

Abstract
Many aspects of cellular behavior are defined by the content of information provided by association of the extracellular matrix (ECM) and with cell membrane receptors. When cultured in the presence of laminin-containing ECM and prolactin (Prl), normal mammary epithelial cells express the milk protein β-casein. We have previously found that the minimal ECM- and Prl-responsive enhancer element BCE-1 was only active when stably integrated into chromatin, and that trichostatin A (TSA), a reagent that leads to alterations in chromatin structure, was able to activate the integrated enhancer element. We now show that endogenous β-casein gene, which is controlled by a genetic assembly that is highly similar to that of BCE-1 and which is also activated by incubation in ECM and Prl, is instead inhibited by TSA. We provide evidence that the differing response of β-casein and BCE-1 to TSA is neither due to an unusual effect of TSA on mammary epithelial cells, nor to secondary consequences from the expression of a separate gene, nor to a particular property of the BCE-1 construct. As a component of this investigation, we also showed that ECM mediated rapid histone deacetylation in mammary epithelial cells. These results are discussed in combination with previous work showing that TSA mediates the differentiation of many types of cancer cells but inhibits differentiation of some nonmalignant cell types.
Keywords: tissue specificity, histone H4, basement membrane, extracellular matrix, chromatin structure
To investigate the mechanisms involved in the differentiation of mammary epithelial cells, we have used an assay in which cells are cultured in recombinant basement membrane (rBM), a laminin-containing extracellular matrix (ECM) that simulates the normal micro-environment of mammary epithelial cells [Bissell et al., 1999]. When incubated in rBM and in the presence of lactogenic hormones, phenotypically normal mammary epithelial cells organize into polarized, alveolar structures reminiscent of those found in lactating mammary glands [Barcellos-Hoff et al., 1989]. Complete reorganization is generally accomplished in 4–5 days, and is accompanied by the transcriptional repression of pro-growth genes, including c-myc, cyclin D1, and Id1 [Boudreau et al., 1995; Desprez et al., 1995; Boudreau et al., 1996], and the activation of genes associated with differentiated mammary epithelium, including lactoferrin, β-casein, and whey acidic protein [Roskelley et al., 1994; Lin et al., 1995]. We previously investigated the genetic determinants of rBM dependent transcriptional activation in mammary epithelial cells and identified BCE-1, a 160 bp rBM-responsive minimal enhancer element derived from the bovine β-casein gene [Schmidhauser et al., 1990, 1992]. Characterization of BCE-1 using site-specific mutagenesis revealed binding sites for C/EBPβ and Stat5 [Myers et al., 1998], transcription factors that play essential roles in mammary gland development and differentiation [Liu et al., 1997; Lekstrom-Himes and Xanthopoulos, 1998].
We found that BCE-1 was active only when stably integrated into chromatin, although pharmacological inhibitors of histone deacetylase enzymes such as trichostatin A (TSA) could activate BCE-1 in the absence of rBM or hormones [Myers et al., 1998]. These results suggested that information from the ECM also impacts on the architecture of the chromatin, since many aspects of chromatin structure are determined by the acetylation state of its histone subunits [Taddei et al., 2001]. A number of developmental processes have been linked to changes in the state of histone acetylation [Mannervik et al., 1999; Litt et al., 2001], including the differentiation of muscle, blood, and immune cells [Puri et al., 1997; Blobel, 2000; Goodman and Smolnik, 2000; McMurry and Krangel, 2000]. Abnormalities of regulation or function of histone acetylase and deacetylase enzymes can lead to developmental abnormalities [Almouzni et al., 1994; Petrij et al., 1995], generally increased tumor susceptibility [Giles et al., 1998; Gayther et al., 2000], and a specific predisposition to promyelocytic leukemia [Grignani et al., 1998; Lin et al., 1998].
Here, we have used our rBM assay to probe the mechanisms by which signals from the ECM lead to expression of β-casein through alterations in chromatin structure. We will provide evidence that the activation of β-casein is controlled by rBM-mediated changes in histone acetylation levels, and we will discuss these results in the context of previous observations relating chromatin architecture and cellular differentiation.
METHODS
Cell Culture and Differentiation Assays
The mouse mammary epithelial cell lines CID-9 [Schmidhauser et al., 1990,1992] and Eph4 [Reichmann et al., 1989], and their transfected progenies were maintained in DMEM/F12 (Life Technologies, Gaithersburg, MD) supplemented with 5% fetal bovine serum and 5 μg/ml insulin (Sigma, St. Louis, MO) (growth medium). Cells were induced to differentiate in DMEM/F12 supplemented with 5 μg/ml insulin, 1 μg/ml hydrocortisone (Sigma) and/or 3 μg/ml prolactin (Prl) (ID# AFP 10677C, NIDDK, NIH, Bethesda, MD) (differentiation medium), as previously described [Schmidhauser et al., 1990,1992]. Culture on nonadhesive substrata was as previously described [Roskelley et al., 1994]. Reconstituted basement membrane (Matrigel, Collaborative Biomedical Products, Bedford, MA) was given in the form of a 1.5% overlay in the medium. Conditioned medium was prepared in chemically defined serum-free medium containing insulin (5 μg/ml), transferrin (5 μg/ml), and selenium (5 ng/ml) (Sigma). Sodium butyrate (Sigma) and TSA (Wako Pure Chemical Industry Ltd, Richmond, VA) were prepared as 1,000× stock solutions in water and ethanol, respectively, and added as described.
