

Summary
Superantigens (SAGs) interact with host immune receptors to induce a massive release of inflammatory cytokines that can lead to toxic shock syndrome and death. Bacterial SAGs can be classified into five distinct evolutionary groups. Group V SAGs are characterized by the α3-β8 loop, a unique ∼15 amino acid extension that is required for optimal T cell activation. Here, we report the X-ray crystal structures of the Group V SAG staphylococcal enterotoxin K (SEK) alone and in complex with the TCR hVβ5.1 domain. SEK adopts a unique TCR binding orientation relative to other SAG-TCR complexes which results in the α3-β8 loop contacting the apical loop of framework region 4 (FR4), thereby extending the known TCR recognition site of SAGs. These interactions are absolutely required for TCR binding and T cell activation by SEK and dictate the TCR Vβ domain specificity of SEK and other Group V SAGs.
Introduction
Bacterial superantigens (SAGs) comprise a large family of disease-associated proteins that are produced primarily by Staphylococcus aureus and Streptococcus pyogenes1. SAGs function by simultaneously interacting with class II major histocompatibility complex (MHC) and T cell receptor (TCR) molecules on antigen presenting cells and T lymphocytes, respectively2. Contrary to processed antigenic peptides, SAGs bind to MHC molecules outside of their peptide binding grooves and interact only with the Vβ domains of TCRs, resulting in the stimulation of up to 20 percent of the entire T cell population. In this way, SAGs initiate a systemic release of inflammatory cytokines that results in various immune-mediated diseases including a condition known as toxic shock syndrome (TSS) that can ultimately lead to multi-organ failure and death. SAGs have also been classified as Category B Select Agents by the U.S. Centers for Disease Control and Prevention. Despite the intense research efforts that have been directed toward the characterization of SAGs, therapeutics capable of neutralizing SAG-mediated T cell activation in humans are clinically unavailable3; 4.
The binding sites on MHC molecules with which SAGs interact are diverse and can be classified into three distinct groups: (1) a zinc-mediated site on the MHC β subunit that extends over the MHC-bound peptide; (2) a site on the MHC α subunit entirely peripheral to the MHC-bound peptide; and (3) a partially overlapping site on the MHC α subunit that extends over the MHC-bound peptide. Each of these three binding modes has been characterized crystallographically5; 6; 7. Crystal structures of several SAGs with their respective TCR Vβ ligands have revealed that SAG-Vβ interactions are also structurally diverse8; 9; 10. These structures have allowed for the construction of models of the ternary MHC-SAG-TCR T cell signaling complexes, which due to structural diversity in both SAG-MHC and SAG-TCR interactions exhibit substantially heterogeneous supramolecular architectures, all of which nonetheless allow for efficient T cell activation.
More than 30 distinct SAG serotypes from both staphylococci and streptococci belong to the pyrogenic toxin SAG family11. Although they are all believed to share a conserved tertiary structure, five distinct evolutionary Groups (I through V) have been proposed for these toxins due to their pyhlogenetic relationships1 and key differences in how the characterized representatives for each Group engage their host receptors12.
Within this classification, toxic shock syndrome toxin-1 (TSST-1) from S. aureus is the only Group I SAG and is also unique in that it binds MHC through an N-terminal, low-affinity binding domain that is peptide-dependent7; 13 and engages the TCR Vβ through two independent regions within both complementarity determining region (CDR) 2 and framework region (FR) 39, that together exhibit positive cooperative binding14. Group II contains both staphylococcal and streptococcal SAGs (including SEB, SEC and SpeA) that also bind the MHC α-chain through an N-terminal, low-affinity binding domain; however, in contrast to Group I, this binding is peptide-independent6. Group II SAGs engage the TCR Vβ through mostly conformation-dependent mechanisms that are largely independent of specific Vβ amino acid side chains8; 10; 15. Group III SAGs contain only staphylococcal SAGs (such as SEA), and these toxins are able to crosslink MHC molecules16; 17 through a low-affinity site similar to Group II18, as well as a high-affinity, zinc-dependent MHC binding interface located within the β-grasp domain of the SAG19. There is currently little available information regarding how Group III SAgs engage Vβ. Group IV SAGs are restricted to only streptococcal members (such as SpeC), and these toxins contain a high-affinity MHC binding domain similar to that of Group III20. The structure of SpeC in complex with human Vβ2.1 revealed that this SAG engages each of the TCR Vβ hypervariable loops, including each of CDR loops 1 through 3 and HV421. Based on these collective characteristics, it is clear that the SAG evolutionary Groups I through IV each display key differences in how they engage their host receptors.
Group V SAGs (including SpeI, SEI and SEK) are the least characterized of all of these toxins. A crystal structure of SEI in complex with HLA-DR1 showed that this group of SAGs binds to class II pMHC molecules in a similar fashion as do Group IV SAGs22. A key feature of Group V SAgs is the presence of a loop extension between the third α-helix and the eighth β-sheet (hereafter referred to as the α3-β8 loop). This ∼15-amino acid extension is not found in the other SAG Groups1 and is not involved in pMHC interactions. Instead, we have recently shown that the α3-β8 loop of SpeI is functionally important for the activation of T cells12. In particular, the presence of several Gly residues within this loop are necessary for optimal T cell stimulation, suggesting that flexibility and/or the ability to adopt an otherwise unattainable conformation are key structural features of this loop.
