

Abstract
Deregulated c-Myc is associated with a wide range of human cancers. In many cell types, overexpression of c-Myc potently promotes cell growth and proliferation concomitant with the induction of apoptosis. Secondary genetic events that shift this balance either by increasing growth and proliferation or limiting apoptosis are likely to cooperate with c-Myc in tumorigenesis. Here, the authors have performed large-scale insertional mutagenesis in Eµ-c-myc mice that, through mdm2 loss of function mutations, are sensitized to apoptosis. The authors chose to use this genetic background based on the hypothesis that the high level of apoptosis induced by c-Myc overexpression in MDM2-deficient mice would act as a rate-limiting barrier for lymphoma development. As a result, it was predicted that the spectrum of retroviral insertions would be shifted toward loci that harbor antiapoptotic genes. Nine novel common insertion sites (CISs) specific to mice with this sensitized genetic background were identified, suggesting the presence of novel antiapoptotic cancer genes. Moreover, cross-comparing the data to the Retroviral Tagged Cancer Gene Database, the authors identified an additional 23 novel CISs. Here, evidence is presented that 2 genes, ppp1r16b and hdac6, identified at CISs, are bona fide cellular oncogenes. This study highlights the power of combining unique sensitized genetic backgrounds with large-scale mutagenesis as an approach for identifying novel cancer genes.
Keywords: Myc, MDM2, p53, apoptosis, lymphoma
Introduction
Overexpression of the c-myc oncogene is associated with a wide variety of human cancers including lymphoma, leukemia, breast, and prostate.1 Deregulated c-myc can potently promote cell growth and proliferation that is likely to be a driving force in c-Myc–induced tumorigenesis. Concomitant with increased cell proliferation, however, overexpression of c-Myc in some cell types, particularly lymphocytes, can induce apoptosis that may serve to limit the expansion of cells harboring oncogenic c-myc. Indeed, overexpression of c-myc under control of the Ig heavy chain enhancer (Eµ) in mice results in increased proliferation of pro/pre-B cells that is initially counterbalanced by increased apoptosis.2 The late onset of lymphomagenesis in Eµ-c-myc transgenic mice suggests that the deregulation of c-myc alone is not sufficient for lymphoma development.3 Rather, secondary mutations that cooperate with oncogenic c-myc by shifting the balance in favor of proliferation over apoptosis are likely required to promote lymphomagenesis.
c-Myc–induced apoptosis in B cells is mediated predominantly through the p19arf-MDM2-p53 pathway. Overexpression of c-Myc in cultured B cells induces expression of p19arf (human p14arf), which in turn antagonizes the function of MDM2, a key negative regulator of p53.4-7 Thus, by increasing p19arf levels, Myc can disrupt MDM2-p53 interactions, thereby stimulating the transactivation and apoptotic functions of p53. Perturbations in the p19arf-MDM2-p53 pathway significantly affect the onset of lymphomagenesis in Eµ-c-myc mice. B cells lacking p19arf or p53 are resistant to c-Myc–induced apoptosis, and mice null for either of these genes succumb rapidly to lymphomagenesis.8 Moreover, 80% of lymphomas that develop in wild-type Eµ-c-myc mice contain mutations in the p19arf-MDM2-p53 pathway.9 Overexpression of the potent antiapoptotic protein Bcl-2 abrogates loss of p53 in lymphomas from Eµ-c-myc mice, suggesting that it is predominantly the apoptotic function of p53 that is required to suppress c-Myc–induced lymphomagenesis.10 The activation of the apoptotic function of p53 in response to deregulated Myc is additionally likely to be modified by collaborative pathways triggered in response to p53-independent signals. Analysis of 2 MYC mutants, T58A and P57S, frequently found in human Burkitt’s lymphoma, revealed that despite the ability of these point mutations to drive aberrant proliferation and activate the p19arf-MDM2-p53 pathway, the apoptotic function of p53 was compromised through the selective failure of these mutants to induce the proapoptotic protein, Bim.11 Genetic and/or epigenetic changes that raise the threshold level for c-Myc–induced apoptosis are therefore predicted to potently cooperate with deregulated c-myc in lymphomagenesis.
Insertional mutagenesis using the slow transforming Moloney murine leukemia virus (M-MuLV) has historically been a successful strategy for identifying oncogenes that contribute to the development of leukemia and lymphoma (reviewed in Jonkers and Berns12), particularly in cooperation with c-myc.13 M-MuLV preferentially infects murine lymphocytes and promotes lymphomagenesis due to deregulation (typically gene activation) of cellular genes at sites of proviral insertion. Given the low probability of identical M-MuLV insertions (<1 × 10−10) into the mouse genome, the identification of a common insertion site (CIS) in independently isolated lymphomas obtained from multiple mice is highly suggestive of a cancer gene locus. Using Eµ-c-myc mice with sensitized genetic backgrounds, a strong selective pressure can be imposed for proviral insertions that deregulate genes that act in a particular pathway or that have a similar function. For example, retroviral mutagenesis when performed in Eµ-c-myc; pim-/- pim2-/- mice resulted in the identification of a novel gene, pim3,14 with significant homology to pim1 and pim2, suggesting that in the absence of pim1 or pim2, a strong selective pressure is imposed for compensatory insertions. Insertional mutagenesis, when performed in different sensitized genetic backgrounds, continues to uncover new cancer genes, thereby demonstrating that this approach has not yet reached saturation (for examples, see references14-18).
To identify new cancer genes that cooperate with c-myc in lymphomagenesis, we took an integrated genetic-genomic approach that combines retroviral insertional mutagenesis using M-MuLV with genetically modified mice that are sensitized to c-Myc–induced apoptosis. We predicted that a genetic background in which B cells are sensitized to c-Myc–induced apoptosis would impose a strong selective pressure for M-MuLV insertions at loci harboring genes that limit apoptosis. We also expected to identify novel oncogenes that could drive a stronger proliferative response or otherwise promote tumor progress (e.g., via angiogenesis).
Results
Increased apoptosis in Eµ-c-myc mice with reduced levels of MDM2
Given that the stability and function of the p53 protein is kept under tight negative regulation by binding to its proximal inhibitor, MDM2,4,5,19,20 we hypothesized that a loss of MDM2 function would sensitize cells to c-Myc–induced apoptosis. To generate a genetic background sensitized to c-Myc–induced apoptosis, we therefore took advantage of the availability of wild-type, null (mdm2Δ7-12),21 and hypomorphic (mdm2puro)22 alleles of mdm2 to decrease the level of MDM2 and therefore increase p53 activity in vivo. Prior studies have demonstrated that the level of mdm2 RNA and protein in all tissues examined including spleen and thymus is reduced approximately 20%, 50%, and 70% in mdm2+/puro, mdm2+/Δ7-12, and mdm2puro/Δ7-12 mice, respectively, relative to the level in wild-type tissues.22 Concomitant with the loss of a wild-type level of MDM2, the transactivation and apoptotic functions of p53 are constitutively elevated in tissues from mice with subphysiological levels of MDM2.22,23 To verify that subphysiological levels of MDM2 sensitized B cells to c-Myc–induced apoptosis in vivo, the apoptotic index of B220+ cells in the bone marrow of 4- to 5-week-old Eµ-c-myc mice expressing 1 of 4 levels of MDM2 (mdm2+/+>mdm2+/puro>mdm2+/Δ7-12>mdm2puro/Δ7-12), prior to the onset of any overt disease, was quantified by flow cytometric analysis (Fig. 1A). The percentage of B220+ cells with a sub-G1 DNA content, a frequently used measure of apoptotic cells, was higher in the bone marrow of Eµ-c-myc; mdm2+/Δ7-12 mice as compared to Eµ-c-myc with wild-type MDM2 levels or nontransgenic mdm2+/Δ7-12 control mice. These results are in agreement with previous studies indicating that a 50% reduction in MDM2 sensitized splenic B cells to c-Myc–induced apoptosis.24 B220+ cells in the bone marrow of Eµ-c-myc; mdm2+/puro mice, which express 80% of the wild-type level of MDM2, also had a higher apoptotic index than cells obtained from either Eµ-c-myc; mdm2+/+ or nontransgenic mdm2+/puro mice (Fig. 1A). Thus, even a 20% reduction in MDM2 is sufficient to sensitize B cells to c-Myc–induced apoptosis.
Figure 1.
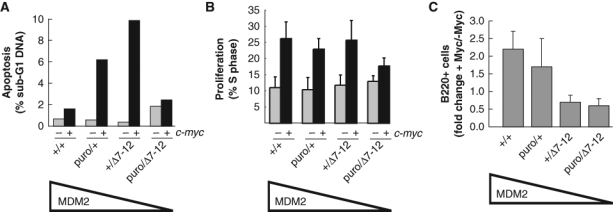
Preneoplastic phenotype of B220+ cells in the bone marrow of Eµ-c-myc mice with subphysiological levels of MDM2. Bone marrow cells were isolated from mice at 4 to 5 weeks of age prior to signs of overt disease and stained for the B cell surface marker B220 and for DNA content with propidium iodide. For mice of the indicated genotype, graphed are the percentages of B220+ B cells in the bone marrow with (A) a sub-G1 DNA content indicative of apoptosis (n = 2) or which (B) are in S-phase based on propidium iodide staining (n = 3-4). (C) Graphed is the fold change in the percentage of preneoplastic B220+ B cells in the bone marrow of Eµ-c-myc transgenic mice relative to nontransgenic mice that differ in the level of MDM2 (n = 5-9).
Surprisingly, overexpression of c-Myc did not appreciably increase the apoptotic index of B220+ cells from the bone marrow of Eµ-c-myc; mdm2puro/Δ7-12 mice compared to nontransgenic control mice (Fig. 1A). However, consistent with prior studies,22 the number of apoptotic B220+ cells of the bone marrow of mdm2puro/Δ7-12 mice is increased compared to wild-type mice, even in the absence of Myc overexpression, thus reflecting the high basal p53 activity present in tissues of mdm2puro/Δ7-12 mice in which MDM2 is expressed at only 30% of the level present in wild-type mice. As mdm2puro/Δ7-12 mice are exquisitely sensitive to stress stimuli that activate p53 (e.g., ionizing radiation),22 we surmise that c-myc overexpression potently induces p53 activity in Eµ-c-myc; mdm2puro/Δ7-12 mice, leading to the rapid elimination of B cells with elevated levels of c-Myc.