Antibodies and Western Blots
We used the following antisera: for β-casein, a murine monoclonal anti-rat β-casein IgG (1:2,000 dilution) (Dr. Kaetzel, Cleveland, OH); for flag-tagged proteins, a mouse monoclonal anti-flag tag IgG (1:1,000 dilution) (Kodak, New Haven, CT); for P/CAF, a rabbit polyclonal anti-human P/CAF (1:1,000 dilution) (Dr. Yang and Dr. Nakatani, NIH); for histone H4 acetylated lysines, a rabbit polyclonal anti-acetylated histone H4 (Upstate Biotechnology Inc., Lake Placid, NY); for E-cadherin, a mouse monoclonal anti-human E-cadherin IgG (1:1,000 dilution) (Transduction Laboratories, Lexington, KY). Cell lysates extracted in RIPA buffer (1% NP-40, 0.5% deoxycholate, 0.2% SDS, 150 mM sodium chloride, 50 mM Tris-HCl, pH 7.4 containing a cocktail of proteases inhibitors from Calbiochem, La Jolla, CA) were mixed in 10× reducing Laemmli buffer and resolved on prepared 12 or 4–20% gradient gels (BioRad, Richmond, CA) using standard SDS–PAGE protocols. Proteins were transferred to Immobilon-P nitrocellulose filters, immuno-probed, and detected by enhanced chemiluminescence (ECL, Amersham, Arlington Heights, IL). Blocking solution was purchased from Pierce Chemical Co. (Rockford, IL). Extracts were quantitated for protein concentration (BioRad). Scanning and densitometric analysis of E-cadherin were performed when required as a measure of equal sample loading of epithelial cells [Wang et al., 1998] using an Eagle Eye II image analysis system (Stratagene, La Jolla, CA).
Histone Preparations and Triton-Acetic Acid-Urea Gel Analysis
Histones were acid-extracted from isolated nuclei after lysis with NP-40. The acid-soluble histone fraction was acetone-precipitated and resuspended in water as previously described [Arts et al., 1995]. Purified histones were quantified using a Bradford assay (BioRad), and then separated on triton-acetic acid-urea gels [Arts et al., 1995]. Gels were stained with Coomassie blue and dried before analysis.
Reverse Transcriptase PCR (RT-PCR)
For each RT reaction, 2.5 μg of total cellular RNA, prepared using TRIzol (Life Technologies) according to the manufacturer’s instructions, was reverse transcribed at 37°C for 3 h with 20 U of reverse transcriptase (Boehringer Mannheim, Indianapolis, IN). Optimal parameters for MgCl2 concentrations, annealing temperatures, and cycle number were empirically determined for each PCR reaction. CAT PCR amplifications were performed with 5′ primer 5′-GCC CGC CTG ATG AAT GCT CA-3′, and 3′ primer 5′-CGC CCC GCC CTG CCA CTC ATC G-3′. Beta-casein PCR amplifications were performed with 5′ primer 5′-ATG AAG GTC TTC ATC CTC GCC-3′, and 3′ primer 5′-TTA GAC AGA AAC GGA ATG TTG TGG-3′. P/CAF PCR amplifications were performed with 5′ primer 5′-CGA ATC GCC GTG AAG AAA GC-3′, and 3′ primer 5′-GGG GTT TCT TTT CCA AAG AGC-3′. Tissue-plasminogen activator PCR amplifications were performed with 5′ primer (5′-TGG ACT GGC TTT CCC ATT GC-3′), and 3′ primer (5′-CCA GCT TGA TGG CAT TTG GC-3′). As a control for total RNA integrity, actin RT-PCR experiments were performed with 5′ primer 5′-GCT GGT CGT CGA CAA CGG CT-3′, and 3′ primer 5′-ATG ACC TGG CCG TCA GGC-3′. Removal of all genomic DNA was verified for every RNA preparation by PCR amplifying actin sequences before cDNA synthesis. The resulting amplified fragments were analyzed on 1.5% ethidium bromide-stained agarose gels with an Eagle Eye II image analysis system (Stratagene).
Plasmids, Transfections and Reporter Gene Analysis
Expression analysis of reporter constructs was performed as described previously [Schmidhauser et al., 1990,1992; Myers et al., 1998]. The expression plasmid for the human open reading frame cDNA of p/CAF, including a N-terminal flag tag, was from Dr. Yang and Dr. Nakatani, NIH [Yang et al., 1996]. Stable and transient transfections, cell harvest, and CAT assays were performed essentially as described previously [Myers et al., 1998]. Cells were cultured in growth medium at a density of 1.5 × 106 per 100 mm tissue culture plastic dish 1 day prior to transfection. Ten micrograms of test plasmid and 1 μg of RSV/neo were co-transfected by the lipofectin-based method (Gibco BRL, Gaitherburg, MD) according to manufacturer’s instructions, and cells were selected using a growth medium with or without geniticin. Pooled populations of stable transfections were analyzed in functional differentiation assays. For the CAT assays, cell extracts normalized for standard protein levels (Bradford assay) were incubated in the presence of 0.9 mM acetylcoenzyme A (Sigma) and 63 μM fluorophore Bodipy 1-deoxychloramphenicol (Molecular Probes, Eugene, OR) for 8 h at 37°C, and extracted with ethyl acetate. The thin layer chromatography plates from the CAT assays were analyzed with an Eagle Eye II image analysis system (Stratagene). pSVCAT reporter plasmid (Stratagene) was used as a positive control for CAT activity.
RESULTS
TSA Inhibits Production of Endogenous β-Casein
We previously characterized BCE-1 as a minimal enhancer element derived from the bovine β-casein gene. In these studies, we found that a BCE-1-CAT construct could be activated in mouse mammary epithelial cells either by differentiation conditions that included rBM and Prl or by TSA, but only when the reporter construct was stably integrated into chromatin [Schmidhauser et al., 1990,1992; Myers et al., 1998]. Here, we examined the effect of these stimuli on the expression of the endogenous β-casein gene. Using the CID-9 mouse mammary epithelial cell line employed in the earlier studies, we found that addition of TSA attenuated production of endogenous β-casein (Fig. 1). Assessing the degree to which TSA treatment inhibited expression of β-casein is complicated by the long half-life of the β-casein protein [Lee et al., 1985] since this assay requires 4–5 days, but toxicity considerations precluded the use of TSA for more than just the final 24 h. Nevertheless, the difference between the response of the endogenous β-casein and the BCE-1 minimal enhancer to TSA was striking, especially given (1) the similar structure of the two promoter/enhancers [Rosen et al., 1999], (2) that both respond identically to different combinations of rBM and Prl [Schmidhauser et al., 1992], and (3) that TSA is often used as a differentiation agent [Marks et al., 2000], and β-casein is a marker of mammary epithelial cell differentiation [Roskelley et al., 1995]. We performed a series of experiments to dissect the basis of the differential response of β-casein and BCE-1 to TSA.