In order to further define the functional relevance of the α3-β8 loop unique to Group V SAGs, we have undertaken structural, energetic and functional analyses of SEK binding to one of its known TCR ligands, the human Vβ domain 5.1 (hVβ5.1). Here, we report the X-ray crystal structures of SEK alone, as well as in complex with hVβ5.1. These structures show that the loop is not altered in conformation upon TCR binding. Residues within the α3-β8 loop make intermolecular contacts with hVβ5.1 and extend the known TCR Vβ domain binding site for SAGs into the FR4 region. Using surface plasmon resonance (SPR) molecular interaction analysis and functional assays employing an engineered hVβ5.1+ Jurkat T cell line, we document the functional importance of these contact residues. The SEK-hVβ5.1 crystal structure also shows that SEK adopts a distinct orientation in binding the TCR Vβ domain relative to other SAG-TCR interactions. As a result, the majority of the interface comes from a contiguous stretch of residues comprising the SEK N-terminus and α1 α-helix.
Results
SEK is structurally homologous to other Group V superantigens
We have determined the X-ray crystal structure of SEK in its unbound state (Figure 1A). The crystal was grown with a single-site mutant of SEK, SEK-C73S, in which the sole cysteine residue in SEK was mutated to serine in order to reduce disulfide-linked aggregation and promote crystallization. This residue lies outside of the molecular interface formed with hVβ5.1 (see below) and does not affect SEK structure or function in any detectable way. The structure was solved by molecular replacement methods using the structure of SpeI as a model12. The structure has been refined to a resolution of 1.56 Å. Data collection, processing and refinement statistics are shown in Table 1.
Figure 1.
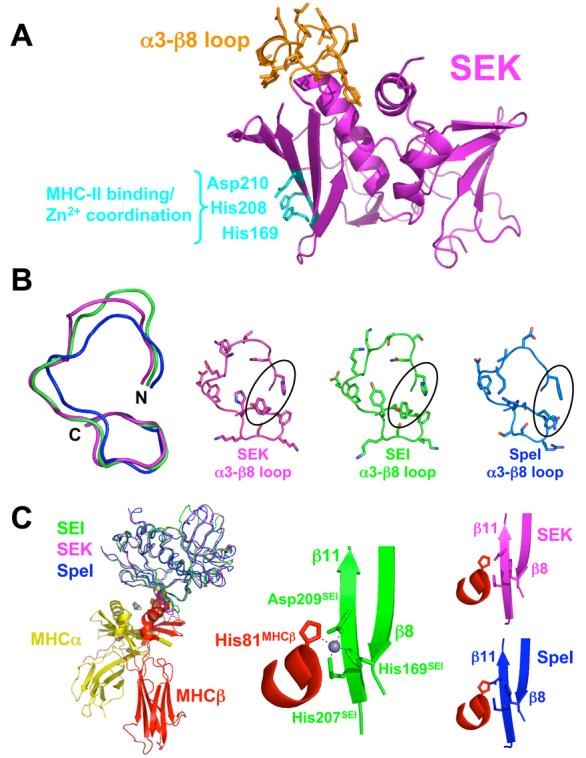
Structural similarity of SEK and other Group V bacterial superantigens. (A) Structure of SEK in the unbound state. The α3-β8 loop is in orange and the residues responsible for Zn2+ coordination and MHC class II binding are in cyan. (B) Structural comparison of the α3-β8 loop domains of SEK, SEI and SpeI. Superposition of the main chains of the α3-β8 loops (left panel). Molecular detail of side chain positions in the α3-β8 loops (right panels). SEK, SEI and SpeI are in magenta, green and blue, respectively. The residues in SEK that interact with TCR, His142 and Tyr158, as well as corresponding residues in SEI and SpeI are encircled. (C) Structural comparison of the MHC binding site of SEI and the putative MHC binding sites of SEK and SpeI. Superposition of SEK and SpeI with SEI from the SEI-MHC class II crystal structure22 (left panel). Close-up view of the Zn2+ coordination between SEI residues His169, His207 and Asp209 and the MHC β subunit residue His81 (middle panel). Close-up views of the putative SAG-MHC interface formed by the superposed SEK and SpeI structures (right panels). The MHC α subunit is in yellow, MHC β subunit is in red, zinc ion in gray, and the SAG colors are as in panel B.
Table 1.
Structure Determination and Refinement Statistics
| Data Collection and processing | ||
|---|---|---|
| SEK | SEK–hVβ5.1 | |
| Space group | P21 | P212121 |
| Resolution (Å) | 1.56 | 2.40 |
| Molecules/asymmetric unit | 2 SEK | 1 SEK/1 hVβ5.1 |
| Observations | 226,878 | 176,078 |
| Unique reflections | 63,074 | 21,526 |
| Completeness (%)a | 99.8 (99.0) | 99.9 (100.0) |
| Redundancya | 3.6 (2.8) | 8.2 (7.9) |
| Rsym (%)b | 8.6 (17.9) | 8.7 (34.1) |
| Mean I/σ(I) | 30.9 (5.9) | 25.9 (6.1) |
| Refinement | ||
| Rcryst (%)c | 16.3 | 21.9 |
| Rfree (%)d | 20.2 | 24.0 |
| Protein residues | 434 | 462 |
| Average B values | ||
| SEK | 9.9 | 32.8 |
| hVβ5.1 | 37.7 | |
| Water molecules | 655 | 152 |
| Ramachandran plot | ||
| Core (%) | 87.0 | 84.5 |
| Allowed (%) | 12.2 | 13.5 |
| Generous (%) | 0.8 | 0.7 |
| Disallowed (%) | 0 | 1.2e |
| RMS deviations from ideality | ||
| Bonds (Å) | 0.008 | 0.007 |
| Angles (°) | 1.23 | 1.69 |
| Molecular Characterization | ||
| Intermolecular contacts | ||
| Hydrogen Bonds | 14 | |
| van der Waals interactions | 75 | |
| Buried surface areas | ||
| ΔASAtotal (Å) | 1572 | |
| ΔASApolar (Å) | 920 | |
| ΔASAapolar (Å) | 658 | |
| ΔASAapolar/ΔASApolar (%) | 42 | |
| Shape complementarity | ||
| SC | 0.70 | |
Values in parentheses are for the highest resolution shell (1.62-1.56 Å for SEK; 2.49-2.40 Å for SEK-hVβ5.1).