In contrast to changes in apoptosis, increased proliferation, a hallmark typically associated with c-Myc overexpression, was largely unaffected in Eµ-c-myc mice with subphysiological levels of MDM2 (Fig. 1B). Together, these results indicate that the primary effect of reducing the level of MDM2 in Eµ-c-myc mice is to sensitize B cells to c-Myc–induced apoptosis.
MDM2 deficiency restricts c-Myc–driven expansion of preneoplastic B cells
To determine the consequences of a reduced level of MDM2 on expansion of the B cell compartment in preneoplastic Eµ-c-myc mice, B220+ B cells in the bone marrow of Eµ-c-myc mice expressing 1 of 4 levels of MDM2 were quantified by flow cytometry. Preneoplastic Eµ-c-myc mice with a wild-type level of MDM2 exhibited a characteristic 2-fold increase in B220+ B cells in the bone marrow (Fig. 1C).25,26 Similarly, the population of B220+ B cells present in the bone marrow compartment of Eµ-c-myc; mdm2+/puro mice was also increased approximately 2-fold relative to nontransgenic control mice. Thus, overexpression of c-Myc was capable of expanding the pool of preneoplastic cells in Eµ-c-myc; mdm2+/puro mice despite the moderately increased apoptotic index of the B220+ cell population. In contrast, further reducing the level of MDM2 in Eµ-c-myc mice to 50% or less than wild-type MDM2 levels severely limited the B220+ B cell expansion. Flow cytometric analysis of the B220+ population revealed that the bone marrow of Eµ-c-myc; mdm2+/Δ7-12 and Eµ-c-myc; mdm2puro/Δ7-12 mice actually contained fewer B220+ B cells than was observed in nontransgenic mdm2+/Δ7-12 and mdm2puro/Δ7-12 control mice. Thus, a 50% to 70% decrease in MDM2 profoundly limits expansion of preneoplastic B cells harboring oncogenic c-Myc that is attributed to the elevated level of apoptosis observed in mice with this genetic background.
M-MuLV/c-myc–induced lymphomagenesis in MDM2-deficient mice
The heightened sensitivity of MDM2-deficient B cells to deregulated c-Myc suggests that the acquisition of secondary genetic changes that limit c-Myc–induced apoptosis is likely to be rate limiting for the promotion of lymphomagenesis in mice of this genetic background. To identify novel c-Myc cooperating genes that may possess antiapoptotic activity, we performed a large-scale insertional mutagenesis screen with the lymphotropic slow transforming retrovirus M-MuLV. Eµ-c-myc mice that differed in the level of MDM2 expression were infected at birth with M-MuLV and monitored for lymphoma development. We observed that the level of MDM2 profoundly affected the onset of M-MuLV/c-myc–induced lymphomagenesis. M-MuLV–infected Eµ-c-myc mice with a wild-type level of MDM2 rapidly developed lymphoma with a 100% incidence and a median survival of 69 days. In contrast, M-MuLV–infected Eµ-c-myc; mdm2puro/Δ7-12 mice that express approximately 30% of the wild-type level of MDM2 developed lymphoma with a median survival of 155 days, with 20% of Eµ-c-myc; mdm2puro/Δ7-12 mice remaining tumor free up to 300 days postinfection with M-MuLV (Fig. 2). Lymphomagenesis was also significantly delayed in M-MuLV–infected Eµ-c-myc; mdm2+/puro and Eµ-myc; mdm2+/Δ7-12 mice (median survival 82 and 88 days, respectively) compared with M-MuLV–infected Eµ-c-myc (P < 0.0001), demonstrating that even a moderate reduction in MDM2 is sufficient to delay tumor formation. Examination of cell surface markers by immunostaining and flow cytometry revealed that the majority of tumors analyzed, independent of mdm2 genotype, were B220+ CD43+ IgM-/lo pre–B cell lymphomas, indicating that level of MDM2 did not alter tumor type. Thus, the primary effect of a subphysiological level of MDM2 was to decrease the rate of M-MuLV/c-myc–induced lymphomagenesis.
Figure 2.
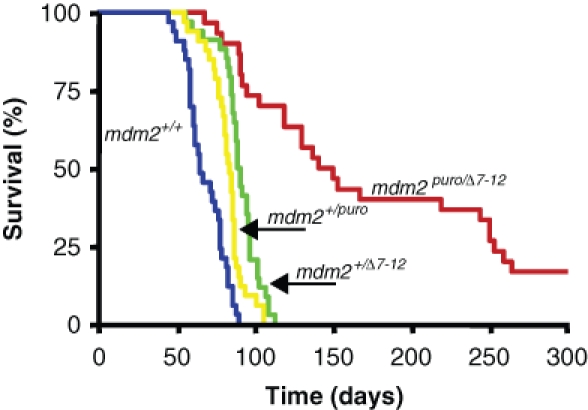
MDM2 deficiency delays M-MuLV–induced lymphomagenesis in Eµ-c-myc mice. Kaplan-Meier survival curve of M-MuLV–infected Eµ-c-myc mice that express one of 4 levels of mdm2: wild type (100%; n = 32), mdm2+/puro (80%; n = 35), mdm2+/Δ7-12 (50%; n = 37), or mdm2puro/Δ7-12 (30%; n = 29). Survival is plotted as days postinfection with M-MuLV.
Through a subphysiological level of MDM2-delayed tumorigenesis in M-MuLV–infected Eµ-c-myc mice, the introduction of M-MuLV was tumor promoting in this genetic background. Previous studies demonstrate that only 80% of Eµ-c-myc; mdm2+/Δ7-12 mice in the absence of M-MuLV infection develop lymphoma, with an extended median survival of approximately 310 days.24 In contrast, our studies revealed that following M-MuLV infection, 100% of Eµ-c-myc; mdm2+/Δ7-12 mice developed lymphoma with a median survival of 88 days (Fig. 2). Taken together, these findings indicate that M-MuLV accelerates lymphomagenesis almost 4-fold in the infection of Eµ-c-myc; mdm2+/Δ7-12 mice. This rapid acceleration of lymphomagenesis is consistent with the ability of M-MuLV to act as a genome-wide mutagen and suggests that proviral insertions at cancer gene loci are activating cellular oncogenes that cooperate with c-myc to promote lymphomagenesis in this sensitized genetic background.
Identification of candidate cancer genes by high-throughput insertion site analyses
To uncover novel cancer gene loci in M-MuLV–infected Eµ-c-myc mice with a less than wild-type level of MDM2, linker-mediated polymerase chain reaction (LM-PCR) and large-scale sequencing were used to identify mouse genomic DNA adjacent to the inserted proviral genome. A small number of insertion site sequences was also obtained by inverse PCR (I-PCR). Each lymphoma contained on average 4 retroviral insertion sites (RISs). Insertion site sequences were mapped to the mouse genome using the UCSC genome mm8 browser (http://www.genome.ucsc.edu/). Using this approach, 247 RISs from 74 individual lymphomas (25 from Eµ-myc; mdm2+/puro mice, 30 from Eµ-myc; mdm2+/Δ7-12 mice, and 19 from Eµ-c-myc; mdm2puro/Δ7-12 mice) were unambiguously mapped on the mouse genome. RISs were classified as forming a CIS using the criteria established by Suzuki et al. (2002)27 as 2, 3, or 4 or more insertions within 30 kb, 50 kb, or 100 kb, respectively. Within these stringent parameters, 33% (82/247) of RISs were found in multiple tumors forming 18 CISs (Table 1). The large percentage of retroviral insertions found within CIS clusters is consistent with that observed in other retroviral screens16,28 and likely reflects the powerful selection within lymphocytes for retroviral insertions that are advantageous for growth or survival.
Table 1.
CISs Identified in Lymphomas from Eµ-c-Myc Mice with Subphysiological Levels of mdm2
| RTCGDd | ||||||||
|---|---|---|---|---|---|---|---|---|
| Total | ||||||||
| CIS no. | Chromosome positiona (Chr [Mb]) | NCBI accession | Gene description | Eµ-c-Mycb (low mdm2) | Eµ-c-Mycc (wild-type mdm2) | RIS | Tn-IS | |
| CIS with known cancer genes | 1.1 | 10 [20] | NM_033597 | myb | 11 | 2 | 61 | 0 |
| 1.2 | 5 [107] | NM_010278 | gfi1 | 6 | 3 | 78 | 0 | |
| 1.3 | 17 [29] | NM_008842 | pim1 | 6 | 2 | 34 | 0 | |
| 1.4 | 2 [18] | NM_007552 | pcgf4/bmi1 | 6 | 4 | 17 | 0 | |
| 1.5 | 2 [117] | NM_011246 | rasgrp1 | 3 | 4 | 31 | 3 | |
| 1.6 | 11 [69] | NM_011640 | trp53 | 3 | 0 | 4 | 0 | |
| 1.7 | 2 [152] | NM_009743 | bcl-2l1/bcl-x | 3 | 0 | 7 | 0 | |
| 1.8 | 16 [92] | NM_009821 | runx1 | 2 | 2 | 9 | 0 | |
| 1.9 | 19 [37] | NM_008245 | hhex | 2 | 0 | 48 | 0 | |
| CIS with candidate cancer genes | 1.10 | 5 [76] | NM_010612 | kdr/flk-1/vegfr2 | 3 | 0 | 10 | 2 |
| 1.11 | 11 [74] | NM_001002764 | smg6 | 2 | 0 | 9 | 0 | |
| 1.12 | 9 [71] | NM_011544 | tcf12, heb | 3 | 0 | 4 | 0 | |
| 1.13 | 2 [158] | NM_153089 | ppp1r16b | 2 | 0 | 4 | 0 | |
| 1.14 | 6 [72] | NM_138592 | usp39 | 2 | 0 | 3 | 0 | |
| 1.15 | 13 [54] | NM_009946 | cplx2 | 2 | 0 | 2 | 0 | |
| 1.16 | 10 [80] | NM_008655 | gadd45β | 3 | 0 | 1 | 0 | |
| 1.17 | 13 [118] | NM_010330 | emb | 2 | 0 | 0 | 0 | |
| 1.18 | 9 [119] | NM_153287 | axud1 | 2 | 0 | 0 | 0 | |
Chromosomal location of common insertion site (CIS) based on UCSC genome mm8 browser. The chromosome number is followed by the position in megabases [Mb].