Fig. 1.
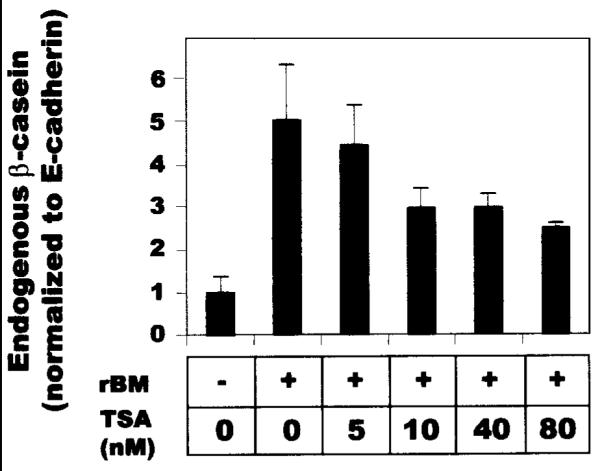
TSA inhibits differentiation-dependent production of endogenous β-casein. CID-9 cells were cultured in differentiation conditions for 4 days, and then the medium was supplemented with TSA at the indicated concentrations for an additional 24 h. Cell extracts were analyzed by Western blot and probed for β-casein. Band intensity was assessed by densitometry and normalized to E-cadherin.
TSA Mediates Activation of BCE-1 and Hyperacetylation of Histone H4
Investigations of the mammary epithelial, tissue-specific MMTV enhancer/promoter had indicated a complex, bimodal activation relationship in response to TSA [Bartsch et al., 1996]. It was possible that both BCE-1 and β-casein were activated and/or repressed by TSA, but at different time points or with different concentrations of TSA. This possibility was tested with CID-9 cells stably transfected with the BCE-1-CAT reporter construct. Induction of CAT activity by TSA became apparent after 8 h and was maximal by 24 h (Fig. 2A). Continued expression of CAT was dependent upon the sustained presence of TSA, as a progressive loss of CAT activity was observed in cells treated for 10 h with TSA, then washed and incubated in medium lacking TSA (Fig. 2B). Furthermore, TSA stimulated CAT activity both on plastic and on ECM. On plastic, CAT expression was induced 100-fold by TSA (Fig. 2C), while cells grown on ECM, in which BCE-1 was already active, showed a less substantial, but still additive, effect (Fig. 2D). To verify that TSA did function in mammary epithelial cells to mediate increases in histone acetylation levels, we examined the specific histone acetylation response in CId-9 cells. We found that 5 h of TSA treatment was sufficient to induce histone H4 hyperacetylation (Fig. 3A), and that when individually tested, lysines 5, 8, and 12 of histone H4 were all found to be hyperacetylated by TSA treatment (Fig. 3B; other histone subunits were not found to be acetylated by these treatments, data not shown).
Fig. 2.

Activation of the BCE-1-CAT reporter construct is potentiated by incubation with rBM. CID-9 mouse mammary epithelial cells stably transfected with the BCE-1-CAT construct (CID-BCE cells) were grown in differentiation medium on plastic substrate (A, B, C) or on rBM (D) for 5 days. TSA was added to the culture medium to 165 nM either (A) for various times up to 24 h before harvest, or (B) for 10 h and then followed by culture in differentiation medium for various times up to 24 h. For (C) and (D), TSA was added at the indicated concentrations 24 h before harvest. The fluorographs show the thin layer chromatography of the CAT assays. The graphs indicate the integrated density of the fluorographs, expressed in relative arbitrary units.
Fig. 3.

TSA mediates acetylation of histone-H4 subunits in mammary epithelial cells. CID-BCE cells were plated on plastic substratum and incubated in differentiation medium for 5 days, then TSA was added at 165 nM for 5 h before harvest. A: Cells were assayed by Western blot for total acetylated histone H4. B: Histone proteins were purified from cell extracts and electro-phoresed on triton-acetic acid-urea gels; lysines 5, 8, and 12 of histone H4 were found to be hyperacetylated. Equal amounts of extract were used on each lane of these gels, and E-cadherin was used as a loading control. E-cad, E-cadherin.
Activation of BCE-1 by TSA Treatment Does Not Require Protein Synthesis
Other studies have shown that TSA-mediated transcriptional activation can occur indirectly, through mechanisms involving activation of intermediate genes [Zhang et al., 2001], and it was possible that activation of BCE-1 (and/or histone acetylation) could be a secondary effect. To test this possibility, we examined the activation of BCE-1 in the presence of the protein synthesis inhibitor cycloheximide. We found that neither histone hyperacetylation nor activation of BCE-1 was affected by inhibition of protein synthesis (Fig. 4).
Fig. 4.

Neither histone acetylation nor activation of BCE-1 by TSA requires protein synthesis. A: CID-BCE cells were cultured on plastic substrata in differentiation medium for 5 days, then treated with 165 nM TSA and/or 0.25 μg/ml cycloheximide for an additional 24 h. B: Activation of the BCE-1 reporter gene activation was unaffected by cycloheximide. Cells were cultured in the presence or absence of 165 nM TSA and in the absence or presence of 0.25 to 1 μg/ml cycloheximide, and then isolated RNA was analyzed by RT-PCR (CAT mRNA and Actin mRNA) and protein lysates were analyzed by CAT assay (CAT activity). Ac-H4, acetylated histone H4; CHXD, cycloheximide.