Rsym = Σ|((Ihkl − I(hkl))|/(ΣIhkl), where I(hkl) is the mean intensity of all reflections equivalent to hkl by symmetry
Rcryst = Σ∥FO| − |FC|/Σ|FO∥, where FC is the calculated structure factor
Rfree is calculated as Rcryst using 5.1% (SEK) and 4.7% (SEK-hVβ5.1) of the reflections chosen randomly and omitted from the refinement calculations.
Residues in the disallowed region include: Ser134hVβ5.1; Ala136hVβ5.1; Thr202hVβ5.1; Glu225hVβ5.1; and Ser73SEK;
The structure of SEK conforms to the classical bacterial SAG fold that is comprised of N-terminal β-barrel and C-terminal β-grasp domains. All residues in the structure exhibited unambiguous electron density, including the α3-β8 loop. The conformation of this loop is very similar to the analogous loops of SEI and SpeI (Figure 1B), two other Group V SAGs for which crystal structures have been determined. The structure of the putative MHC binding site on SEK is also structurally homologous to that of SEI (Figure 1C), and thus, SEK likely binds to MHC in a similar fashion as does the latter 22. The zinc-dependency of SEK binding to the high affinity MHC class II binding site was confirmed by an aggregation assay using LG-2 antigen presenting cells (data not shown).
The α3-β8 loop does not change conformation upon TCR binding
We have also determined the X-ray crystal structure of SEK bound to its TCR Vβ domain ligand hVβ5.1 (Figure 2A). This structure was solved by molecular replacement methods using the structures of SEK in the unbound state and of the β chain of the 3A6 TCR 23 and refined to a resolution of 2.4 Å. Data collection, processing and refinement statistics for this structure are shown in Table 1. Characterization of the SEK-hVβ5.1 molecular interface reveals that it is more akin to those SAG-TCR complexes formed by Group I (i.e., TSST-1) and Group IV (i.e., SpeC) SAGs than by Group II (i.e., SEB) SAGs, in terms of intermolecular contacts, buried suraface area and shape complementarity (Table 1).
Figure 2.
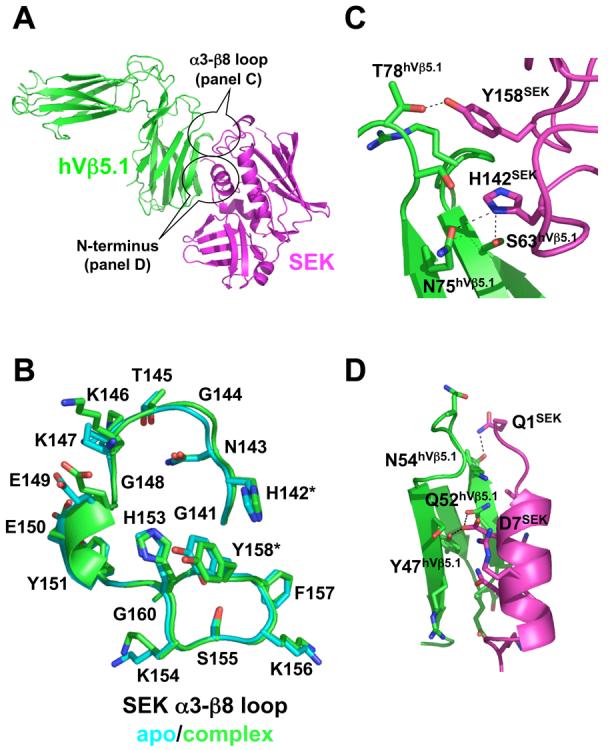
Structure of the SEK-hVβ5.1 complex. (A) Cartoon representation of the interaction between SEK and hVβ5.1. SEK is in magenta, hVβ5.1 is in green. The two regions of SEK, the α3-β8 loop and the N-terminus, that contact the TCR are encircled. (B) Comparison of SEK α3-β8 loop structures in the unbound (cyan) and hVβ5.1-bound (green) crystal structures. The residues that make intermolecular contacts with hVβ5.1, His142 and Tyr158, are demarcated by asterisks. (C) Close-up view of the interface formed by the α3-β8 loop of SEK and the FR3 and FR4 loops of hVβ5.1. (D) Close-up view of the interface formed by the N-terminus of SEK and the CDR2 loop of hVβ5.1. In both panels C and D, the side chains of only those residues that make intermolecular contacts are shown and side chain to side chain hydrogen bonds are shown as dashed lines.