Number of M-MuLV retroviral insertions at the indicated locus identified in tumors from Eµ-c-myc mice with a subphysiological level of mdm2 (Eµ-c-myc; mdm2+/puro, Eµ-c-myc; mdm2+/Δ7-12 mice, and Eµ-c-myc; mdm2puro/Δ7-12; n = 74, this study).
Number of M-MuLV retroviral insertion at the indicated locus identified in tumors from Eµ-c-myc mice with wild-type levels of mdm2 (n = 21; Mikkers et al. [2002]14). Data obtained from Retroviral Tagged Cancer Gene Database (RTCGD).
Total number of retroviral (RIS) and transposon (Tn-IS) insertions at the indicated locus deposited in the RTCGD as of August 2006.
Insertion sites (currently more than 5,000) from a growing number of retroviral, as well as transposon-based, insertional mutagenesis screens have been recently compiled to form the Retroviral Tagged Cancer Gene Database (RTCGD).29 A complete list of RISs generated from this study will be deposited in the RTCGD (http://RTCGD.ncifcrf.gov). Through comparison with RTCGD, CISs identified in our study formed 2 classes of CISs. The first class consists of CISs identified previously in retroviral insertional mutagenesis screens and for which a known cancer gene is located (sites 1.1-1.9; Table 1). For the majority (8 of 9) of the CISs in Class 1, the cancer gene has been experimentally confirmed to be a cellular proto-oncogene. These proto-oncogenes include myb, gfi1, pim1, bmi1, rasgrp1, bcl-x, runx1, and hhex.30-37 The one exception is a CIS within the p53 tumor suppressor gene. As the frequency of insertion within both alleles of a gene is <1 × 10−10, insertional activation of tumor suppressors is a rare event in insertional mutagenesis screens. However, for some genes, both copies need not be inactivated for abrogation of function. The p53 gene, for example, is known to be haplo insufficient for tumor suppression, and a 50% reduction in gene dosage accelerates tumor formation.38 In addition, tetramerization of p53 through interaction at the C-termini is necessary for p53 function. Therefore, p53 mutants lacking the N-terminal trans-activation domain can be dominant negative to wild-type p53 through the formation of inactive p53 tetramers.39 All M-MuLV insertions identified in this screen and the RTCGD occur within intronic regions of p53 and therefore could potentially interfere with p53 function through either mechanism.
As the genetic background used in the present study is sensitized to c-Myc–induced apoptosis, it is significant that >50% of the cancer genes in class 1 are known to attenuate apoptosis through their overexpression (e.g., bmi1, myb, pim1, bcl-x, gfi1) or loss-of-function (e.g., p53). The identification of retroviral insertions within p53 in lymphomas from multiple Eµ-c-myc mice with subphysiological levels of MDM2 (all 3 were Eµ-c-myc; mdm2puro/Δ7-12) is particularly striking as a review of the RTCGD reveals that similar M-MuLV insertions have not been identified in tumors from Eµ-c-myc mice with a wild-type level of MDM2 (RTCGD). These results support our contention that a genetic background that is deficient in MDM2 imposes a selective pressure for retroviral insertions near genes that regulate apoptosis.
The second class of CISs we identified consists of M-MuLV insertions near or within genes that have not been identified previously as CISs in tumors from M-MuLV–infected Eµ-c-myc mice with a wild-type level of MDM2 and that have been observed only infrequently in insertional mutagenesis screens performed with different genetic backgrounds (sites 1.10-1.18; Table 1). The chromosomal location and orientation of M-MuLV insertion were used to identify candidate cancer genes at these 9 loci. For many of the putative target genes in class 2 (e.g., usp39, emb, axud1), little is known about their biological function. Importantly, at least 2 loci harbor genes previously implicated in the negative regulation of apoptosis. For example, gadd45β mRNA expression is rapidly induced in response to genotoxic stress, and overexpression of Gadd45β in vitro can block cell death following treatment with tumor necrosis factor alpha and the DNA-damaging agents daunorubicin and cisplatinum.40 Furthermore, cells from gadd45β-deficient mice are significantly more susceptible to UVC-induced apoptosis than wild-type cells.41,42 Likewise, knockdown of ppp1r16b (to be discussed below) by siRNA elicited a 4-fold increase in apoptosis in Hela cells, supporting a role for Ppp1r16b in promoting cell survival.43 The role of these genes in tumorigenesis remains to be determined.
Integral analysis of retroviral insertions sites
Although 33% of the RISs identified in Eµ-c-myc mice with subphysiological levels of MDM2 mapped to CIS clusters, the majority of insertion sites were represented singly. As it is unlikely that our insertional mutagenesis has reached saturation, many such single insertion sites may, in fact, occur at cancer gene loci that would be revealed as CISs within a larger set of retroviral insertion site data. Therefore, to further evaluate the RISs identified in Eµ-c-myc mice with a subphysiological level of MDM2 as potential cancer gene loci, we integrally analyzed our insertion data with that of the RTCGD, which contains approximately 5,750 RISs (accessed August 2006) from more than 20 different insertional mutagenesis screens.29 As the probability of falsely labeling a genomic locus as a CIS increases with a large number of RISs, we statistically modeled the frequency of random insertion events to determine the statistical significance of multiple retroviral insertions (2 to 10+ hits) within a defined window size (1, 5, and 10 kb; Table 2). Ignoring possible effects of chromatin structure or sequence context, proviral insertions into the genome were assumed to follow a Poisson distribution. From these calculations, we chose to apply a 5-kb window with which to identify novel CISs that have a high probability of being a true nonrandom CIS. Using this criteria, 23 new CISs were formed by a single insertion identified in the present Myc/MDM2 screen and a single insertion identified in another tumor model (Table 3). Based on the tight clustering of insertion sites, genes located at these loci (e.g., p42pop, mtss1, emid1; Fig. 3A) represent strong candidates for novel cancer genes. In addition, 13 single RISs identified in tumors from Eµ-c-myc mice with subphysiological levels of MDM2 contributed to rare CISs (Table 4). In almost all cases, these CISs were formed by multiple insertion sites identified in a small number of tumors from different genetic backgrounds. For example, CISs mapping to the genes tcfap4 and vars1 are each composed of 4 RISs identified in 4 distinct genetic backgrounds. We hypothesize that such infrequently identified CISs are loci that harbor weak oncogenes or possible cancer-modifying genes. Ultimately, however, the role of any putative cancer gene in tumorigenesis must be validated experimentally.
Table 2.
Statistical Significance of CISs Based on Simulation of Random Proviral Insertions
| Predicted no. of random CISsa | P valueb | |||||
|---|---|---|---|---|---|---|
| CIS | 10 kb | 5 kb | 1 kb | 10 kb | 5 kb | 1 kb |
| 2-hits | 802 | 712 | 142 | NS | 0.0483 | 0.0351 |
| 3-hits | 202 | 174 | 36 | NS | 0.0479 | 0.0348 |
| 4 hits | 38 | 31 | 6 | 0.0489 | 0.0462 | 0.0322 |
| 5-hits | 13 | 4 | 1 | 0.0475 | 0.0458 | 0.0317 |
| 6-hits | 6 | 2 | 1 | 0.0469 | 0.0442 | 0.0298 |
| 7-hits | 2 | 1 | 0 | 0.0447 | 0.0435 | 0.0294 |
| 8-hits | 0 | 0 | 0 | 0.0438 | 0.0423 | 0.0276 |
| 9-hits | 0 | 0 | 0 | 0.0422 | 0.0418 | 0.0253 |
| 10+ | 0 | 0 | 0 | 0.0416 | 0.0411 | 0.0215 |
The number of common insertion sites (CISs) occurring by chance within a data set of 5,750 retroviral insertions was simulated using the nonstationary Poisson process using the indicated windows (kb).
Probability that a CIS occurs nonrandomly. NS, not significant.
Table 3.
Novel CISs Generated from an Integral Analysis of the Myc/MDM2 Screen and the Retroviral Tagged Cancer Gene Database
| CIS no. | Chromosome positiona (Chr [Mb]) | Candidate gene | Putative functionb | Genetic backgroundc | Distance (bp)d |
|---|---|---|---|---|---|
| 2.1 | 8 [126] | Tubb3 | Polymerization of microtubules; cell motility | Cdkn2a-/- | 2 |
| 2.2 | 3 [107] | Kcna3 | Potassium ion transport | BXH2 | 17 |
| 2.3 | 11[116] | Foxj1 | Transcription factor; cell polarity; patterning | Cdkn2a-/- | 29 |
| 2.4 | 4 [132] | BC013712 | Hypothetical protein | p27Kip-/- | 31 |
| 2.5 | 3 [88] | 2810403A07Rik | Hypothetical protein | BXH2 | 34 |
| 2.6 | 2 [31] | BC034076 | ECM protein; cell migration and basement invasions | BXH2 | 78 |
| 2.7 | 4 [133] | CD52 | Extracellular membrane protein, GPI anchor binding activity | NIH/Swiss | 83 |
| 2.8 | 7 [18] | P42pop | Regulation of Pol II transcription | BXH2 | 128 |
| 2.9 | 5 [115] | Pxn | Focal adhesion; lamellipodium biogenesis and cell motility | Blm(ko/ko) | 137 |
| 2.10 | 7 [126] | Sh2b1 | Cell motility and lamellipodium biogenesis; signaling | PDGFB_MMLV | 239 |
| 2.11 | 18 [36] | Psd2 | Unknown function; pleckstrin and Sec7 domain containing | BXH2 | 336 |
| 2.12 | 8[87] | CalR | Cortical actin cytoskeleton organization and biogenesis | AKxD | 348 |
| 2.13 | 5[72] | Cnga1 | Potassium ion transport; cGMP binding | Eµ-c-Myc; Pim1-/-; Pim2-/- | 651 |
| 2.14 | 11 [5] | Emid1 | Phosphate transport | Blm(ko/ko) | 955 |
| 2.15 | 17 [27] | Def6 | Pleckstrin-homology domain containing; calcium binding | Blm(ko/ko) | 1,182 |
| 2.16 | 12 [102] | Cpsf2 | Cleavage and polyadenylation specific factor | Blm(ko/ko) | 1,864 |
| 2.17 | 2 [28] | gfi1B | Transcription factor | Eµ-c-Myc; Pim1-/-; Pim2-/- | 2,178 |
| 2.18 | 15 [38] | Azin1 | Activator of ornithine decarboxylase activity | BXH2 | 2,204 |
| 2.19 | 8 [123] | Gse1 | Function unknown | PDGFB_MMLV | 3,317 |
| 2.20 | 9 [89] | Tmed3 | Transmembrane protein; intracellular transport | NIH/Swiss | 3,526 |
| 2.21 | 15 [58] | Mtss1 | Organization and polymerization of actin filaments | AKxD | 3,254 |
| 2.22 | 19 [45] | Fbxw4 | Wnt signaling; ubiquitination | p27Kip-/- | 4,170 |
| 2.23 | 6[8] | Ica1 | Neurotransmitter transport | Blm(ko/ko) | 4,310 |
Chromosomal location of common insertion site (CIS) based on UCSC genome mm8 browser. The chromosome number is followed by the position in megabases [Mb].