TSA Inhibits rBM-Mediated Transcription of Endogenous β-Casein
Incubation of mammary epithelial cells with rBM is associated with cell rounding, a morphological reorganization that appears to be a prerequisite for expression of β-casein [Roskelley et al., 1994]. To determine if the inhibition of β-casein expression by TSA was due to an alteration of cell rounding processes, we used an alternative assay. When grown on poly-HEMA, a nonadhesive substrata, mammary epithelial cells become rounded; in this state, the cells are poised for expression of milk proteins, so that exposure to either rBM or laminin results in the induction of β-casein in less than 24 h [Roskelley et al., 1994; Muschler et al., 1999]. The use of this modified assay allowed us to separate the effects of rBM on morphological reorganization and gene expression. The CID-9 cells used in Figure 1 and previously [Schmidhauser et al., 1990,1992; Myers et al., 1998] were inappropriate for this assay, as they produce sufficient endogenous ECM to autonomously activate the expression of β-casein in pre-rounded cells [Pujuguet et al., 2000]. For this reason, we used Eph4 cells, another phenotypically normal mammary epithelial cell line [Reichmann et al., 1989] that is readily transfectable and that also expresses β-casein in the presence of rBM and Prl. When stably transfected into Eph4 cells, BCE-1-driven reporter constructs were also activated either by rBM and Prl or by TSA (data not shown). Exposure of pre-rounded Eph4 cells to rBM activated transcription of the β-casein gene, as assessed by RT-PCR (Fig. 5A, lane 2). However, simultaneous exposure to TSA blocked rBM-dependent expression of β-casein in a manner that was both cumulative (Fig. 5A) and reversible (Fig. 5B). This effect was not due to a generalized repression of transcription, as expression of actin was unaffected, and the tissue plasminogen activator (tPA), an unrelated gene, was found to be induced by simultaneous exposure to rBM and TSA. Neither was the decreased expression of the β-casein transcript due to excessive cell death, as assessed by an alimar blue viability assay (Fig. 5C).
Fig. 5.

TSA selectively inhibits transcription of β-casein. A: TSA effect is cumulative. Eph4 cells were cultured on poly-HEMA to induce morphological change, then incubated in the presence of Prl and the presence or absence of rBM for 24 h. Additionally, some samples (lanes 3+4) were incubated with 80 nM TSA for the times indicated. B: TSA effect is reversible. As for (A), except in lane 4, the cells were incubated in the presence of rBM and TSA for 24 h, then the cells were washed and incubated with rBM for an additional 24 h. Expression of β-casein, tissue plasminogen activator (t-PA), or actin was assessed by RT-PCR. C: Minimal cytotoxicity was associated with poly-HEMA assay and TSA incubation, as revealed by alimar blue exclusion assay. C, control untreated cells; V, EtOH-treated only.
Alternative BCE-1 Reporter Constructs Were Also Activated by TSA
As BCE-1 is a minimal enhancer element [Myers et al., 1998], the discrepancy between the endogenous β-casein gene and BCE-1 could have been due to an inhibitory effect exerted by cis-acting elements present in the endogenous gene, but absent in the minimal enhancer. To examine this hypothesis, two reporter constructs, previously generated during the optimization of BCE-1 [Schmidhauser et al., 1992], were stably transfected into Eph4 cells. BCE-3815 contains 3815 bp upstream of the β-casein gene, while BCE-2605 is a construct of intermediate length. All three BCE constructs were stably transfected into Eph4 cells and found to be activated by treatment with TSA (Fig. 6), or by growth on rBM and Prl (Schmidhauser et al., 1992 and data not shown). These results suggest that the difference between the activation properties of BCE-1 and the endogenous β-casein is not due to cis-acting regulatory elements that were removed during enhancer minimization. Another possibility was that some aspect of the CAT reporter gene was directly activated by TSA, but we found that a BCE-1-luciferase reporter construct responded similarly to BCE-1-CAT when incubated with TSA or in the presence of rBM and Prl (data not shown).
Fig. 6.
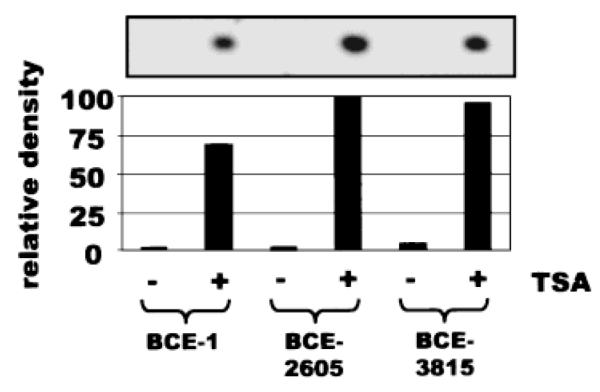
TSA activates other β-casein reporter constructs. Eph4 cells were stably transfected with either the BCE-1-CAT construct (BCE-1) or with constructs containing the CAT gene cloned behind the 2605 bp upstream of the bovine β-casein gene (BCE-2605) or the 3815 bp upstream of the bovine β-casein gene (BCE-3815). Cells were cultured on plastic for 5 days in differentiation medium and treated with TSA at 165 nM, 24 h prior to harvest and assayed for CAT activity. The fluorograph shows the thin layer chromatography of the CAT assays. The graph indicates the integrated density of the fluorograph, expressed in relative arbitrary units.
Overexpression of Histone Acetyltransferase Inhibits rBM-Mediated Activation of β-Casein
To test the possibility that repression of β-casein by TSA could be the consequence of activities other than inhibition of histone deacetylase enzymes, we generated stable transfectants of Eph4 cells that constitutively expressed the p300/CBP-associating factor (p/CAF) histone acetyltransferase [Yang et al., 1996]. Expression of p/CAF was verified by RT-PCR (Fig. 7A) and by immunofluorescence (data not shown). Overexpression of p/CAF did not prevent rBM-mediated cell rounding or growth arrest (data not shown), but did interfere with production of β-casein (Fig. 7B). These results suggest that increased histone acetylation is a key event for suppression of endogenous β-casein expression.
Fig. 7.