Our recent structural and functional analysis of the Group V SAG SpeI 12 showed that the α3-β8 loop of this SAG, and by analogy those of other Group V SAGs, was necessary for optimal T cell activation. In the unbound structure of SpeI, the α3-β8 loop is well-ordered, built into unambiguous electron density and exhibits relatively low temperature factors, but is also glycine-rich. Thus, we proposed that one possible mechanism by which this loop contributes to TCR engagement was through conformational changes induced upon binding. A comparison of the α3-β8 loop in SEK when bound or not to hVβ5.1, however, shows that this loop is conformationally static (Figure 2B). There are essentially no main chain or side chain movements between the unbound and bound states of SEK. Thus, the α3-β8 loop of SEK is pre-ordered in a conformation that is amenable for TCR engagement.
The TCR recognition site for superantigens is extended by the α3-β8 loop
Two distinct regions of SEK, the α3-β8 loop and the N-terminus, form intermolecular contacts with hVβ5.1. Only two residues within the α3-β8 loop, His142 and Tyr158 contact TCR, and each of these exhibits both hydrogen bonding and van der Waals interactions with residues in hVβ5.1 (Figure 2C). Surprisingly, these contacts are made to residues not only in the apical loop of FR3, which we predicted from our previous analysis of SpeI12, but also with residues in the apical loop of FR4. This region of the TCR has never before been implicated in SAG binding. Additionally, a contiguous stretch of residues forming the N-terminus of SEK at positions 1-16, make a variety of hydrogen bonding and van der Waals interactions with hVβ5.1 exclusively from the CDR2 loop (Figure 2D).
SEK adopts a unique TCR binding orientation relative to other SAG-TCR complexes
SEK adopts a unique orientation when engaging the TCR Vβ domain relative to representative SAGs from Groups I, II and IV (Figure 3A). Like TSST-1, SEK, due to the extension by the α3-β8 loop, is positioned higher (i.e., closer to the Cβ domain) on the Vβ domain than are SEB and SpeC (Figure 3A, upper panels). Despite its higher position on the Vβ domain, SEK, is aligned in the same plane as are SEB and SpeC, whereas TSST-1 is rotated approximately 45 degrees (Figure 3A, lower panels).
Figure 3.
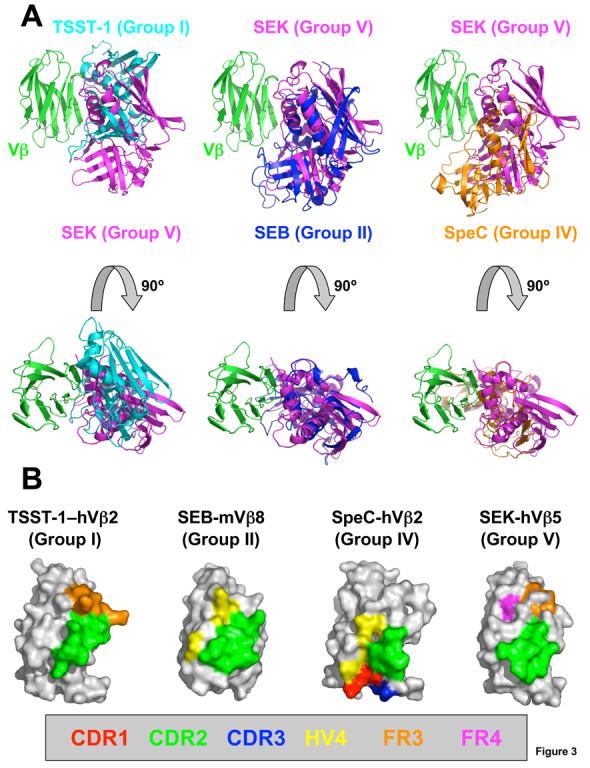
Diverse TCR engagement by bacterial SAGs. (A) Superposition of the SEK-hVβ5.1 crystal structure with the TSST-1–hVβ2.1 (left panels), SEB-mVβ8.2 (middle panels) and SpeC-hVβ2.1 (right panels) complexes. The hVβ2.1 and mVβ8.2 molecules have been removed for clarity. SEK is in magenta, TSST-1 is in cyan, SEB is in blue, SpeC is in orange and Vβ is in green. (B) TCR Vβ domain molecular surface buried by various SAGs. Hypervariable and framework region surface residues buried in the interface formed by TSST-1, SEB, SpeC and SEK are color-coded as follows: CDR1 (red); CDR2 (green); CDR3 (blue); HV4 (yellow); FR3 (orange); and FR4 (magenta). The total buried surface areas for each SAG-TCR complex are: 1917 Å2 (TSST-1–hVβ2.1)9; 1268 Å2 (SEB-mVβ8.2)10; 1818 Å2 (SpeC-hVβ2.1)8; and 1572 Å2 (SEK-hVβ5.1).
The distinct orientations with which each of these representative SAGs from Groups I, II, IV and V engage the TCR Vβ domain result in unique patterns of hypervariable and framework region surfaces that are buried (Figure 3B). Binding to the TCR Vβ CDR2 loop is a requirement for all bacterial SAGs, and the proportion of the SAG-TCR interface that is contributed by the CDR2 loop is invariably the greatest in any SAG-TCR complex, relative to any other single hypervariable or framework region. In this way, SEK is similar to other SAGs. When the Group V SpeI crystal structure is superimposed onto the SEK-hVβ5.1 crystal structure, SpeI residues Asn13, Ser77 and Arg198, all of which were shown to be important for human T cell proliferation12, all surround the CDR2 loop.
