOI] [PubMed] [Google Scholar]
- Zhang X, Winkles JA, Gongora MC, Polavarapu R, Michaelson JS, Hahm K, Burkly L, Friedman M, Li XJ, Yepes M. TWEAK-Fn14 pathway inhibition protects the integrity of the neurovascular unit during cerebral ischemia. J Cereb Blood Flow Metab. 2007;27:534–544. doi: 10.1038/sj.jcbfm.9600368. [DOI] [PubMed] [Google Scholar]
- Zhao Z, Burkly LC, Campbell S, Schwartz N, Molano A, Choudhury A, Eisenberg RA, Michaelson JS, Putterman C. TWEAK/Fn14 interactions are instrumental in the pathogenesis of nephritis in the chronic graft-versus-host model of systemic lupus erythematosus. J Immunol. 2007;179:7949–7958. doi: 10.4049/jimmunol.179.11.7949. [DOI] [PubMed] [Google Scholar]
- Zheng Z, Yenari MA. Post-ischemic inflammation: molecular mechanisms and therapeutic implications. Neurol Res. 2004;26:884–892. doi: 10.1179/016164104X2357. [DOI] [PubMed] [Google Scholar]