Overexpression of p/CAF inhibits differentiation-dependent production of endogenous β-casein. Eph4 cells were stably transfected with constitutive expression plasmids containing the p/CAF cDNA along with a plasmid containing a selection marker (p/CAF transfectants) or with just the plasmid containing a selection marker (control transfectants). A: RT-PCR analysis of RNA extracted from control (lane 1) or p/CAF (lane 2) transfectants. Lane 3 is a PCR amplification of the p/CAF plasmid, as a size control. B: Control (lane 1, 3, 5) or p/CAF (lane 2, 4, 6) transfectants were grown for 5 days in the absence or presence of rBM or Prl, and whole-cell extracts were analyzed for β-casein protein expression by Western blot; E-cadherin was used as a loading control.
rBM Induces Widespread Chromatin Deacetylation
That increased histone acetylation was antagonistic to rBM-mediated activation of β-casein, suggested that rBM could act in the opposite fashion as TSA. When we examined this possibility, we found that exposure of Eph4 cells to rBM led to a decrease of histone H4 acetylation. TSA treatment produced the expected opposite result, demonstrating that a full dynamic range of histone acetylation is possible in these cells (Fig. 8). This experiment suggests that some component of the rBM matrix leads to deacetylation of the histone H4 subunits, and that this signal may be required for subsequent transcription of the β-casein gene. Moreover, this is the first demonstration that signals from the ECM can have a general effect on histone acetylation, and presumably, on chromatin structure as well.
Fig. 8.
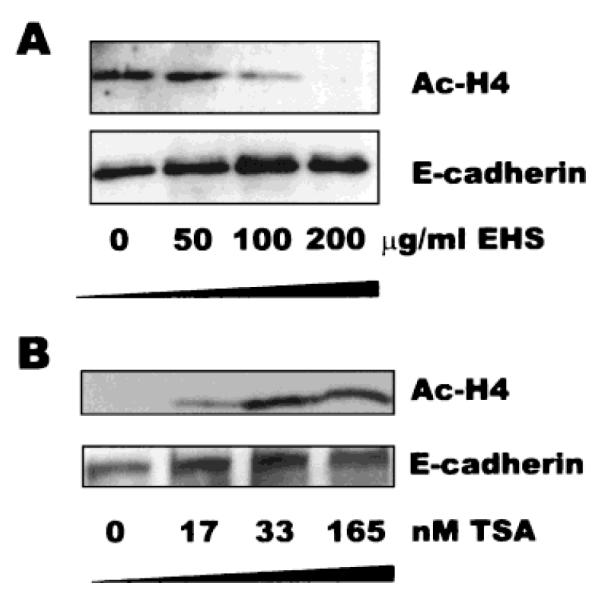
ECM induces deacetylation of histone H4 in Eph4 cells. A: rBM was added to culture medium at the indicated concentration for 5 h prior to harvest, and equivalent amounts of cell extract were analyzed by Western blot for acetylated histone H4. B: TSA was added to culture medium at the indicated concentration for 5 h prior to harvest, and equivalent amounts of cell extract were analyzed by Western blot for acetylated histone H4. E-cadherin was used as a loading control for both experiments.
DISCUSSION
Many investigations have probed the relationship between the activation or repression of a single gene and changes in histone acetylation status, and we are now beginning to understand some of the basic mechanisms by which chromatin structure regulates gene expression. Increasingly, however, it is becoming apparent that adaptive changes in gene expression involve simultaneous activation and repression of many genes. Now the challenge is to characterize the mechanisms that produce these complex changes. In this work, we have used an assay system in which normal mouse mammary epithelial cells are grown on a laminin-containing, three-dimensional matrix in the presence of lactogenic hormones. Under these conditions, the cells assume the properties of differentiated cells, organizing into tissue structures and producing milk proteins. We have shown that either pharmacological inhibitors of histone deacetylases or overexpression of a histone acetylase enzyme is sufficient to inhibit rBM-dependent expression of the endogenous β-casein gene. These results point to a model in which regulation of tissue-specific gene expression in differentiated cells is maintained, in part, through control of chromatin structure and histone acetylation by information from the microenvironment. This cell-based assay can be used for investigating the mechanisms by which extracellular information is transduced into structural changes of chromatin.
We have provided evidence that the differential transcriptional effects of rBM and TSA on the expression of the endogenous β-casein gene are due to differential effects on histone acetylation. This conclusion, however, does not explain why the BCE-1 enhancer element is activated by both conditions. It is possible that positional effects are responsible for the difference; the β-casein locus is fixed, while the integration site of the BCE-1-reporter construct is variable. Such positional effects have been suggested for the MMTV promoter [Lambert and Nordeen, 1998], another mammary epithelial cell-specific construct that has been shown to be activated by distinct changes in chromatin architecture [Truss et al., 1995]. Characterization of MMTV activation has distinguished separate stages of transcription factor binding, coactivator recruitment, and chromatin rearrangement [Sheldon et al., 1999; Belikov et al., 2000], and investigations of MMTV promoter activation have revealed much about mechanisms of transcriptional activation in vivo [McNally et al., 2000]. Our results with BCE-1 and the endogenous β-casein promoter suggest that a comparison of these two systems may be useful to dissect the complex processes by which cell differentiation controls transcriptional regulation.
We have found that exposure of mammary epithelial cells to rBM results in histone H4 deacetylation. Although a variety of pharmacological inhibitors of histone deacetylases have been instrumental to probe the mechanisms by which histone acetylation correlates with gene activation [Marks et al., 2000], no reagents that result in deacetylation have yet been found. If this property of rBM applies to other cell types, then it could represent a tool that is complementary to deacetylase inhibitors for studying the effects of chromatin structure on gene expression.