Involvement of Vβ domain regions beyond the CDR2 loop, however, plays a significant role in the TCR Vβ domain specificity and cross-reactivity of a SAG (Figure 3B)14; 24; 25. SEK and TSST-1, due to their higher position on the Vβ domain, engage one or more framework region apical loops, at the expense of contacting the hypervariable elements. SEK buries significant molecular surface belonging to both the FR3 and FR4, while TSST-1 contacts only residues from FR3. The lower relative positions of SEB and SpeC on the Vβ domain result in their engagement of hypervariable elements at the expense of binding the apical loops of the framework regions. SEB buries molecular surface belonging to HV4, while SpeC contacts residues from CDR1, CDR3 and HV4.
Contact residues in the α3-β8 loop are required for TCR binding
Using surface plasmon resonance (SPR) analysis, we determined the affinity of the wild type SEK-hVβ5.1 complex to be 6 μM (Figure 4A). This is within the range of affinities (10−7 – 10−4 M) of all known SAG-TCR interactions.
Figure 4.
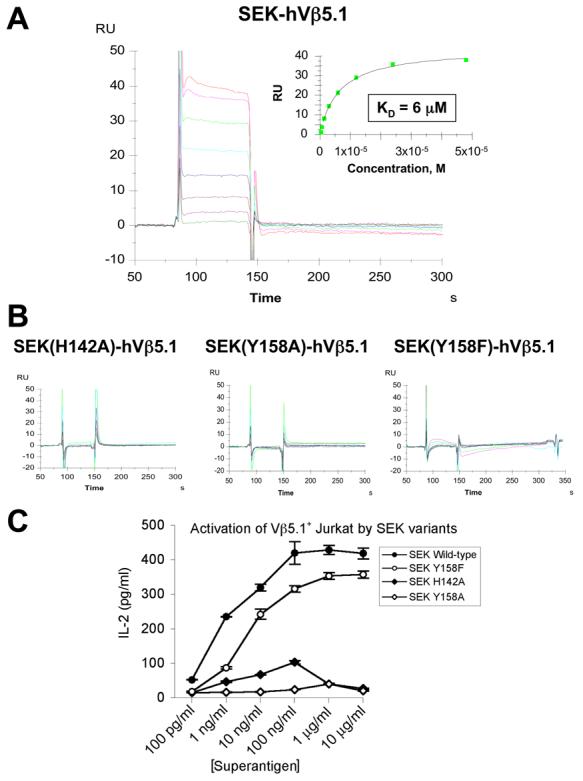
Analysis of SEK-hVβ5.1 binding and SEK-mediated T cell activation. (A) SPR analysis of the wild type SEK-hVβ5.1 interaction. Non-linear regression analysis of maximal responses versus concentration is shown in the inset plot. (B) SPR analyses of hVβ5.1 interactions with the SEK(H142A), SEK(Y158A) and SEK(Y158F) mutants. (C) IL-2 secretion by eJRT3-5.1 cells incubated with various concentrations of wild type SEK and the SEK(H142A), SEK(Y158A) and SEK(Y158F) mutants presented by LG-2 cells. Data shown are the average ± SEM.
In order to determine whether the intermolecular contacts made by residues in the α3-β8 loop that we observed in our SEK-hVβ5.1 crystal structure are energetically significant, we analyzed the capacity of SEK variants with mutations at either His142 or Tyr158 to bind hVβ5.1 by SPR analysis. His 142 makes three hydrogen bonds (one of which is to the hVβ5.1 main chain) and three van der Waals contacts with residues in both the apical loops of FR3 and FR4 of hVβ5.1 (Figure 2C). An alanine mutation at this position completely abrogated binding to hVβ5.1 (Figure 4B, left panel). Tyr158 makes a single hydrogen bond and ten van der Waals contacts to residues in the apical loop of the hVβ5.1 FR4 (Figure 2C). An alanine mutation at this position also resulted in no hVβ5.1 binding (Figure 4B, middle panel). A phenylalanine mutation of Tyr158, which removes the hydrogen bond formed between the hydroxyl groups of Tyr158SEK and Thr78hVβ5.1, as well as a single van der Waals contact, showed minimal SPR responses at higher concentrations (up to 50 μM, ∼10-fold the wild type KD; Figure 4B, right panel), indicating dramatically reduced binding.
The α3-β8 loop contact residues are functionally critical
In order to verify the functional importance of the α3-β8 loop for the activation of T cells, we performed stimulation assays using an engineered hVβ5.1+ Jurkat T cell line with the wild type and mutant SEK molecules. The functional responses (Figure 4C) for wild type SEK, SEK(H142A), SEK(Y158A) and SEK(Y158F) all corroborate the results of the SPR binding analysis. As expected, wild type SEK produced significant dose-dependent secretion of interleukin-2 (IL-2) starting at concentrations as low as 100 pg/ml. T cell stimulation by both of the alanine mutants of His142 and Tyr158 resulted in very little IL-2 production, even at concentrations as high as 10 μg/ml. In accordance with the reduced, but not fully abrogated binding of the SEK(Y158F), T cell stimulation by this mutant resulted in significant IL-2 production at concentrations as low as 1 ng/ml, albeit diminished relative to wild type SEK.
