

Summary
Large variations in the proportion of intragenic deletion in the dystrophin gene have been observed in different populations. Although dystrophin gene deletion was extensively studied all over the world, only few studies were done on Egyptian population and there was no account on the dystrophin gene duplication. In this study, we present our results on the pattern of deletion of the dystrophin gene together with the usage of quantitative polymerase chain reaction (PCR) as a method for duplication analysis within the dystrophin gene in Egyptian patients. Forty one Duchene/Becker muscular dystrophy patients were included in this study. The diagnosis was based on detailed clinical assessment, serum creatine kinase (CK) level, neurophysiologic study and muscle biopsy for histopathological analysis. DNA was extracted from ten milliliter peripheral blood according to basic protocol, and multiplex polymerase chain reaction for dystrophin gene using both Chamberlin and Beggs sets of primers amplifying eighteen exons covering the two main dystrophin gene hot spots. In addition primers from Abbs set were used when it was necessary to check the exon borders. DNA from cases with no detectable deletion was analyzed for dystrophin gene duplication using quantitative PCR technique. We had a percentage of 61.1% deletion which is higher than data from previous Egyptian studies and most of the deletion was localized in the major hotspot region between exons 44 and 52 and we had 5% of the cases with duplication. Our results were compared with previous studies from Egypt and with studies from different populations especially with data recorded in the Middle East and North Africa.
Keywords: Duchenne muscular dystrophy, multiplex PCR, quantitative PCR
Introduction
Duchenne muscular dystrophy (DMD) and its milder allelic form Becker muscular dystrophy (BMD) both exhibit X-linked recessive mode of inheritance and affect more than two thirds of the total number of patients suffering from muscular dystrophy. Males carrying the mutated gene are affected while females become carriers. Due to the extremely large size of the gene (2.4 Mb) 65% of DMD patients exhibit deletions while 6% exhibit duplication within the dystrophin gene (1, 2).
Deletions are mainly clustered in two high frequency deletion regions, one in the 5’ end (centromeric) portion of the gene and the other in the 3’ half of the gene (3, 4). Mutations that disrupt the open reading frame cause DMD resulting in no truncated protein gene product, where those which maintain it cause BMD resulting in abnormal, but partially functional protein product (5, 6). Although the dystrophin gene has been analyzed extensively all over the world, only a few studies have been reported on Egyptian patients (7, 8).
Accordingly, our aim of this study was to extensively study the pattern of deletion and duplication mutation within the dystrophin gene in Egyptian DMD and BMD patients and the use of the quantitative analysis.
Patients and methods
Since 1999, 100 patients with progressive muscular dystrophy have been referred to our laboratory of Neurogentics, Neurology Department Ain Shams University from all over the country. All patients were subjected to full clinical examination, family pedigree, serum CK levels, EMG and muscle biopsy for histopathological analysis.
Inclusion criteria: we selected our patients according to the clinical criteria of DMD/BMD proposed by Emery in 1991. DMD patients were diagnosed according to the age of onset where symptoms are present before the age of 5 years and loss of unassisted ambulation before the age of 12 years. DMD cases were usually differentiated from BMD by the age at which the patient became wheelchair-bound. Some patients showed common features and were categorized as undetermined DMD/BMD. Patients with family history of autosomal recessive inheritance were excluded. Twenty normal cases were included as control group.
Dystrophin gene testing
Genomic DNA was isolated from 10 ml peripheral blood, using the standard protocol. The frequency and distribution of deletions in the dystrophin gene were assessed by multiplex PCR amplification of 18 exons of the dystrophin gene using two sets of primers (9, 10) flanking exons pm-3-4-6-8-12-13-16-17-19-43-44-45-46-47-50-51-52-60 covering the major hot spot of the dystrophin gene. In addition primers from Abbs set(11) were used when it was necessary to check the exon borders (16-41-32-42-34). DNA from the normal male controls served as positive control and reaction without a template DNA as a negative control were included in each set of the PCR reactions. PCR products were subjected to 3% nusceive gel electrophoresis.
Quantitative PCR
All DNA samples which didn’t show deletion in multiplex PCR were subjected for quantitative PCR for duplication detection. Six sets of primers each including 3 primers of Chamberlain and Beggs were used (45-48-19) (17-51-8) (12-44-4) (Pm-3-43) (50-13-6) (47-60-52) with both a normal male and a normal female as positive control. PCR products were run simultaneously in 1.5% nusceive gel electrophoresis, for detecting the duplicated exons.