Predicted functional information obtained from the Mouse Genome Informatics (MGI) Database (www.informatics.jax.org/).
A single retroviral insertion site (RIS) was identified in an Eµ-c-myc mouse with a subphysiological level of mdm2 and a mouse of the indicated genetic background.
Distance between the 2 single retroviral insertions.
Figure 3.

Schematic illustration of selected provirally tagged genes. Arrows denote position and transcriptional orientation of the provirus. Double arrows represent proviral insertions identified by the Myc/MDM2 screen (present study). Solid arrows represent proviral insertions identified by screens performed in other genetic backgrounds in which mdm2 is wild type. (A) Subset of novel common insertions sites generated from overlap of Myc/MDM2 screen and Retroviral Tagged Cancer Gene Database. (B) Tight clustering of proviral insertions at the 5′ of the hdac6 transcriptional start site. One retroviral insertion site (RIS; double arrow), 167 bp 5′ of the hdac6 exon 1, was identified in a tumor from an Eµ-c-myc; mdm2+/Δ7-12 mouse. Four additional RISs (solid arrows) at this locus have also been identified in 3 different genetic backgrounds: Blm+/+ (2 insertions), CD2-Myc/Runx2 transgenic (1 insertion), and AKxD (1 insertion).
Table 4.
Rare CISs Identified through Integral Analysis of the Myc/MDM2 Screen and the Retroviral Tagged Cancer Gene Database
| CIS No. | Chromosome positiona (Chr [Mb]) | Candidate gene | Putative functionb | RISc | Genetic back- grounds | Distanced (bp) |
|---|---|---|---|---|---|---|
| 3.1 | X[7] | Hdac6 | Histone deacetylase activity; cell migration | 4 | 3 | 270 |
| 3.2 | 19 [4] | Frat1 | Regulation of glycogen synthase kinase-3 | 3 | 3 | 1,431 |
| 3.3 | 7[140] | Athl1 | Carbohydrate metabolism; transferase activity | 3 | 2 | 1,526 |
| 3.4 | 16 [4] | Tcfap4 | Transcription factor | 4 | 4 | 1,778 |
| 3.5 | 4 [116] | 9530048O09Rik | Hypothetical protein | 3 | 3 | 2,542 |
| 3.6 | 4 [32] | Bach2 | B cell–specific transcription factor; heterodimerizes with MafK | 3 | 3 | 2,569 |
| 3.7 | 17 [34] | Vars2 | tRNA aminoacylation for protein translation | 4 | 4 | 2,659 |
| 3.8 | 7[129] | Brwd2 | Bromodomain and WD repeat domain containing; function unknown | 3 | 2 | 3,713 |
| 3.9 | 5 [143] | ActB | Actin cytoskeleton; cell motility | 6 | 5 | 4,033 |
| 3.10 | 5 [123] | Rhof | GTPase; actin filament organization | 5 | 4 | 5,197 |
| 3.11 | 10 [79] | TcfE2a | Transcription factor, B cell maturation | 11 | 8 | 5,214 |
| 3.12 | 2 [90] | Sfpi1 | Transcription factor; lymphocyte differentiation | 7 | 5 | 5,501 |
| 3.13 | 5 [140] | MafK | Transcription factor; heterodimerizes with Bach2 | 5 | 3 | 8,177 |
Chromosomal location of common insertion site (CIS) based on UCSC genome mm8 browser. The chromosome number is followed by the position in megabases [Mb].
Predicted functional information obtained from the Mouse Genome Informatics (MGI) Database (www.informatics.jax.org/).
Total number of retroviral insertion sites (RISs) identified at the indicated locus.
Distance within the indicated number of RISs are located.
Ppp1r16b and hdac6 are deregulated by M-MuLV insertion
To identify candidate genes at individual CISs that are deregulated by M-MuLV insertion, we used quantitative RT-PCR to measure the mRNA expression levels of genes adjacent to sites of proviral insertion. We focused our analysis on 2 CISs.
CIS 1.13 (Table 1) is composed of RIS sequence tags obtained from tumors #82 and 101 (both Eµ-c-myc; mdm2puro/Δ7-12). In both tumors, the RIS was mapped to intron 3 of the gene ppp1r16b. Based on sequence homology, Ppp1r16b is predicted to be a negative regulator of protein phosphatase-1 (PP1), an abundant cellular serine/threonine phosphatase with pleiotropic roles in diverse cellular processes including metabolism, transcription, and apoptosis (reviewed in Ceulemans and Bollen44). The cellular function of Ppp1r16b is largely unknown. However, ppp1r16b was identified in a large-scale screen of an RNAi screen of kinases and phosphatases, as a gene whose inhibition resulted in cell death.43 Based on the finding that ppp1r16b can function to promote cell survival, we chose to investigate CIS 1.13 in more detail.
We performed quantitative RT-PCR analysis of ppp1r16b mRNA in a panel of tumors to determine if retroviral insertion at CIS 1.13 deregulated gene expression. Ppp1r16b expression was found to be modestly elevated in tumors #82 and #101 with the relevant viral insertions compared to the majority of tumors analyzed, thereby suggesting that M-MuLV integration had altered ppp1r16b gene expression (Fig. 4). Moreover, ppp1r16b expression was also elevated in several tumors in addition to #82 and #101, suggesting that ppp1r16b expression may be induced through other collaborative pathways during lymphomagenesis. Additional analyses failed to identify another gene within 100 kb of ppp1r16b whose expression was altered in tumors with the relevant viral insertions. As proviral insertion may enhance expression of genes at distances greater than 100 kb, it remains formally possibly that a gene outside the examined region is the primary target of M-MuLV insertion. Although the change in Ppp1r16b is quite modest, the tight clustering of M-MuLV insertions specifically in a background sensitized to apoptosis in conjunction with the identification of Ppp1r16b as a prosurvival factor43 together suggest that ppp1r16b may have oncogenic properties that merit further investigation.
Figure 4.
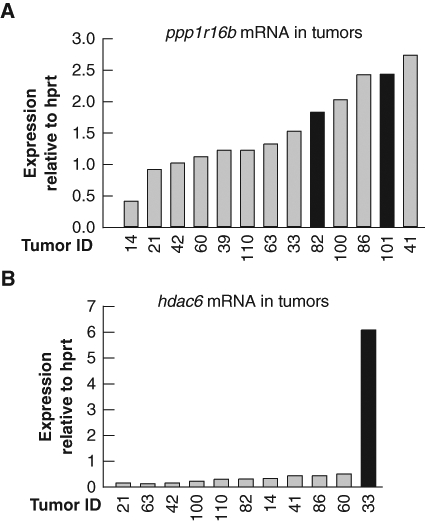
Ppp1r16b and hdac6 are candidate cancer genes. Expression of (A) ppp1r16b and (B) hdac6 mRNA in a panel of M-MuLV/c-myc–induced lymphomas. Quantitative RT-PCR analysis was performed using primers specific to either ppp1r16b or hdac6 and normalized to expression of hprt. Bars in black represent tumors with known retroviral insertions at the indicated locus. For each panel of tumors, gene expression analyses were confirmed using a second set of primers to the indicated gene.
A second locus investigated in more detail is CIS 3.1 (Table 4) located on the X chromosome. We identified a single retroviral insertion 5′ of histone deacetylase 6 (hdac6) in tumor #33 (Eµ-c-myc; mdm2+/Δ7-12). Searching the RTCGD, we noted that 4 RISs at the hdac6 locus had been previously identified in 3 separate retroviral screens performed on mice with diverse genetic backgrounds. All 4 RISs mapped within 300 bp of the transcriptional start site of hdac6 (Table 4, Fig. 3B). Such tight clustering of RIS is highly suggestive of an oncogenic locus. Although we were unable to assess the effect of retroviral insertion at the hdac6 locus in tumors from other screens, quantitative RT-PCR analysis revealed that the level of hdac6 mRNA was indeed significantly increased in the tumor #33 from the Myc/MDM2 screen with the relevant RIS (Fig. 4B). Pim2, a known oncogene,45 is also located on chromosome X, approximately 70 kb from hdac6. Although enhancer elements within the M-MuLV long terminal repeat (LTR) region can stimulate gene expression over large distances, pim2 expression was not increased in tumor #33 (data not shown). Thus, the predominant effect of M-MuLV insertion at CIS 3.1 is the deregulation of hdac6 expression.