Pharmacological inhibitors of histone deacetylases are capable of inducing or potentiating the differentiation of tumor cells [Minucci et al., 1997; Marks et al., 2000]; this property is so general that it can be used as a screen to identify novel inhibitors [Jung et al., 1999]. Accordingly, these compounds have been proposed for treatment of various cancers [Hosugi et al., 1999; Saunders et al., 1999; Butler et al., 2000]. However, recent studies have found that the same compounds also interfere with transcriptional regulation relating to muscle- and hematopoeitic-specific gene expression [Koipally et al., 1999; Miska et al., 1999], and also the differentiation of normal hepatic stellate cells into myofibroblasts [Niki et al., 1999]. We now show that inhibitors of histone deacetylases and overexpression of histone acetylase and deacetylase also block mammary-specific gene expression.
We have previously shown that normal and malignant mammary epithelial cells respond differently to ECM [Petersen et al., 1992], and that normalization of ECM signaling in malignant breast cells can cause a reversion to a normal phenotype [Weaver et al., 1997; Wang et al., 1998]. Since inhibitors of histone deacetylases also revert some tumor cells, and since, as described here, ECM produces changes in histone acetylation to regulate normal tissue-specific gene expression, we propose that the functional reversions mediated by normalization of ECM signaling are mediated by changes of the patterns of histone acetylation that return the chromatin structure to that of a normal, differentiated cell. Investigations of this hypothesis are currently underway.
ACKNOWLEDGMENTS
We thank Xiang-Jiao Wang and Yoshihiro Nakatani (NIH, Bethesda, MD) for the p/CAF expression vector. We also thank Eva Turley, John Muschler, Johanne LeBeyec, Karen Schmeichel, and Paul Kaufman for helpful suggestions and discussion. This work was supported by the U.S. Department of Energy, Office of Biological and Environmental Research and the National Institutes of Health, by a Distinguished Hollaender Postdoctoral fellowship to DR, and by support from the French League Against Cancer and the Breast Cancer Research Program of California to PP.
Grant sponsor: U.S. Department of Energy, Office of Biological and Environmental Research; Grant number: DE-AC03-76SF00098; Grant sponsor: National Institutes of Health; Grant number: CA-57621-02.
REFERENCES
- Almouzni G, Khochbin S, Dmitrov S, Wolffe AP. Histone acetylation influences both gene expression and development of Xenopus laevis. Dev Biol. 1994;165:654–659. doi: 10.1006/dbio.1994.1283. [DOI] [PubMed] [Google Scholar]
- Arts J, Lansink M, Grimbergen J, Toet KH, Kooistra T. Stimulation of tissue-type plasminogen activator gene expression by sodium butyrate and trichostatin A in human endothelial cells involves histone acetylation. Biochem J. 1995;310:171–176. doi: 10.1042/bj3100171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barcellos-Hoff MH, Aggeler J, Ram TG, Bissell MJ. Functional differentiation and alveolar morphogenesis of primary mammary cultures on reconstituted basement membrane. Development. 1989;105:223–235. doi: 10.1242/dev.105.2.223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartsch J, Truss M, Bode J, Beato M. Moderate increase in histone acetylation activates the mouse mammary tumor virus promoter and remodels its nucleosome structure. Proc Natl Acad Sci USA. 1996;93:10741–10746. doi: 10.1073/pnas.93.20.10741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belikov S, Gelius B, Almouzni G, Wrange Ö . Hormone activation induces nucleosome positioning in vivo. EMBO J. 2000;19:1023–1033. doi: 10.1093/emboj/19.5.1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bissell MJ, Weaver VM, Lelièvre SA, Wang F, Petersen OW, Schmeichel KL. Tissue structure, nuclear organization, and gene expression in normal and malignant breast. Cancer Res. 1999;59(Suppl.):1757s–1764s. [PubMed] [Google Scholar]
- Blobel GA. CREB-binding protein and p300: molecular integrators of hematopoietic transcription. Blood. 2000;95:745–755. [PubMed] [Google Scholar]
- Boudreau N, Sympson CJ, Werb Z, Bissell MJ. Suppression of ICE and apoptosis in mammary epithelial cells by extracellular matrix. Science. 1995;267:891–893. doi: 10.1126/science.7531366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boudreau N, Werb Z, Bissell MJ. Suppression of apoptosis by basement membrane requires three-dimensional tissue organization and withdrawal from the cell cycle. Proc Natl Acad Sci USA. 1996;93:3509–3513. doi: 10.1073/pnas.93.8.3509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler LM, Agus DB, Scher HI, Higgins B, Rose A, Cordon-Cardo C, Thaler HT, Rifkind RA, Marks PA, Richon VM. Suberoylanilide hydroxamic acid, an inhibitor of histone deacetylase, suppresses the growth of prostate cancer cells in vitro and in vivo. Cancer Res. 2000;60:5165–5170. [PubMed] [Google Scholar]
- Desprez PY, Hara E, Bissell MJ, Campisi J. Suppression of mammary epithelial differentiation by the helix-loop-helix protein Id-1. Mol Cell Biol. 1995;15:3398–3404. doi: 10.1128/mcb.15.6.3398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gayther SA, Batley SJ, Linger L, Bannister A, Thorpe K, Chin SF, Daigo Y, Russell P, Wilson A, Sowter HM, Delhanty JDA, Ponder BAJ, Kouzarides T, Caldas C. Mutations truncating the EP300 acetylase in human cancers. Nat Genet. 2000;24:300–303. doi: 10.1038/73536. [DOI] [PubMed] [Google Scholar]