Immunohistochemical study
All cases with no deletion or duplication were subjected for immunohistochemical study for their muscle biopsy using dystrophin antibodies against: amino terminal, carboxyl terminal and rod domain (NCL-DYS1 between amino acids 1181 and 1388, NCL-DYS2 between 3669 and 3685, and NCL-DYS3 between 321 and 494; Novocastra, UK) to confirm DMD/BMD diagnosis and exclude cases with positive dystrophin protein.
Results
Our study conducted a total of 41 heparinised peripheral blood samples which were obtained from 41 clinically diagnosed unrelated DMD/BMD patients with prior informed consent. Of these, 21 were DMD and 14 BMD patients, and 6 not determined patients. Four DMD cases were wheel chair-bound before 12 years of age. One DMD patient had a brother died at age of 17 years (case number 5). One BMD brother died at age of 19 years (case number 24). One patient with non determined phenotype had a brother died at age of 36 years (case number 37). One non determined patient was still ambulant at age 19 years while his brother was bed ridden at 12 years (case number 36). Table 1 describes the patients & the results.
Table 1. Phenotypic genotypic correlation.
| Serial | Phenotype | Age (ys) | Age of wheelchair bound | X linked family history | Intragenic deletion | Intragenic duplication | Immunohistochemicalanalysis |
| 1 | DMD | 12 | 11 | + | 50 FS | ||
| 2 | DMD | 6 | SA | - | - | - | Dystrophin intact |
| 3 | DMD | 10 | SA | - | - | - | Dystrophin intact |
| 4 | DMD | 14 | SA | + | - | - | -ve dystrophin (DMD) |
| 5 | DMD | 7 | SA | + | 44-48 FS | ||
| 6 | DMD | 12 | 11 | + | 3-6 FS | ||
| 7 | DMD | 4 | SA | - | 44 FS | ||
| 8 | DMD | 10 | SA | - | - | - | Dystrophin intact |
| 9 | DMD | 15 | 11 | - | 51 FS | ||
| 10 | DMD | 10 | SA | - | Pm-4 FS | ||
| 11 | DMD | 8 | SA | - | 51 FS | ||
| 12 | DMD | 12 | 10 | - | 17-19 FS | ||
| 13 | DMD | 7 | SA | - | 45-51 FS | ||
| 14 | DMD | 9 | SA | - | 51 FS | ||
| 15 | DMD | 10 | SA | - | 48-50 FS | ||
| 16 | DMD | 11 | SA | - | 48-52 FS | ||
| 17 | DMD | 17 | SA | - | 50 FS | ||
| 18 | DMD | 7 | SA | - | 44 FS | ||
| 19 | DMD | 9 | SA | - | 51 FS | ||
| 20 | DMD | 10 | SA | + | - | - | Mosaic app (BMD) |
| 21 | DMD | 4 | - | - | - | ||
| 22 | BMD | 10 | SA | - | 51-52 In frame | ||
| 23 | BMD | 21 | SA | - | 45-48 In frame | ||
| 24 | BMD | 14 | SA | + | 48-50 In frame | ||
| 25 | BMD | 18 | SA | - | 48 In frame | ||
| 26 | BMD | 15 | SA | - | - | 52 | |
| 27 | BMD | 14 | SA | - | - | - | |
| 28 | BMD | 18 | SA | - | - | 50 | |
| 29 | BMD | 12 | SA | - | 48 In frame | ||
| 30 | BMD | 16 | SA | + | 45-51Inframe | ||
| 31 | BMD | 21 | SA | - | 44-45 In frame | ||
| 32 | BMD | 27 | SA | - | - | - | Dystrophin intact |
| 33 | BMD | 21 | SA | + | 3-4 In frame | ||
| 34 | BMD | 21 | SA | - | - | - | Dystrophin intact |
| 35 | BMD | 19 | SA | - | - | - | Faint & patchy (BMD) |
| 36 | ND | 17 | SA | + | 44-48 In frame | ||
| 37 | ND | 15 | SA | + | 52-60 In frame | ||
| 38 | ND | 12 | SA | - | - | - | Mosaic app (BMD) |
| 39 | ND | 10 | SA | - | - | - | -ve dystropin (DMD) |
| 40 | ND | 7 | SA | - | - | - | -ve dystrophin (DMD) |
| 41 | ND | 12 | SA | - | - | - | Faint & patchy (BMD) |
ND: not determined phenotype; SA: still ambulant; Pm: promoter exon; FS: frame shift (DMD); In frame (BMD)
The control blood samples were collected from unrelated normal healthy individuals (5 males and 5 females) who had no family history of any muscle disease.