Ppp1r16b and hdac6 accelerate c-Myc–induced lymphomagenesis
Candidate cancer genes ppp1r16b and hdac6 were first tested for their ability to transform NIH3T3 cells in vitro. We found that neither ppp1r16b nor hdac6 overexpression was capable of promoting focus formation in NIH3T3s (data not shown). Next, we further tested the oncogenic potential of ppp1r16b and hdac6 using a bone marrow transplantation assay developed by Schmitt et al. (2002).10 To specifically determine whether overexpression of these candidate oncogenes could cooperate with c-Myc in vivo, hematopoietic stem cells (HSCs) derived from Eµ-c-myc mice were infected with a murine stem cell virus (MSCV) engineered to express either ppp1r16b or hdac6 and transplanted into lethally irradiated nontransgenic recipients. As controls, recipient mice were transplanted with Eµ-c-myc–derived HSCs that had been infected with either empty MSCV or a retrovirus that expresses the antiapoptotic protein bcl-2. Lethally irradiated mice surviving 14 days posttransplantation were considered successfully transplanted and were subsequently monitored for tumor formation.
Twenty-five percent of the recipient mice reconstituted with Eµ-c-myc HSCs infected with empty MSCV developed tumors within 200 days following transplantation (4 of 14; Fig. 5A), in agreement with that observed in prior studies.10 The introduction of a cooperating oncogene into Eµ-c-myc HSCs is predicted to promote lymphomagenesis in recipient mice following bone marrow transplantation. Indeed, the introduction of bcl-2, which encodes a known inhibitor of c-Myc–induced apoptosis, into Eµ-c-myc HSCs potently promoted lymphomagenesis in recipient mice following bone marrow transplantation (6 of 6, average survival 42 ± 3 days; P < 0.005 relative to MSCV controls). Likewise, 100% of mice transplanted with ppp1r16b-expressing Eµ-c-myc HSCs developed lymphomas with a significantly decreased latency (6 of 6, average survival 108 ± 24 days; P = 0.003 relative to MSCV controls). The majority of mice that received hdac6-expressing Eµ-c-myc HSCs also succumbed more quickly to tumorigenesis (9 of 11, average survival 132 ± 28 days; P = 0.01) than MSCV control mice. Histological analyses revealed that lymphomas arising in mice transplanted with ppp1r16b- or hdac6-expressing Eµ-c-myc HSCs frequently involved the peripheral lymph nodes with dissemination into organs such as liver and lung (Fig. 5B), consistent with a more aggressive tumor type. These results demonstrate that ppp1r16b and hdac6 are bona fide cellular oncogenes.
Figure 5.
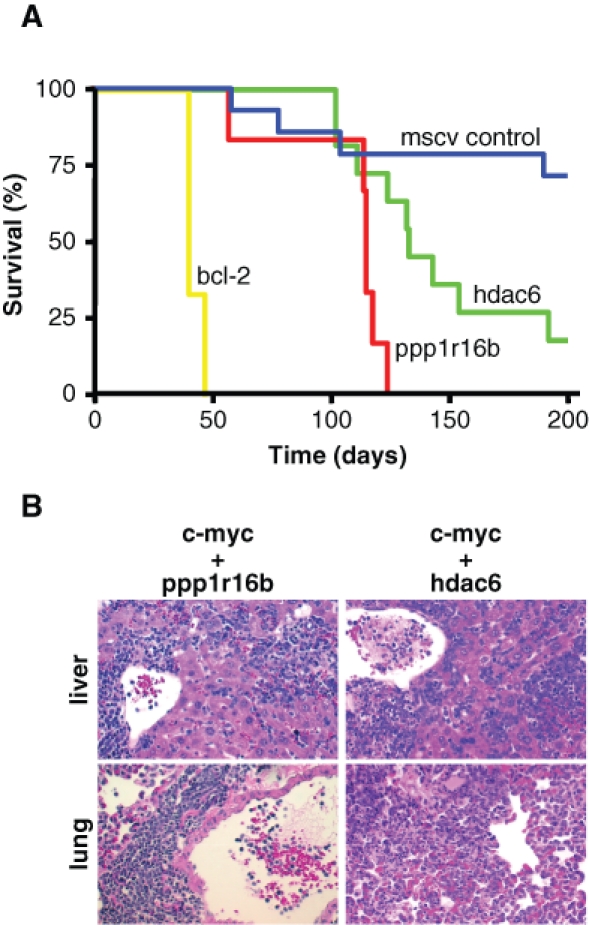
Ppp1r16b and hdac6 promote aggressive lymphomas. (A) Mice reconstituted with Eµ-c-myc hematopoietic stem cells (HSCs) infected with the indicated murine stem cell virus retroviruses were monitored for tumor onset and sacrificed when moribund. Presented is a Kaplan-Meier curve showing the percentage of mice surviving at various days following bone marrow reconstitution. (B) Hematoxylin/eosin–stained sections illustrating infiltration into the peripheral organs of mice transplanted with Eµ-c-myc HSCs infected with either ppp1r16b or hdac6 indicative of disseminated disease (20x).
Discussion
In this study, we have used retroviral insertional mutagenesis with M-MuLV to identify novel cancer genes involved in the pathogenesis of B cell lymphoma. To promote the identification of novel cancer genes, we took advantage of multiple mdm2 alleles that allow the level of MDM2, a direct negative regulator of the p53 tumor suppressor protein, to be reduced in vivo. A subphysiological level of MDM2 increased the sensitivity of B cells to oncogene-driven apoptosis and delayed M-MuLV/c-myc–induced lymphomagenesis. As inhibition of c-Myc–induced apoptosis is predicted to be rate limiting for tumor formation in mice with a subphysiological level of MDM2, we predicted that M-MuLV CISs uniquely identified in this genetic background would be enriched for proto-oncogenes encoding negative regulators of apoptosis. Supporting this idea, more than 50% of the well-characterized CISs we identified contain cancer genes that inhibit apoptosis when overexpressed. Moreover, at least 2 CISs uniquely identified in the Myc/MDM2 screen also harbor genes (gadd45β and ppp1r16b) that have been previously implicated in the regulation of cell death. However, as discussed below, a surprising number of the genes identified near CIS in our study have also been implicated in cell motility, suggesting a more complex mechanism for cooperativity.
It has been estimated that 1% of the genes encoded by the human genome may be involved in tumor formation.46 Therefore, to expand on our initial analysis of CISs identified in the Myc/MDM2 screen, we took advantage of the RTCGD as a tool for cancer gene discovery. By integrally analyzing RISs from the Myc/MDM2 screen with those sites previously deposited in the RTCGD, we identified 23 additional novel CISs. In addition, we identified 13 single RISs from the Myc/MDM2 screen that contributed to CISs composed of 5 or fewer RISs identified in diverse genetic backgrounds, thereby strengthening the significance of these poorly represented CISs as potential cancer gene loci.
A bottleneck to cancer gene discovery through insertional mutagenesis has been the confirmation of the oncogenic (or, less frequently, tumor-suppressive) activity of candidate cancer genes identified at CISs. To date, more than 500 CISs have been identified through various insertional mutagenesis screens. However, for only a small number of CISs has the candidate cancer gene been demonstrated to play a causal role in tumorigenesis. Here, we have used an in vivo bone marrow transplant assay to test putative cancer genes for their oncogenic potential. Using this assay, we have validated that the genes ppp1r16b and hdac6 are bona fide oncogenes, capable of promoting lymphomagenesis in cooperation with c-Myc. To the best of our knowledge, hadc6 is the first example of a gene identified solely through an integrative analysis of the RTCGD that has been confirmed to be a bona fide oncogene.
Although both Ppp1r16b and Hdac6 accelerated lymphomagenesis in cooperation with c-Myc, neither did so with the potency of the antiapoptotic protein Bcl-2. This finding suggests that these proto-oncogenes may exert their oncogenic effect(s) at a late stage of lymphomagenesis or by modifying the cellular response to c-myc overexpression. Furthermore, we noted that mice transplanted with hdac6-expressing Eµ-c-myc HSCs died more stochastically (between 102 and 192 days posttransplantation) than observed with either ppp1r16b or bcl-2, suggesting that genetic events in addition to hdac6 and c-myc overexpression may be required for lymphomagenesis. Our future studies are aimed at elucidating the function of Hdac6 and Ppp1r16b in lymphomagenesis, and we are currently developing transgenic mice as a more robust model for these mechanistic studies.
The mechanism(s) by which newly identified oncogenes promote tumorigenesis remains to be elucidated but may be inferred by our current understanding of their biological function. Ppp1r16b, originally identified as a gene whose expression is inhibited by TGF-β signaling, is preferentially expressed in endothelial and hematopoietic cells.47 Ppp1r16b binds to the cellular phosphatase PP1 and based on homology with known PP1 interacting proteins is predicted to negatively regulate PP1 activity. SiRNA-mediated inhibition of ppp1r16b in Hela cells resulted in a 4-fold decrease in viability, suggesting a role for Ppp1r16b in cell survival.43 A role of Ppp1r16b in human cancer is further suggested by the demonstration that Ppp1r16b can bind to and promote the dephosphorylation of the nonintegrin laminin receptor LAMR1 (also known as 67LR).48 LAMR1 has been clearly implicated in tumor progression, and its expression is increased in a wide variety of cancers, including high-grade non-Hodgkin’s lymphoma.49 Increased release of soluble LAMR1 into the extracellular matrix parallels increased expression levels and has been shown to elicit conformational changes in laminin. The interaction of tumor cells with modified laminin, in turn, leads to intracellular changes in the dynamic state of actin-containing motility structures that promote cell attachment, migration, and invasion.50,51 Thus, by regulating the association of LAMR1 with laminin, Ppp1r16b may promote cell motility through the intracellular reorganization of actin.