- Giles RH, Peters DJM, Breuning MH. Conjunction dysfunction: CBP/p300 in human disease. Trends Genet. 1998;14:178–183. doi: 10.1016/s0168-9525(98)01438-3. [DOI] [PubMed] [Google Scholar]
- Goodman RH, Smolnik S. CBP/p300 in cell growth, transformation, and development. Genes Dev. 2000;14:1553–1577. [PubMed] [Google Scholar]
- Grignani F, De Matteis S, Nervi C, Tomassoni L, Gelmetti V, Cioce M, Fanelli M, Ruthardt M, Ferrara FF, Zamir I, Seiser C, Grignani F, Lazar MA, Minucci S, Pelicci PG. Fusion proteins of the retinoic acid receptor-alpha recruit histone deacetylase in promyelocytic leukaemia. Nature. 1998;391:814–818. doi: 10.1038/35901. [DOI] [PubMed] [Google Scholar]
- Hosugi H, Towatari M, Hatano S, Kitamura K, Kiyoi H, Kinoshita T, Tanimoto M, Murate T, Kawashima K, Saito H, Naoe T. Histone deacetylase inhibitors are the potent inducer/enhancer of differentiation in acute myeloid leukemia: a new approach to anti-leukemia therapy. Leukemia. 1999;13:1316–1324. doi: 10.1038/sj.leu.2401508. [DOI] [PubMed] [Google Scholar]
- Jung M, Brosch G, Kölle D, Scherf H, Gerhäuser C, Loidl P. Amide analogues of trichostatin A as inhibitors of histone deacetylase and inducers of terminal cell differentiation. J. Med Chem. 1999;42:4669–4679. doi: 10.1021/jm991091h. [DOI] [PubMed] [Google Scholar]
- Koipally J, Renold A, Kim J, Georgopoulos K. Repression by Ikaros and Aiolos is mediated through histone deacetylase complexes. EMBO J. 1999;18:3090–3100. doi: 10.1093/emboj/18.11.3090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert JR, Nordeen SK. Steroid-selective initiation of chromatin remodeling and transcriptional activation of the mouse mammary tumor virus promoter is controlled by the site of promoter integration. J Biol Chem. 1998;273:32708–32714. doi: 10.1074/jbc.273.49.32708. [DOI] [PubMed] [Google Scholar]
- Lee EY, Lee WH, Kaetzel CS, Parry G, Bissell MJ. Interaction of mouse mammary epithelial cells with collagen substrata: regulation of casein gene expression and secretion. Proc Natl Acad Sci USA. 1985;82:1419–1423. doi: 10.1073/pnas.82.5.1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lekstrom-Himes J, Xanthopoulos KG. Biological role of the CCAAT/enhancer-binding protein family of transcription factors. J Biol Chem. 1998;273:28545–28548. doi: 10.1074/jbc.273.44.28545. [DOI] [PubMed] [Google Scholar]
- Lin CQ, Dempsey P, Coffey C, Bissell MJ. Extracellular matrix regulates whey acidic protein gene expression by suppression of TGF-alpha in mouse mammary epithelial cells: studies in culture and in transgenic mice. J Cell Biol. 1995;129:1115–1126. doi: 10.1083/jcb.129.4.1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin RJ, Nagy L, Inoue S, Shao W, Miller WH, Evans RM. Role of the histone deacetylase complex in acute promyeolcytic leukaemia. Nature. 1998;391:811–814. doi: 10.1038/35895. [DOI] [PubMed] [Google Scholar]
- Litt MD, Simpson M, Recillas-Targa F, Priouleau MN, Felsenfeld G. Transitions in histone acetylation reveal boundaries of three separately regulated neighboring loci. EMBO J. 2001;20:2224–2235. doi: 10.1093/emboj/20.9.2224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Robinson GW, Wagner KU, Garrett L, Wynshaw-Boris A, Hennighausen L. Stat5a is mandatory for adult mammary gland development and lactogenesis. Genes Dev. 1997;11:179–186. doi: 10.1101/gad.11.2.179. [DOI] [PubMed] [Google Scholar]
- Mannervik M, Nibu Y, Zhang H, Levine M. Transcriptional control in development. Science. 1999;284:606–609. doi: 10.1126/science.284.5414.606. [DOI] [PubMed] [Google Scholar]
- Marks PA, Richon VM, Rifkind RA. Histone deacetylase inhibitors: inducers of differentiation or apoptosis of transformed cells. J Natl Cancer Inst. 2000;92:1210–1216. doi: 10.1093/jnci/92.15.1210. [DOI] [PubMed] [Google Scholar]
- McMurry MT, Krangel MS. A role for histone acetylation in the developmental regulation of VDJ recombination. Science. 2000;287:495–498. doi: 10.1126/science.287.5452.495. [DOI] [PubMed] [Google Scholar]
- McNally JG, Müller WG, Walker D, Wolford R, Hager GL. The glucocorticoid receptor: rapid exchange with regulatory sites in living cells. Science. 2000;287:1262–1265. doi: 10.1126/science.287.5456.1262. [DOI] [PubMed] [Google Scholar]
- Minucci S, Horn V, Bhattacharyya N, Russanova V, Ogryzko VV, Gabriele L, Howard BH, Ozato K. A histone deacetylase inhibitor potentiates retinoid receptor action in embryonal carcinoma cells. Proc Natl Acad Sci USA. 1997;94:11295–11300. doi: 10.1073/pnas.94.21.11295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miska EA, Karlsson C, Langley E, Nielsen SJ, Pines J, Kouzarides T. HDAC4 deacetylase associates with and represses the MEF2 transcription factor. EMBO J. 1999;18:5099–5107. doi: 10.1093/emboj/18.18.5099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muschler J, Lochter A, Roskelley CD, Yurchenco P, Bissell MJ. Division of labor among the alpha6beta4 integrin, beta1 integrins, and an E3 laminin receptor to signal morphogenesis and beta-casein expression in mammary epithelial cells. Mol Biol Cell. 1999;10:2817–2828. doi: 10.1091/mbc.10.9.2817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers CA, Schmidhauser C, Mellentin-Michelotti J, Fragoso G, Roskelley CD, Casperson G, Mossi R, Pujuguet P, Hager G, Bissell MJ. Characterization of BCE-1, a transcriptional enhancer regulated by prolactin and extracellular matrix and modulated by the state of histone acetylation. Mol Cell Biol. 1998;18:2184–2195. doi: 10.1128/mcb.18.4.2184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niki T, Rombouts K, De Bleser P, De Smet K, Rogiers V, Shuppan D, Yoshida M, Gabbiani G, Geerts A. A histone deacetylase inhibitor, trichostatin A, suppresses myofibroblastic differentiation of rat hepatic stellate cells in primary culture. Hepatology. 1999;29:858–867. doi: 10.1002/hep.510290328. [DOI] [PubMed] [Google Scholar]