Dystrophin gene deletion was detected in twenty two patients, as shown in (Table 1) and (Fig. 1a-b), while two patients showed duplication, at exon 52 in patient number 26 and exon 50 in patient number 28 as shown in (Table 1) and (Fig. 2). Twelve patients showed neither deletion nor duplication and the dystrophin protein was affected in the immunohistochemical study while five patients had neither deletion nor duplication and the dystrophin protein was intact as shown in (Table 1) and (Fig. 3).
Figure 1.
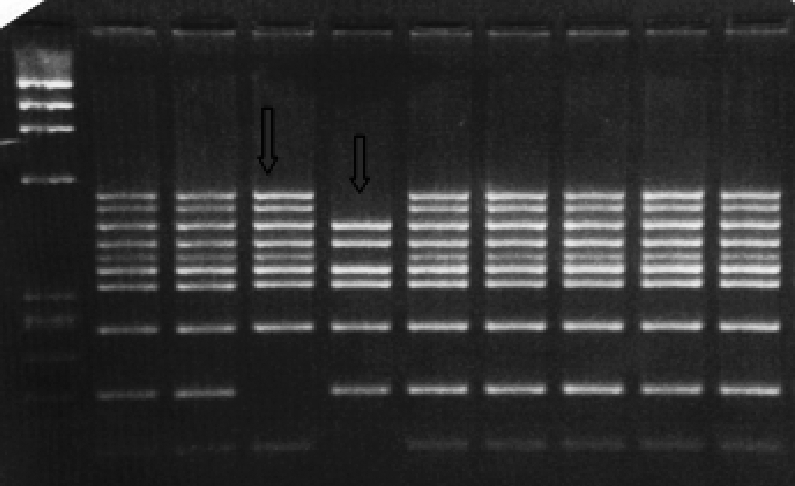

Multiplex PCR amplification of genomic DNA from unrelated male patients using two sets of primers Chamberlain (set A) and Beggs (set b) as described patients and methods and PCR products were electrophoresed on 3% nusceive gels containing ethidium bromide.
Set A: DNA samples amplified using Chamberlain set of primers: Lane 1: DNAmarker, Lane 2-9: DMD\BMD patients, Lane 10: control male. Lanes 4&5 display deletions of one or several exons.
Set B: DNA samples amplified using Beggs set of primers: Lane 1: DNA marker, Lane 2-9: DMD\BMD patients, Lane 10: control male. Lanes 4 & 5: display deletions of one or several exons.
Figure 2.
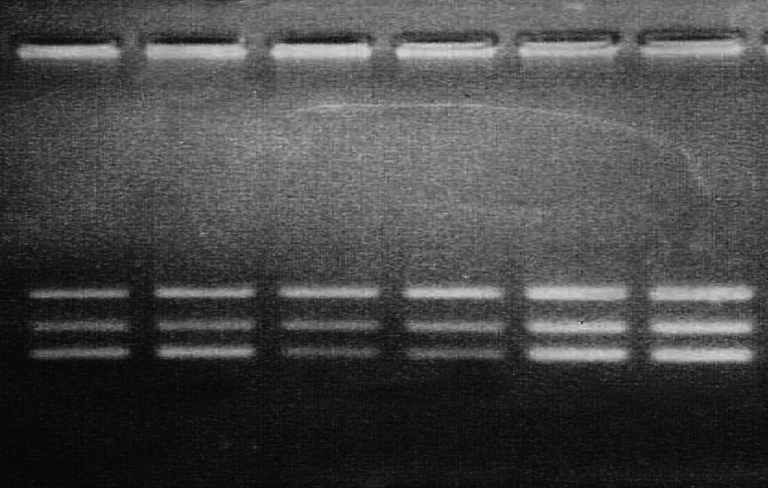
Quantitative PCR amplification of genomic DNA from unrelated male patients, using Beggs set of primers ( Bc) amplifying exons 47–60–52 as described in patients and methods and PCR products were electrophoresed on 1.5%nusceive gels containing ethidium bromide DNA samples amplified using Beggs (Bc) set of primers amplifying exons 47–60–52: Lane 1–2:case 28, male BMD patient showing duplication in band 52, Lane 3–4: control male, Lane 5–6: control female.