The SEK-dependent T cell signaling complex
In order to activate T cells, SAGs must bind simultaneously to both MHC class II molecules and TCRs. By superimposing SAG-MHC and SAG-TCR X-ray crystal structures, models of MHC-SAG-TCR ternary signaling complexes have been generated for TSST-17; 9, SEB6; 10 and SpeC5; 8, representative SAGs for Group I, II and IV SAGs, respectively (Figure 5A-C). Using the SEK and SEK-hVβ5.1 complex structures reported here, we have generated a model of the MHC-SEK-TCR T cell signaling complex (Figure 5D), which is likely representative of all Group V SAGs. The supramolecular architecture of this SEK-dependent ternary complex is very similar to that made by SpeC. Because SEK engages the TCR Vβ domain such that it can bind to the FR apical loops, while SpeC engages the Vβ domain such that it binds all of the CDR loops, the angle formed between the axis of the MHC-displayed peptide and the long axis the TCR Vβ domains is more acute in the SEK- versus SpeC-dependent complexes (i.e., these axes are approximately 25 degrees closer to parallel in the SEK-dependent ternary complex than in the SpeC-dependent ternary complex).
Figure 5.
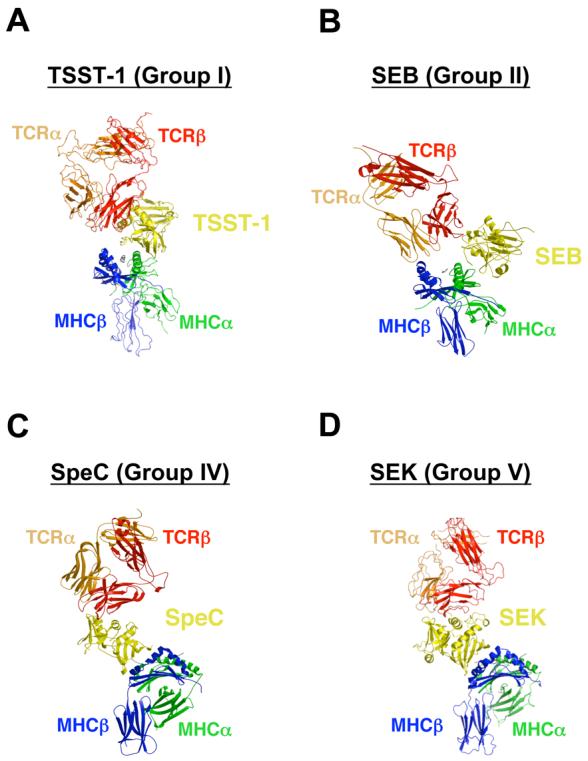
MHC-SAG-TCR ternary signaling complexes mediated by (A) TSST-1, (B) SEB, (C) SpeC, and (D) SEK. Colors are as follows: MHC α subunit, green; MHC β subunit, blue; antigenic peptide, gray; TCR α chain, orange; TCR β chain, red; SAGs, yellow. For clarity, the MHC-SAG-TCR complexes mediated by SpeC (panel C) and SEK (panel D) are rotated approximately 90 degrees clockwise about the vertical axis of the page relative to those mediated by TSST-1 (panel A) and SEB (panel B).
Discussion
The activation of T cells by SAGs is dependent on both their interactions with MHC class II and TCR molecules. The manner in which SAGs bind to MHC has been investigated in great detail5; 6; 7 and it is now clear that these interactions are restricted to three distinct binding modes: (1) a zinc-mediated site on the MHC β subunit that extends over the antigenic peptide; (2) a site on the MHC α subunit entirely peripheral to the displayed peptide; and (3) a partially overlapping site on the MHC α subunit that extends over the MHC-bound peptide.
There appears to be even greater diversity in the way that SAGs engage TCR (Figure 3A). While each SAG-TCR complex investigated by structural analysis to date involves extensive SAG contacts with the CDR2 loop of the TCR, representative SAGs from each SAG Group contact distinct combinations of hypervariable and framework regions contribute to these SAG-TCR interfaces. Moreover, the interactions outside of the CDR2 loop have been shown to be important for the TCR specificity of individual SAGs14; 24; 25.
The structural analysis of SEK (Figure 1) and its complex with hVβ5.1 (Figure 2) presented here reveal that SEK is no different in this sense. As a representative of Group V SAGs, SEK engages its TCR Vβ domain ligand in a manner that is distinct from representatives of Group I, II, and IV SAGs (Figure 3). In particular, SEK makes intermolecular contacts with residues in the apical loops of both FR3 and FR4, the latter of which has never before been observed to play a role in SAG-TCR interactions and was heretofore not known to have any effect on the SAG-mediated activation of T cells.
The molecular surface of hVβ5.1 belonging to FR4 that is buried by SEK makes numerous hydrogen bonds and van der Waals interactions with two residues, His 142 and Tyr158, in the α3-β8 loop of SEK. Our previous studies 12 had implicated the α3-β8 loop, which is unique to Group V SAGs, as being required for the efficient activation of T cells. Indeed, binding and functional analyses of wild type and mutant SEK presented here (Figure 4) show that each of these two residues in the SEK α3-β8 loop is absolutely critical for stimulation of hVβ5.1+ T cells.
Beyond being required for the SEK-hVβ5.1 interaction, contacts between Group V α3-β8 loop and Vβ FR4 residues appear to dictate the TCR Vβ specificity of these SAGs. The α3-β8 loops of SEK and SEI are essentially identical in conformation and both of these SAGs have the same Vβ-contacting His and Tyr residues at positions 142 and 158, respectively (Figure 1B). SEI, like SEK, predominantly stimulates hVβ5.1+ T cells 26. SpeI, conversely, does not stimulate hVβ5.1+ T cells, even though this Group V SAG is highly homologous to both SEK and SEI12. While the positions of SpeI residues that are analogous to those at positions 142 and 158 of SEK are superimposable, these two residues are Phe and Gln in SpeI (Figure 1B).