HDAC6 is a Class II histone deacetylase. Despite this designation, HDAC6 has never been demonstrated to modify histones in vivo. Rather, perhaps the best-characterized target of deacetylation by HDAC6 is α-tubulin consistent with the cytoplasmic localization of this deacetylase.52 Acetylation/deacetylation of α-tubulin is predicted to play an important regulatory role in the control of cell motility. Indeed, studies by Hubbert et al. (2002)52 have demonstrated that overexpression of HDAC6 results in decreased acetylation of α-tubulin and increased cell migration of NIH3T3 fibroblasts. Though the deacetylase activity of HDAC6 was required for increased cell motility in NIH3T3s, HDAC6 promoted the migration of T cells independent of the deacetylase catalytic domain.53 Thus, the requirement for HDAC6 deacetylase activity for increased cell motility may be cell type dependent. Another role for HDAC6 is in the ubiquitin proteasome system. HDAC6 binds polyubiquitinated proteins through an ubiquitin-binding–Zinc finger domain54 and facilitates the dynein-microtubule–dependent transport of misfolded cellular proteins to the aggresome.55 Loss of HDAC6 has been shown to induce apoptosis due to the accumulation of cytotoxic misfolded proteins.55 Moreover, treatment of Eµ-c-myc lymphomas with vorinostat (suberoylanilide hydroamic acid), an inhibitor of class I and II HDACs, elicited a p53-independent apoptotic response through the regulation of Bim, a component of the intrinsic apoptotic pathway.56 Although HDAC inhibitors show promise for the treatment of human malignancies, the role of individual HDACs in cancer remain unclear. HDAC6, for example, is found at elevated levels in diffuse large B-cell lymphoma (DLBCL) and peripheral T-cell lymphoma (PTCL); however, HDAC6 expression was found to correlate with a favorable prognosis for DLBCL but not for PTCL.57 Further investigation into the function of HDAC6 in cell migration and other biological processes is predicted to yield important insight into the role of HDAC6 in human hematological malignancies.
As cell migration is a prerequisite for tumor metastasis, mutations that foster cell motility are likely to play an important role in the dissemination and invasion of primary tumor cells into secondary sites. It is therefore interesting to note that in addition to ppp1r16 and hdac6, more than 25% of the putative cancer genes (e.g., Tubb3, BC034076, Pxn, Sh2b1, CalR, Mtss1) located at novel CIS generated from our integrative analysis of the Myc/MDM2 screen and the RTCGD encode for proteins implicated in cell motility, or more broadly, in the reorganization of the cytoskeleton. Overexpression of these putative cancer genes may promote the migration of tumor cells with oncogenic c-myc, thereby eliciting a more aggressive tumor phenotype. A role for regulators of cell motility in lymphomagenesis is further supported by a recent loss-of-function screen in which genes involved in cytoskeletal reorganization and cell adhesion were highly enriched in the pool shRNA targets whose knock-down limits the growth of Eµ-c-myc tumor cells.58
A critical step toward improved cancer therapy is the identification of genes deregulated in cancer that can be used as targets for new cancer therapeutics. The power of retroviral insertional mutagenesis to identify new cancer genes has significantly grown in the past few years due to the convergence of new molecular biology tools and publicly accessible databases. Advances in PCR-based methodologies combined with the availability of the complete mouse genome sequence have accelerated the identification of putative cancer genes from years to months. Our results illustrate that retroviral insertional mutagenesis screens have yet to reach saturation and continue to be a useful approach for cancer gene discovery. Moreover, we have illustrated the power of the RTCGD as a tool to identify novel cancer gene loci. This is a virtually untapped resource that may hold the key to identifying rare cancer genes, modifiers of cancer susceptibility, or genetic interactions between cancer genes.
Materials and Methods
Mice and M-MuLV infection
Eµ-c-myc mice (C57BL/6) were first bred to mice heterozygous for the null mdm2 allele (mdm2Δ7-12).21 Eµ-c-myc; mdm2Δ7-12 progeny were subsequently bred to Sv129 mice heterozygous for the hypomorphic mdm2 allele (mdm2puro) to generate C57BL6 x Sv129 F1 progeny that were transgenic for Eµ-c-myc and that expressed 1 of 4 levels of Mdm2 (mdm2+/+>mdm2+/puro>mdm2+/Δ7-12>mdm2puro/Δ7-12). Within 24 hours of birth, all progeny were injected intraperitoneally with M-MuLV (1 × 105 pfu; Dusty Miller, Fred Hutchinson Cancer Research Center [FHCRC]). Mice were genotyped for the Eµ-c-myc transgene using primers dmr375 5′CAG CTG GCG TAA TAG CGA AGA G and dmr376 5′CTG TGA CTG GTG AGT ACT CAA CC and mdm2 as described previously.22 At weaning, mice lacking the Eµ-c-myc transgene were euthanized. Eµ-c-myc transgenic mice infected with M-MuLV were followed for the development of tumors and euthanized when moribund. All animal experiments were done at the FHCRC with IACUC approval.
Identification of genomic sites of M-MuLV integration
Genomic DNA was purified from lymphomas isolated from M-MuLV–infected Eµ-c-myc mice with less than wild-type levels of MDM2. The majority of genomic sites of M-MuLV provirus integration were identified using a modified anchoring PCR protocol developed by Chen and Soriano (2003)59 using the primers PDA-L, PDA-S, AB827, AB949, and DAP.60 In brief, tumor DNA (2 µg) was digested with the blunt-cutter restriction enzyme HpaI for 4 to 6 hours, followed by purification of digested DNA (QIAEXII; Qiagen, Valencia, CA). A pseudo-double-stranded adapter (PDA) was annealed to blunt-ended genomic DNA by incubating 200 ng HpaI-digested tumor DNA overnight with 1 µL 10 µM PDA-L primer, 1 µL 10 µM PDA-S primer, 2 µL 10X ligation buffer (Roche, Indianapolis, IN), 1 µL T4 DNA ligase (Roche, Indianapolis, IN) in 20 µL final volume. Following overnight ligation, samples were incubated at 65°C for 10 minutes to inactive T4 DNA ligase and diluted 10-fold with ddH2O. Two rounds of nested PCR (PCR-I and PCR-II) were performed. PCR-I was performed using primer PDA-L and the primer AB827 (reference) that anneals specifically to the M-MuLV LTR. PCR-I reaction conditions are as follows: 1 µL diluted PDA-ligated tumor genomic DNA, 0.5 µL 10 µm PDA-L, 0.5 µL 10 µm AB827, 0.5 µL 10 mM dNTPs, 0.26 µL DMSO, 2.0 µL 10X PC2 buffer ([50 mM Tris-HCl pH 9.1, 16 mM ammonium sulfate, 3.5 mM MgCl2 and 150 µg/mL BSA], 5.2 µL 5M Betaine, 1.0 µL Rediload [Research Genetics, Huntsville, AL], 0.04 µL LA16 enzyme mix [KlenTaq (Sigma, St. Louis, MO): Pfu (Roche, Indianapolis, IN) = 15:1, v/v], 9 µL ddH20). MoMuLV-flanking sequences were amplified in a GeneAmp 9700 (Applied Biosystems, Foster City, CA). PCR conditions were 5 minutes for 95°C, 30 seconds for 95°C, 45 seconds for 58°C, 2:30 minutes for 72°C (35 cycles), and 10 minutes for 72°C. For nested PCR-II, 1 µL of amplified product from PCR-I that had been diluted 1:100 in ddH20 was used as input template in conjunction with the primer PDA-L and the M-MuLV–specific primer AB949.60 PCR-II reaction conditions were identical to PCR-I. Amplified products were separated on a 2.0% agarose gel, excised from the gel, and heated for 10 minutes at 80°C in100 µL ddH20. Genomic sequence adjacent to M-MoLV insertion was reamplified in PCR-III using amplified product from PCR-II that had been diluted 1:100 in ddH20 as input template. PCR-I reaction conditions are as follows: 1.5 µL diluted PCR-II product, 2.5 µL 10X PCR buffer (Invitrogen, Carlsbad, CA), 0.25 µL 10 µm DAP, 0.25 µL 10 µm AB949, 0.5 µL 10 mM dNTPs, 1.0 µL 50 mM MgCl2, 0.2 µL Taq (Invitrogen), in 25 µL final volume. PCR-III conditions were 5 minutes for 95°C, 30 seconds for 95°C, 45 seconds for 58°C, 60 seconds for 72° (25 cycles), and 10 minutes for 72°C. Amplified products from PCR-III were treated with 2 U exonuclease and 0.1 U shrimp alkaline phosphatase (SAP; NEB, Ipswich, MA). 1.5 µL of exonuclease/SAP-treated PCR was directly sequenced using BigDye terminator mix (Applied Biosystems) with primer DAP.60
A small subset of M-MuLV integration sites were identified using Inverse PCR (I-PCR) as previously described.16 I-PCR products were subcloned into pCR2.1-Topo (Invitrogen) and sequenced using an M13F primer.
Statistical analysis of RISs
We used statistical techniques such as K-means clustering and Partition around Medioids (PAM) to investigate associations between mouse tumor types and their addresses on different mouse chromosomes. The PAM method is preferable to the K-means clustering. This is because PAM is less sensitive to outliers (data values that in our large data set [n = 5,750] deviate by more than 3 standard deviations above or below the mean). However, some of the 2-hits, 10 or more hits do not truly belong to one cluster or cannot be forced into an adjoining cluster. To address this issue, we used a discrete probability distribution, the Poisson process, which is more suitable for this biological study. We assumed that an experimenter will observe a CIS for a period of time and the time at which she or he starts to observe a CIS is at the origin (zero). We assumed that she or he observes CISs for a fixed time period (t), where t is a positive number. The number of events that occur in this fixed interval (0, t) is a discrete random variable X. Therefore, in this experiment, X takes discrete values of 2-hits, 3-hits, 10 or more hits. Now if X is the number of events in the interval of fixed length t, then the probability function for X is
The number of hits (X) is called a Poisson random variable with parameter λ.