Figure 3.
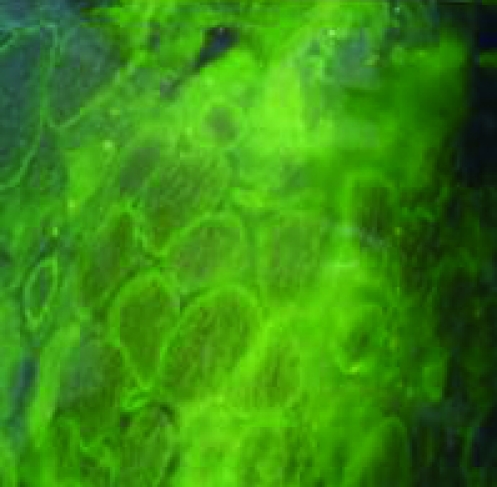
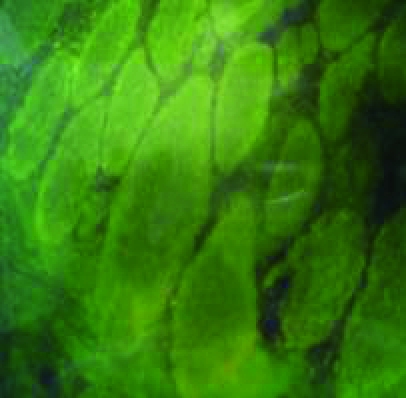
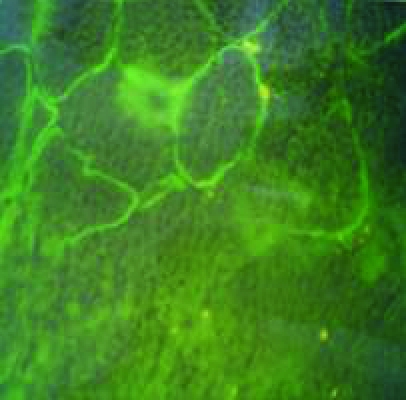

Immunohistochemical study of muscle biopsies using dystrophin antibodies, for cases which had neither deletion nor duplication within the dystrophin gene. a: mosaic appearance of BMD, b: faint dystrophin staining of BMD, c: no staining of DMD, d: normal dystrophin staining.
In order to calculate the percentage of deletion in our study accurately the 5 patients with normal dystrophin were excluded. In the results, 61.1% of patients had dystrophin gene deletion.
The most frequently deleted exon was exon 51. We had one patient with deletion at the promoter site pm and one patient had deletion of exon 60. In our study fourteen patients had deletion in the major hotspot region between exons 44 and 52 (63.6%). Nine patients had the deletion confined to the distal hot spot (exons pm and 19) (40.9%) (Table 2).
Table 2. Frequency distribution of deletions in different exons.
| Numer of duplications | Number of deletions | |||
| Exons tested | Single exon | Multiple exons | Total | |
| Pm | 1 | 1 | ||
| 3 | 3 | 3 | ||
| 4 | 2 | 2 | ||
| 6 | 1 | 1 | ||
| 8 | ||||
| 12 | ||||
| 13 | ||||
| 17 | 1 | 1 | ||
| 19 | 1 | 1 | ||
| 43 | ||||
| 44 | 2 | 3 | 5 | |
| 45 | 6 | 6 | ||
| 46 | 5 | 5 | ||
| 47 | 5 | 5 | ||
| 48 | 8 | 8 | ||
| 49 | 5 | 5 | ||
| 50 | 1 | 2 | 5 | 7 |
| 51 | 4 | 7 | 11 | |
| 52 | 1 | 3 | 3 | |
| 60 | 1 | 1 | ||
| Total | 2 | 8 | 57 | 65 |
The clinical severity of the disorder in patients with deletions was tested against the “reading frame hypothesis”. The border type and the effect of deletion on the translational reading frame. Deletions that created a frame-shift mutation in the protein coding region caused DMD, while inframe deletions caused BMD.