This extension of the TCR recognition site to FR4 through critical contacts with SEK α3-β8 loop residues, provides essential data with which to refine our understanding of SAG-TCR specificity and cross-reactivity. Crystal structures of SEB, SpeA and SpeC with their TCR ligands8; 10, have shown that TCR increased specificity for these SAGs correlates with the amount of buried surface, the number of side chain to side chain hydrogen bonds, the engagement of increasing numbers of CDR loops and the targeting of CDR loops that have incorporated non-canonical residue insertions. The recent crystal structure of TSST-1, the most specific SAG known, bound to its sole TCR ligand, hVβ2.1, showed that TSST-1 derives its specificity primarily by targeting unique residues within the apical loop of FR3, at the expense of contacting any other CDR loop besides CDR29.
Likewise, SEK, which activates T cells bearing primarily hVβ5.1, as well as hVβ5.2 and hVβ6.727, appears to derive its specificity, at least in part, through interactions with residues in FR3 and FR4, namely at positions 63 and 75, with which His142 forms side chain to side chain hydrogen bonds (Figure 2C). These include a serine residue at position 63 in all three of these Vβ domains and either an asparagine residue in hVβ5.1 and hVβ5.2 or a threonine residue in hVβ6.7, which can presumably satisfy the same hydrogen bonding constraints observed in the SEK-hVβ5.1 complex structure. The SEK-Vβ domain interfaces are extensive (1572 Å2 for the SEK-hVβ5.1 complex), however, and the numerous other residues within these interfaces are likely to contribute significantly to both binding and specificity. Only an extensive mutagenesis, structural and energetic analysis would allow for the determination of the molecular basis of fine specificity of SEK-Vβ domain interactions, such as we have done for TSST-1–Vβ and SpeC-Vβ interactions9; 14; 25.
TCR hVβ5 and hVβ6 domains, and especially hVβ5.1, have been shown to be overrepresented in patients with Crohn's disease, a severe inflammatory bowel syndrome28. These TCRs are also overrepresented in juvenile arthritis and periodontitis29; 30. Beyond those SAGs that belong to Group V, only SEE, a Group III SAG, stimulates hVβ5.1+ T cells. However, with no α3-β8 loop the orientation of SEE on hVβ5.1 is likely to be markedly different than that of SEK, and the interaction is unlikely to involve the FR3 and FR4 apical loops of the Vβ domain. No other SAGs are known to stimulate hVβ5.2+ or hVβ6.7+ T cells.
The unique binding orientation, relative to other SAG-TCR complexes, by which SEK interacts with hVβ5.1 has two important ramifications for T cell activation. First, it results in several regions of both SEK and hVβ5.1 that are not involved in any other SAG-TCR complex, including the N-terminus and the α3-β8 loop of the former and residues in FR4 of the latter, forming the majority of the molecular interface. Second, the MHC-SAG-TCR signaling complex mediated by SEK, although more similar to that of SpeC than of other SAGs due to their common interaction with MHC class II molecules, exhibits a different relative juxtaposition of the TCR and MHC molecules. The recent discovery of a Gα11-dependent T cell signaling pathway used by SAGs that is distinct from the Lck-dependent pathway used by antigenic peptide-MHC complexes suggests that SAGs can use a G protein-coupled receptor as a co-receptor on T cells31. Thus, the relative orientations of SAG, MHC and TCR in SAG-mediated signaling complexes that we observe in the MHC-SEK-TCR ternary complex, may have significant functional consequences for the engagement of this co-receptor and subsequent triggering of this alternative and SAG-specific T cell signaling pathway.
Materials and Methods
Protein production
Like SpeI, SEK contains a single cysteine residue that promoted a monomer-dimer equilibrium at protein concentrations needed for crystallization. It had been previously observed for SpeI that mutation of its cysteine residue to a serine resulted in the growth of diffraction-quality crystals12, and thus, an identical strategy was followed for SEK mutagenesis, expression, purification and crystallization. Briefly, a C73S mutant of SEK was made following the QuikChange site-directed mutagenesis protocol (Stratagene). SEK-C73S was cloned into a modified pET28a expression vector (Novagen), in which the enterokinase cleavage site had been replaced by a tobacco etch virus (TEV) cleavage site leaving the N-terminal His6 purification tag intact, expressed in E. coli BL21(DE3) cells, and purified on a nickel affinity column. Recombinant, auto-inactivation resistant His7::TEV was used to cleave the His6::SEK-C73S fusion protein, and the cleaved purification tag and the His7::TEV were removed by an additional passage over the nickel affinity column. The resulting product, which contained no extra residues on either terminus, was further purified by size exclusion chromatography in 10 mM Tris, pH 7.5, 25 mM NaCl. For SEK-hVβ5.1 co-crystallization and binding analysis, the SEK-C73S mutant described above was used and the TCR β chain from the 3A6 TCR (hVβ5.1) was expressed, refolded in vitro and purified as described previously23.
Crystallography
Crystals of SEK were grown by sitting drop vapor diffusion in 10% polyethylene glycol (PEG) 6000, 2 M NaCl at room temperature. A complete data set to a nominal resolution of 1.56 Å was collected at CHESS, beam line A-1 and processed using HKL200032. A molecular replacement solution for SEK was found using the program PHASER33 with SpeI (PDB accession code 2ICI) as a search model.