- Petersen OW, Ronnov-Jessen L, Howlett AR, Bissell MJ. Interaction with basement membrane serves to rapidly distinguish growth and differentiation patterns of normal and malignant human breast epithelial cells. Proc Natl Acad Sci USA. 1992;89:9064–9068. doi: 10.1073/pnas.89.19.9064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrij F, Giles RH, Dauwerse HG, Saris JJ, Hennekam RCM, Masuno M, Tommerup N, van Ommen GJB, Goodman RH, Peters DJ, Breuning MH. Rubinstein-Taybi syndrome caused by mutations in the transcriptional co-activator CBP. Nature. 1995;376:348–351. doi: 10.1038/376348a0. [DOI] [PubMed] [Google Scholar]
- Pujuguet P, Simian M, Liaw J, Timpl R, Werb Z, Bissell MJ. Nidogen-1 regulates laminin-1-dependent mammary-specific gene expression. J Cell Sci. 2000;113:849–858. doi: 10.1242/jcs.113.5.849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puri PL, Sartorelli V, Yang XJ, Hamamori Y, Ogryzko VV, Howard BH, Kedes L, Wang JYJ, Graessmann A, Nakatani Y, Levrero M. Differential roles of p300 and PCAF acetyltransferases in muscle differentiation. Mol Cell. 1997;1:5–45. doi: 10.1016/s1097-2765(00)80005-2. [DOI] [PubMed] [Google Scholar]
- Reichmann E, Ball R, Groner B, Friis RR. New mammary epithelial and fibroblastic cell clones in coculture form structures competent to differentiate functionally. J Cell Biol. 1989;108:1127–1138. doi: 10.1083/jcb.108.3.1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosen JM, Wyszomeierski SL, Hadsell D. Regulation of milk protein gene expression. Annu Rev Nutr. 1999;19:407–436. doi: 10.1146/annurev.nutr.19.1.407. [DOI] [PubMed] [Google Scholar]
- Roskelley CD, Desprez PY, Bissell MJ. Extracellular matrix-dependent tissue-specific gene expression in mammary epithelial cells requires both physical and biochemical signal transduction. Proc Natl Acad Sci USA. 1994;91:12378–12382. doi: 10.1073/pnas.91.26.12378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roskelley CD, Srebrow A, Bissell MJ. A hierarchy of ECM-mediated signaling regulates tissue specific gene expression. Curr Opin Cell Biol. 1995;7:736–747. doi: 10.1016/0955-0674(95)80117-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saunders N, Dicker A, Popa C, Jones S, Dahler A. Histone deacetylase inhibitors as potential anti-skin cancer agents. Cancer Res. 1999;59:399–404. [PubMed] [Google Scholar]
- Schmidhauser C, Bissell MJ, Myers CA, Casperson GF. Extracellular matrix and hormones transcriptionally regulate bovine beta-casein 5′ sequences in stably transfected mouse mammary cells. Proc Natl Acad Sci USA. 1990;87:9118–9122. doi: 10.1073/pnas.87.23.9118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidhauser C, Casperson GF, Myers CA, Sanzo KT, Bolten S, Bissell MJ. A novel transcriptional enhancer is involved in the prolactin- and extracellular matrix-dependent regulation of beta-casein gene expression. Mol Biol Cell. 1992;3:699–709. doi: 10.1091/mbc.3.6.699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheldon LA, Smith CL, Bodwell JE, Munck AU, Hager GL. Ligand binding domain mutation in the mouse glucocorticoid receptor functionally links chromatin remodeling and transcription initiation. Mol Cell Biol. 1999;19:8146–8157. doi: 10.1128/mcb.19.12.8146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taddei A, Maison C, Roche D, Almouzni G. Reversible disruption of pericentric heterochromatin and centromere function by inhibiting deacetylases. Nat Cell Biol. 2001;3:114–120. doi: 10.1038/35055010. [DOI] [PubMed] [Google Scholar]
- Taunton J, Hassig CA, Schreiber SL. A mammalian histone deacetylase related to the yeast transcriptional regulator Rpd3p. Science. 1996;272:408–411. doi: 10.1126/science.272.5260.408. [DOI] [PubMed] [Google Scholar]
- Truss M, Bartsch J, Schulbert A, Hache RJG, Beato M. Hormone induces binding of receptors and transcription factors to a rearranged nucleosome on the MMTV promoter in vivo. EMBO J. 1995;14:1737–1751. doi: 10.1002/j.1460-2075.1995.tb07163.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F, Weaver VM, Petersen OW, Larabell CA, Dedhar S, Briand P, Lupu R, Bissell MJ. Reciprocal interactions between beta1-integrin and epidermal growth factor receptor in three-dimensional basement membrane breast cultures: a different perspective in epithelial biology. Proc Natl Acad Sci USA. 1998;95:14821–14826. doi: 10.1073/pnas.95.25.14821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaver VM, Peterson OW, Wang F, Larabell CA, Briand P, Damsky C, Bissell MJ. Reversion of the malignant phenotype of human breast cells in three-dimensional culture and in vivo by integrin blocking antibodies. J Cell Biol. 1997;137:231–245. doi: 10.1083/jcb.137.1.231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang XJ, Ogryzko VV, Nishikawa J, Howard BH, Nakatani Y. A p300/CBP-associated factor that competes with the adenoviral oncoprotein E1A. Nature. 1996;382:319–324. doi: 10.1038/382319a0. [DOI] [PubMed] [Google Scholar]
- Zhang JS, Wang L, Huang H, Nelson M, Smith DI. Keratin 23 (K23), a novel acidic keratin, is highly induced by histone deacetylase inhibitors during differentiation of pancreatic cancer cells. Genes Chromosomes Cancer. 2001;30:123–135. [PubMed] [Google Scholar]