We compared the deletion rate published in previous Egyptian studies, Arab studies and studies from Morocco and Tunisia, Turkey and Greece as described in (Table 3).
Table 3. Comparison of frequency of deletions in different Ethnic groups.
| Country | % of deletion | Exons tested | Methods | Authors |
| Morocco | 59% | 18 exons | Multiplex PCR (I-II) | Aziza El Sbiti et al., 2002 |
| Kuwait | 86% | 25 exons | Multiplex PCR (I-II-III) | Haider et al., 1998 |
| Turkey | 59% | 18 exons | Multiplex PCR (I-II) | Battalogue et al., 1992 |
| Turkey | 52% | 10 exons | Multiplex PCR (II) | Kuseyri et al., 1993 |
| Greece | 63% | 18 exons | Multiplex PCR (I-II) | Florentin t al., 1995 |
| Egypt | 55% | 18 exons | Multiplex PCR (I-II) | Effat et al., 2000 |
| Egypt | 52% | 10 exons | Multiplex PCR (I) | Elhawary et al., 2004 |
| Present study | 61.1% | 25 exons | Multiplex PCR (I-II-III) |
Discussion
The dystrophin gene is a huge and fascinating gene with a complexity in transcriptional regulation, function, and protein – protein interactions that we are only beginning to fully appreciate. The relation between genotype and phenotype is particularly important not only to diagnostic and counseling viewpoints, but also to the understanding of the pathways and mechanisms that regulate expression.
Improvements in knowledge about these features point the way towards a future treatment for this devastating group of disorders and that’s why a proper molecular analysis of the dystrophin gene is considered crucial for the diagnosis of this disorder. Although dystrophin gene mutation was extensively studied all over the world, only few studies were done on Egyptian population and there was no account on the dystrophin gene duplication.
In this study we combined the use of both multiplex PCR analysis for deletion detection with the quantitative PCR study for duplication detection within the dystrophin gene and further the diagnosis of the cases with no deletion or duplication was confirmed using the immunohistochemical analysis.
In our study the rate of deletion within the dystrophin gene was 61.1%, and as regard the pattern of the deletion and its distribution most of the deleted exons were within the central hotspot of the gene between exons 44-52 (72%) and the rest were scattered between exons pm and 19 (28%). We had one case with deletion of exon 60.
Multiple exons deletion was noticed more than single exon affection which is the same as data received from previous Egyptian studies and from populations.
The rate of dystrophin gene deletion was higher than the results obtained from previous Egyptian studies, 52% (7) and 55% (8). However as regard the deletion distribution within the gene, it was fairly the same except for the deletion of exon pm and exon 60.
We can attribute the higher rate of dystrophin gene deletion in our study to the use of both the quantitative PCR analysis for detecting cases with duplication and also the use of immunohistochemical study using dystrophin antibodies to exclude cases with intact dystrophin which confirmed the diagnosis.
Also the usage of more than one set of primers (set I-II) and occasionally (III) was very important for accurate border checking in case of differentiating between cases of DMD & BMD.
The use of quantitative PCR proved to be very beneficial as a method for detecting any duplicated exons within the dystrophin gene we found one case with duplication at exon 52 and another one with duplication at exon 50 which accounts for 5%.
As regard other populations the rate of deletion in our study was close to the results from recent Turkish and Greek studies (12, 16) (Table 3). This may be due to the related ancient history between the Egyptian, Turkish and Greek populations.
Also the use of more than one set of primers in the studies from showed a higher deletion (12, 13) (Table 3).
In case of the study done in Kuwait on different Arabic populations, the percentage was higher than our results (86%) were they used the three sets of primers (14). While the study done on the Moroccan population revealed percentage less than our results (51.3%) (15).
With that we conclude the importance of including both sets A and B primers to cover most of the gene hotspot for more accurate screening of the deletion within the dystrophin gene, together with the duplication detection as the quantitative PCR proved to be beneficial in our study. Immunohistochemical study for further detection of DMD/BMD patients with point mutation, in the cases with no deletion or duplication is necessary for accurate diagnosis.