For the simulation runs performed on 1 Kb, 5 Kb, and 10 Kb window sizes, an integral method was used in which the distances between 2-hits, 3-hits, 10-hits or more were minimized. To generate random variables from the Poisson probability distribution, we assumed that the time intervals between events (CISs) are from exp (1/λ), which is the number of CISs occurring in a unit time period that is from the Poisson process P (λ), where λ is the average number of CISs in the time period. We generated the random numbers in 1,000 runs by using a uniform probability distribution where U (k) is generated from u (0, 1). The λ is the average number of 2-hit, 3-hit, 10 or more hit insertions in this time period. The u (0, 1) was generated by applying a uniform probability distribution. We have used the continuous uniform random variable on the interval (0, 1) to conduct our simulation for different widow sizes.61,62
The P values are calculated by dividing the number of false positives for each and every insertion site by the total number 2-hit, 3-hit, 10 or more hits. The P values associated with 2-hits to 10-hits or more within a 5-Kb window are all statistically significant when the type I error rate (λ) is 0.05.63
Flow cytometry
Lineage marker analysis of preneoplastic and tumor hematopoietic cells was performed by flow cytometry essentially as described previously.22 Briefly, bone marrow cells were flushed from the femur using phosphate-buffered saline (PBS) supplemented with 3% fetal bovine serum (PF3). Single-cell suspensions from normal and tumor tissues were prepared by pressing tissues between frosted glass slides and dispersing cells in PF3. Red blood cells were lysed in Gey’s solution prior to staining.22 Hematopoietic lineages were identified by staining aliquots of cells with the following antibodies: fluorescein isothiocyanate-conjugated anti-CD4 (clone GK1.5), phycoerythrin- conjugated anti-CD8 (clone 53-6.7), fluorescein isothiocyanate-conjugated anti-CD45R/B220 (clone RA-6B2), phycoerythrin-conjugated anti-immunoglobulin M (clone R6-60.2), and phycoerythrin-conjugated anti-CD43 (clone S7; BD Biosciences, San Jose, CA). Following a 20-minute incubation on ice, cells were washed twice and resuspended in PF3.
For cell cycle analysis, 1 × 106 cells stained for anti-CD45R/B220 as described above were washed in cold PBS and fixed at 4°C in ice-cold 70% ethanol for 1 hour or longer. Following fixation, cells were washed twice in cold PBS, resuspended in PBS containing 0.5 mg/mL RNase A and 50 µg/mL propidium iodide, and incubated at 4°C for 30 minutes in the dark. Cells were counted on an FACSCalibur flow cytometer and data analyzed with CellQuest software (BD Biosciences).
Quantitative RT-PCR
Total RNA from tumor tissue was isolated with Trizol (Invitrogen) as recommended by the manufacturer. Total RNA (1 µg) was reverse transcribed (RT) using Thermoscript RT (Invitrogen) and a random hexamer primer. An aliquot (1/40th) of RT reaction was used for quantitative PCR. Gene-specific primers were designed with the aid of the program Primer3 (http://frodo.wi.mit.edu/cgi-bin/primer3/primer3_www.cgi). In general, primers used for quantitative PCR analysis were annealed at a Tm of 60°C and generated amplified products that were <350 bps and spanned one or more exon junctions. PCR was performed with [32P] dCTP. PCR conditions were 5 minutes for 95°C, followed by cycling at 30 seconds for 95°C, 30 seconds for 60°C, and 1:00 minute for 72°C. The number of cycles used for each gene-specific primer pair was determined empirically by first performing PCR with a control cDNA and evaluating amplification at multiple cycles to identify the linear range of amplification. To control for RT input in the quantitative PCR reaction, PCR was performed in parallel using primers specific for the housekeeping gene HPRT. [32P]dCTP-labeled PCR products were run on a 10% tris-borate-EDTA acrylamide gel and quantified using a Molecular Dynamics PhosphorImager. Primer sequences are available upon request.
Constructs
Full-length ppp1r16b cDNA was PCR amplified from mouse spleen cDNA and cloned into the XhoI site of the retroviral construct MSCV-ires-GFP. The entire ppp1r16b gene was sequenced and determined to be identical to RefSeq NM_153089. Bcl-2 and hdac6 cDNAs were generously provided by David Hockenberry (FHCRC) and Stuart Schreibner (Harvard University), respectively, and subcloned into MSCV-ires-GFP.
Adoptive bone marrow transfer and tumor analysis
Adoptive transfer experiments were performed as described previously.10 Briefly, fetal liver cells (FLCs) obtained from E13.5 Eµ-c-myc transgenic embryos were infected with MSCV-based retroviruses. Following infection, FLCs were washed with PBS and approximately 2 × 106 injected via tail vein into 6- to 8-week-old C57BL6 mice 6 to 12 hours post–lethal irradiation (10 Gy total body dose). Mice were maintained under pathogen-free conditions. Mice surviving 14 days posttransplantation were considered reconstituted and were monitored for tumor formation. Mice were sacrificed when moribund for necropsy. Mice surviving more than 200 days posttransplantation were considered tumor-free and euthanized.
Acknowledgments
We thank Dusty Miller (FHCRC) for titering the M-MuLV virus stock and Bruce Clurman and Harry Hwang (FHCRC) for technical advice and discussion. We also thank Mike Hemann (MIT) for advice and expertise on the bone marrow transplant assays.
Footnotes
The authors declare no conflicts of interest with respect to the publication of this article.
S.M.M. was supported by a postdoctoral fellowship from the American Cancer Society. This work was funded by NIH/NCI grant R37CA57138 to R.N.E.
References
- 1. Nesbit CE, Tersak JM, Prochownik EV. MYC oncogenes and human neoplastic disease. Oncogene 1999;18:3004-16 [DOI] [PubMed] [Google Scholar]
- 2. Jacobsen KA, Prasad VS, Sidman CL, Osmond DG. Apoptosis and macrophage-mediated deletion of precursor B cells in the bone marrow of E mu-myc transgenic mice. Blood 1994;84:2784-94 [PubMed] [Google Scholar]
- 3. Adams JM, Harris AW, Pinkert CA, Corcoran LM, Alexander WS, Cory S, et al. The c-myc oncogene driven by immunoglobulin enhancers induces lymphoid malignancy in transgenic mice. Nature 1985;318:533-8 [DOI] [PubMed] [Google Scholar]
- 4. Kubbutat MH, Jones SN, Vousden KH. Regulation of p53 stability by Mdm2. Nature 1997;387:299-303 [DOI] [PubMed] [Google Scholar]
- 5. Haupt Y, Maya R, Kazaz A, Oren M. Mdm2 promotes the rapid degradation of p53. Nature 1997;387:296-9 [DOI] [PubMed] [Google Scholar]
- 6. Pomerantz J, Schreiber-Agus N, Liegeois NJ, Silverman A, Alland L, Chin L, et al. The Ink4a tumor suppressor gene product, p19Arf, interacts with MDM2 and neutralizes MDM2’s inhibition of p53. Cell 1998;92:713-23 [DOI] [PubMed] [Google Scholar]
- 7. Zindy F, Eischen CM, Randle DH, Kamijo T, Cleveland JL, Sherr CJ, et al. Myc signaling via the ARF tumor suppressor regulates p53-dependent apoptosis and immortalization. Genes Dev 1998;12:2424-33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Schmitt CA, McCurrach ME, de Stanchina E, Wallace-Brodeur RR, Lowe SW. INK4a/ARF mutations accelerate lymphomagenesis and promote chemoresistance by disabling p53. Genes Dev 1999;13:2670-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Eischen CM, Weber JD, Roussel MF, Sherr CJ, Cleveland JL. Disruption of the ARF-Mdm2-p53 tumor suppressor pathway in Myc-induced lymphomagenesis. Genes Dev 1999;13:2658-69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Schmitt CA, Fridman JS, Yang M, Baranov E, Hoffman RM, Lowe SW. Dissecting p53 tumor suppressor functions in vivo. Cancer Cell 2002;1:289-98 [DOI] [PubMed] [Google Scholar]
- 11. Hemann MT, Bric A, Teruya-Feldstein J, Herbst A, Nilsson JA, Cordon-Cardo C, et al. Evasion of the p53 tumour surveillance network by tumour-derived MYC mutants. Nature 2005;436:807-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Jonkers J, Berns A. Retroviral insertional mutagenesis as a strategy to identify cancer genes. Biochim Biophys Acta 1996;1287:29-57 [DOI] [PubMed] [Google Scholar]
- 13. van Lohuizen M, Verbeek S, Scheijen B, Wientjens E, van der Gulden H, Berns A. Identification of cooperating oncogenes in E mu-myc transgenic mice by provirus tagging. Cell 1991;65:737-52 [DOI] [PubMed] [Google Scholar]
- 14. Mikkers H, Allen J, Knipscheer P, et al. High-throughput retroviral tagging to identify components of specific signaling pathways in cancer. Nat Genet 2002;32:153-9 [DOI] [PubMed] [Google Scholar]
- 15. Castilla LH, Perrat P, Martinez NJ, et al. Identification of genes that synergize with Cbfb-MYH11 in the pathogenesis of acute myeloid leukemia. Proc Natl Acad Sci U S A 2004;101:4924-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hwang HC, Martins CP, Bronkhorst Y, Randel E, Berns A, Fero M, et al. Identification of oncogenes collaborating with p27Kip1 loss by insertional mutagenesis and high-throughput insertion site analysis. Proc Natl Acad Sci U S A 2002;99:11293-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Johansson FK, Brodd J, Eklof C, Ferletta M, Hesselager G, Tiger CF, et al. Identification of candidate cancer-causing genes in mouse brain tumors by retroviral tagging. Proc Natl Acad Sci U S A 2004;101:11334-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Uren AG, Kool J, Matentzoglu K, de Ridder J, Mattison J, van Uitert M, et al. Large-scale mutagenesis in p19(ARF)- and p53-deficient mice identifies cancer genes and their collaborative networks. Cell 2008;133:727-41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Honda R, Tanaka H, Yasuda H. Oncoprotein MDM2 is a ubiquitin ligase E3 for tumor suppressor p53. FEBS Lett 1997;420:25-7 [DOI] [PubMed] [Google Scholar]