Acknowledgment
The authors express their sincere thanks to Dr. Hideo Sugita (Honorary President of National Center of Neurology and Psychiatry, NCNP, Japan) for his continuous efforts to support this work, Dr. Eijiro Ozawa (Director General Emeritus, National Institute of Neuroscience, NCNP) for his guidance and encouragement and Dr. Narihiro Minami (NCNP) for his teaching effort.
References
- 1.Koenig M, Hoffman EP, Bertelson CJ, et al. Complete cloning of the Duchenne muscular dystrophy (DMD) cDNA and preliminary genomic organization of the DMD gene in normal and affected individuals. Cell 1987;50:509-11. [DOI] [PubMed] [Google Scholar]
- 2.Mendell JR, Buzin CH, Feng J, et al. Diagnosis of Duchenne dystrophy by enhanced detection of small mutations. Neurology 2001;57:645-50. [DOI] [PubMed] [Google Scholar]
- 3.Den Dunnen JT, Grootscholten PM, Bakker E, et al. Topography of the DMD gene: FIGE and cDNA analysis of 194 cases reveals 115 deletions and 13 duplications. Am J Hum Genet 1989;45:835-47. [PMC free article] [PubMed] [Google Scholar]
- 4.Kim UK, Chae JJ, Lee CC. Molecular diagnosis of Duchenne muscular dystrophy by polymerase chain reaction and micro satellite analysis. Mol Cells 2002;13:385-8. [PubMed] [Google Scholar]
- 5.Monaco AP, Bertelson CJ, Liechti-Gallati S, et al. An explanation for phenotyping differences between patients bearing partial deletions of the Duchenne muscular dystrophy locus. Genomics 1988;2:90. [DOI] [PubMed] [Google Scholar]
- 6.Thanh LT, Nguyen MT, Helliwell TR. Characterization of revertant muscle fibers in Duchenne muscular dystrophy using exon-specific monoclonal antibodies against dystrophin. Am J Hum Genet 1995;56:725-31. [PMC free article] [PubMed] [Google Scholar]
- 7.Elhawary Nasser A, Shawky RM, Hashem N. Frame shift deletion mechanisms in Egyptian Duchenne and Becker muscular dystrophy families. Mol Cells 2004;18:141-9. [PubMed] [Google Scholar]
- 8.Effat LK, El Harouni AA, Amr KS. Screening of dystrophin gene deletions in Egyptian patients with DMD/BMD muscular dystrophies. Dis Markers 2000;16:125-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chamberlain JS, Gibbs RA, Ranier JE, Nguyen PN, Caskey CT. Multiplex PCR for the diagnosis of DMD. In: Innis MA, Gelfand DH, Sninski J, White T, eds. PCR protocols: a guide methods and applications. San Diego: Accademic Press 1990, p. 272-81. [Google Scholar]
- 10.Beggs AH, Koenig M, Boyce FM, et al. Detection of 98% of DMD/BMD gene deletions by polymerase chain reaction. Hum Genet 1990;86:45-8. [DOI] [PubMed] [Google Scholar]
- 11.Abbs S, Yau S, Clark S, Mathew CG. A convenient multiplex PCR system for the detection of dystrophin gene deletions: a comparative analysis with cDNA hybridization shows mistyping by both methods. J Med Genet 1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Battalogue E, Telatar M, Deymeer F, et al. DNA analysis in Turkish Duchenne/Becker muscular dystrophy families. Hum Genet 1992;89:635-9. [DOI] [PubMed] [Google Scholar]
- 13.Kuseyrei GN, Topalogulu H, Yuksel-Apak M. Screening of deletions and RFLP analysis in Turkish DMD/BMD families by PCR. Clin Genet 1993;43:261-6. [DOI] [PubMed] [Google Scholar]
- 14.Haider MZ, Bastaki L, Habib Y. Screening 25 dystrophin gene exons for deletions in Arab children with Duchenne muscular dystrophy. H Heredity 1998;48:61-6. [DOI] [PubMed] [Google Scholar]
- 15.Sbiti A, El Kerch F, Sefiani AA. Analysis of Dystrophin gene deletions by multiplex PCR in Moroccan patients. Journal of biomedicine and biotechnology. J Biomed Biotechnol 2002:158-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Florentine L, Mavrou A, Kekou K, et al. Deletion patterns of Duchenne and Becker muscular dystrophies in Greece. J Med Genet 1995;32:48-51. [DOI] [PMC free article] [PubMed] [Google Scholar]