- 20. Momand J, Zambetti GP, Olson DC, George D, Levine AJ. The mdm-2 oncogene product forms a complex with the p53 protein and inhibits p53-mediated transactivation. Cell 1992;69:1237-45 [DOI] [PubMed] [Google Scholar]
- 21. Jones SN, Roe AE, Donehower LA, Bradley A. Rescue of embryonic lethality in Mdm2-deficient mice by absence of p53. Nature 1995;378:206-8 [DOI] [PubMed] [Google Scholar]
- 22. Mendrysa SM, McElwee MK, Michalowski J, O’Leary KA, Young KM, Perry ME. mdm2 is critical for inhibition of p53 during lymphopoiesis and the response to ionizing irradiation. Mol Cell Biol 2003;23:462-72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Mendrysa SM, O’Leary KA, McElwee MK, Michalowski J, Eisenman RN, Powell DA, et al. Tumor suppression and normal aging in mice with constitutively high p53 activity. Genes Dev 2006;20:16-21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Alt JR, Greiner TC, Cleveland JL, Eischen CM. Mdm2 haplo-insufficiency profoundly inhibits Myc-induced lymphomagenesis. EMBO J 2003;22:1442-50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Iritani BM, Eisenman RN. c-Myc enhances protein synthesis and cell size during B lymphocyte development. Proc Natl Acad Sci U S A 1999;96:13180-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Vaux DL, Adams JM, Alexander WS, Pike BL. Immunologic competence of B cells subjected to constitutive c-myc oncogene expression in immunoglobulin heavy chain enhancer myc transgenic mice. J Immunol 1987;139:3854-60 [PubMed] [Google Scholar]
- 27. Suzuki T, Shen H, Akagi K, Morse HC, Malley JD, Naiman DQ, et al. New genes involved in cancer identified by retroviral tagging. Nat Genet 2002;32:166-74 [DOI] [PubMed] [Google Scholar]
- 28. Suzuki T, Minehata K, Akagi K, Jenkins NA, Copeland NG. Tumor suppressor gene identification using retroviral insertional mutagenesis in Blm-deficient mice. EMBO J 2006;25:3422-31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Akagi K, Suzuki T, Stephens RM, Jenkins NA, Copeland NG. RTCGD: retroviral tagged cancer gene database. Nucleic Acids Res 2004;32:D523-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Eischen CM, Woo D, Roussel MF, Cleveland JL. Apoptosis triggered by Myc-induced suppression of Bcl-X(L) or Bcl-2 is bypassed during lymphomagenesis. Mol Cell Biol 2001;21:5063-70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. George A, Morse HC, III, Justice MJ. The homeobox gene Hex induces T-cell-derived lymphomas when overexpressed in hematopoietic precursor cells. Oncogene 2003;22:6764-73 [DOI] [PubMed] [Google Scholar]
- 32. Hanlon L, Barr NI, Blyth K, Stewart M, Haviernik P, Wolff L, et al. Long-range effects of retroviral insertion on c-myb: overexpression may be obscured by silencing during tumor growth in vitro. J Virol 2003;77:1059-68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Haupt Y, Bath ML, Harris AW, Adams JM. bmi-1 transgene induces lymphomas and collaborates with myc in tumorigenesis. Oncogene 1993;8:3161-4 [PubMed] [Google Scholar]
- 34. Klinger MB, Guilbault B, Goulding RE, Kay RJ. Deregulated expression of RasGRP1 initiates thymic lymphomagenesis independently of T-cell receptors. Oncogene 2005;24:2695-704 [DOI] [PubMed] [Google Scholar]
- 35. Maki K, Yamagata T, Yamazaki I, Oda H, Mitani K. Development of megakaryoblastic leukaemia in Runx1-Evi1 knock-in chimaeric mouse. Leukemia 2006;20:1458-60 [DOI] [PubMed] [Google Scholar]
- 36. Schmidt T, Karsunky H, Gau E, Zevnik B, Elsasser HP, Moroy T. Zinc finger protein GFI-1 has low oncogenic potential but cooperates strongly with pim and myc genes in T-cell lymphomagenesis. Oncogene 1998;17:2661-7 [DOI] [PubMed] [Google Scholar]
- 37. van Lohuizen M, Verbeek S, Krimpenfort P, Domen J, Saris C, Radaszkiewicz T, et al. Predisposition to lymphomagenesis in pim-1 transgenic mice: cooperation with c-myc and N-myc in murine leukemia virus-induced tumors. Cell 1989;56:673-82 [DOI] [PubMed] [Google Scholar]
- 38. Donehower LA, Harvey M, Slagle BL, McArthur MJ, Mongtomery CA, Jr, Butel JS, et al. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature 1992;356:215-21 [DOI] [PubMed] [Google Scholar]
- 39. Chan WM, Siu WY, Lau A, Poon RY. How many mutant p53 molecules are needed to inactivate a tetramer? Mol Cell Biol 2004;24:3536-51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. De Smaele E, Zazzeroni F, Papa S, Nguyen DU, Jin R, Jones J, et al. Induction of gadd45beta by NF-kappaB downregulates pro-apoptotic JNK signalling. Nature 2001;414:308-13 [DOI] [PubMed] [Google Scholar]
- 41. Gupta M, Gupta SK, Balliet AG, Hollander MC, Fornace AJ, Hoffman B, et al. Hematopoietic cells from Gadd45a- and Gadd45b-deficient mice are sensitized to genotoxic-stress-induced apoptosis. Oncogene 2005;24:7170-9 [DOI] [PubMed] [Google Scholar]
- 42. Zhan Q, Lord KA, Alamo I, Hollander MC, Carrier F, Ron D, et al. The gadd and MyD genes define a novel set of mammalian genes encoding acidic proteins that synergistically suppress cell growth. Mol Cell Biol 1994;14:2361-71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. MacKeigan JP, Murphy LO, Blenis J. Sensitized RNAi screen of human kinases and phosphatases identifies new regulators of apoptosis and chemoresistance. Nat Cell Biol 2005;7:591-600 [DOI] [PubMed] [Google Scholar]
- 44. Ceulemans H, Bollen M. Functional diversity of protein phosphatase-1, a cellular economizer and reset button. Physiol Rev 2004;84:1-39 [DOI] [PubMed] [Google Scholar]
- 45. Allen JD, Verhoeven E, Domen J, van der Valk M, Berns A. Pim-2 transgene induces lymphoid tumors, exhibiting potent synergy with c-myc. Oncogene 1997;15:1133-41 [DOI] [PubMed] [Google Scholar]
- 46. Futreal PA, Coin L, Marshall M, Down T, Hubbard T, Wooster R, et al. A census of human cancer genes. Nat Rev 2004;4:177-83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Cao W, Mattagajasingh SN, Xu H, Kim K, Fierlbeck W, Deng J, et al. TIMAP, a novel CAAX box protein regulated by TGF-beta1 and expressed in endothelial cells. Am J Physiol Cell Physiol 2002; 283:C327-37 [DOI] [PubMed] [Google Scholar]
- 48. Kim K, Li L, Kozlowski K, Suh HS, Cao W, Ballermann BJ. The protein phosphatase-1 targeting subunit TIMAP regulates LAMR1 phosphorylation. Biochem Biophys Res Comm 2005;338:1327-34 [DOI] [PubMed] [Google Scholar]
- 49. Carbone A, Gloghini A, Colombatti A, Castronovo V, Menard S. Expression of the monomeric 67-kd laminin-binding protein in human lymphomas as defined by MLuC5 monoclonal antibody and paraffin section immunohistochemistry. Hum Pathol 1995;26:541-6 [DOI] [PubMed] [Google Scholar]
- 50. Berno V, Porrini D, Castiglioni F, et al. The 67 kDa laminin receptor increases tumor aggressiveness by remodeling laminin-1. Endocr Relat Cancer 2005;12:393-406 [DOI] [PubMed] [Google Scholar]
- 51. Menard S, Castronovo V, Tagliabue E, Sobel ME. New insights into the metastasis-associated 67 kD laminin receptor. J Cell Biochem 1997;67:155-65 [PubMed] [Google Scholar]
- 52. Hubbert C, Guardiola A, Shao R, Kawaguchi Y, Ito A, Nixon A, et al. HDAC6 is a microtubule-associated deacetylase. Nature 2002;417:455-8 [DOI] [PubMed] [Google Scholar]
- 53. Cabrero JR, Serrador JM, Barreiro O, Mittelbrunn M, Naranjo-Suarez S, Martin-Cofreces N, et al. Lymphocyte chemotaxis is regulated by histone deacetylase 6, independently of its deacetylase activity. Mol Biol Cell 2006;17:3435-45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Hook SS, Orian A, Cowley SM, Eisenman RN. Histone deacetylase 6 binds polyubiquitin through its zinc finger (PAZ domain) and copurifies with deubiquitinating enzymes. Proc Natl Acad Sci U S A 2002;99:13425-30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Kawaguchi Y, Kovacs JJ, McLaurin A, Vance JM, Ito A, Yao TP. The deacetylase HDAC6 regulates aggresome formation and cell viability in response to misfolded protein stress. Cell 2003;115:727-38 [DOI] [PubMed] [Google Scholar]
- 56. Lindemann RK, Newbold A, Whitecross KF, Cluse LA, Frew AJ, Ellis L, et al. Analysis of the apoptotic and therapeutic activities of histone deacetylase inhibitors by using a mouse model of B cell lymphoma. Proc Natl Acad Sci U S A 2007;104:8071-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Marquard L, Poulsen CB, Gjerdrum LM, de Nully Brown P, Christensen IJ, Jensen PB, et al. Histone deacetylase 1, 2, 6 and acetylated histone H4 in B- and T-cell lymphomas. Histopathology 2009;54:688-98 [DOI] [PubMed] [Google Scholar]
- 58. Meacham CE, Ho EE, Dubrovsky E, Gertler FB, Hemann MT. In vivo RNAi screening identifies regulators of actin dynamics as key determinants of lymphoma progression. Nat Genet 2009;41:1133-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Chen WV, Soriano P. Gene trap mutagenesis in embryonic stem cells. Methods Enzymol 2003;365:367-86 [PubMed] [Google Scholar]
- 60. Scheijen B, Jonkers J, Acton D, Berns A. Characterization of pal-1, a common proviral insertion site in murine leukemia virus-induced lymphomas of c-myc and Pim-1 transgenic mice. J Virol 1997;71:9-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Searle SR, Casella G, McCulloch CE. Variance components. Hoboken, NJ: Wiley Interscience; 2006 [Google Scholar]
- 62. Lewis PAW, Shedler GS. Simulation methods for Poisson processes in nonstationary systems. Proceedings of the 10th Conference on Winter Simulation; 1978 [Google Scholar]
- 63. Gentleman R, Carey VJ, Huber W, Irizarry R, Dudoit S. Bioinformatics and computational biology solutions using R and bioconductor. New York, NY: Springer: 2005 [Google Scholar]