For SEK-hVβ5.1 co-crystallization, the two proteins were mixed in a 1:1 molar ratio at ∼10 mg/ml total protein concentration in 10 mM Tris, pH 7.5, 25 mM NaCl, but due to the relatively low affinity of the complex no further purification of the complex was performed prior to crystallization. Co-crystals were grown by hanging drop vapor diffusion in 6-10 % PEG 20,000, 0.1 M MES, pH 6.0 – 7.0 at room temperature. A complete data set to a nominal resolution of 2.40 Å was collected at NSLS, beam line X6A and processed using HKL200032. A molecular replacement solution for the SEKhVβ5.1 complex was found using the program PHASER 33 with the partially refined SEK structure and the β chain molecule of the 3A6 TCR (PDB accession code 1ZGL) as search models.
The SEK and SEK-hVβ5.1 structures were refined using REFMAC34 and CNS35, respectively, interspersed with manual model building in COOT36. Crystallographic data collection, processing and refinement statistics are listed in Table 1. The atomic coordinates and structure factors have been deposited in the Protein Data Bank (PDB ID codes: 2NTT for SEK; 2NTS for SEK-hVβ5.1).
Binding analysis
Affinity measurements were made using a Biacore 3000 surface plasmon resonance instrument (Biacore, Piscataway, NJ). Wild type SEK, or SEK mutants were immobilized to CM5 sensor chips by standard amine coupling to an immobilization density of approximately 500 response units. TSST-1 was immobilized at an equivalent density as a negative control surface. A concentration gradient of hVβ5.1 was injected over all surfaces and the net maximal responses were recorded for steady-state analysis. Affinity values were derived by non-equilibrium regression analysis using the BiaEvaluation 4.1 software (Biacore, Piscataway, NJ).
Functional analysis
To allow for a functional readout for the SEK mutants specific for the hVβ5.1 chain used in the SEK-hVβ5.1 complex, the Jurkat T cell line JRT3-T3.5 (ATCC) was used. This cell line lacks the endogenous hVβ8.1 chain present in wild type Jurkat T cells37. Recombinant hVβ5.1 was constructed essentially as described 25 where the leader and transmembrane DNA sequences of human hVβ8.1 38 were attached to the 5' and 3' ends of hVβ5.1 cDNA, respectively, to allow for surface expression and pairing of hVβ5.1 with the endogenous hVα1. These modifications to the hVβ5.1 cDNA were performed by sequential megaprimer PCR reactions and the complete hVβ5.1 cDNA was cloned into the unique KpnI and BamHI sites in pBIG2i39. Ten μg of linearized pBIG2i::hVβ5.1 was electroporated into 5 × 106 JRT3-T3.5 cells using 300V and 950μF and stable transfectants were selected using increasing concentrations of hygromycin B. JRT3-T3.5 transfected with pBIG2i alone was used as a negative control. Surface expression of hVβ5.1 paired with endogenous Vα1 was confirmed using FACS analysis with PE conjugated anti-TCR antibody (eBioscience). Transfected JRT3-T3.5 cells (105) were incubated with LG-2 cells (2 × 104) to provide MHC class II, with different concentrations of the various SEK variants in a non-adherent 96-well plate for 16-18 hours. Activation was monitored using ELISA for human IL-2 (BD biosciences).
For aggregation assays, LG-2 cells (1 × 105 cells/ml) were incubated with 1 μg/ml of recombinant SEK proteins in a non-adherent, flat-bottom 96-well plates and monitored for cell aggregation for 7 h. Wells were exposed to 1 μg/ml superantigen and the percentage of cells in aggregates was assessed at 1 h intervals for 6 h. The control consisted of LG-2 cells not exposed to superantigen. The zinc-dependency of the high-affinity MHC class II binding site was tested by the addition of 1mM EDTA at 2 h and then the subsequent addition of 2 mM ZnSO4 at 4 h. The number of cells in aggregates was assessed as a percentage of the total number of cells in the field of view at 100x magnification.
Acknowledgements
We thank the staffs at CHESS beam line A-1 and NSLS beam line X6A. This work was supported by a Canadian Institute of Health Research operating grant and a New Investigator award (to JKM), and by National Institutes of Health grant AI036900 (to RAM).
Abbreviations used
- APC
antigen presenting cell
- CDR
complementarity determining region
- FR
framework region
- hVβ2.1
human T cell receptor β chain variable domain 2.1
- hVβ5.1
human T cell receptor β chain variable domain 5.1
- Ig
immunoglobulin
- IL-2
interleukin-2
- MHC
major histocompatibility complex
- mVβ2.3
murine T cell receptor β chain variable domain 2.3
- mVβ8.2
murine T cell receptor β chain variable domain 8.2
- pMHC
peptide-MHC complex
- RU
resonance units
- SAG
superantigen
- SEB
staphylococcal enterotoxin B
- SEC3
staphylococcal enterotoxin C3
- SEI
staphylococcal enterotoxin I
- SEK
staphylococcal enterotoxin K
- SpeC
streptococcal pyrogenic exotoxin C
- SpeI
streptococcal pyrogenic exotoxin I
- SPR
surface plasmon resonance
- TCR
T cell receptor
- TSS
toxic shock syndrome
- TSST-1
toxic shock syndrome toxin-1
- V
variable
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.McCormick JK, Yarwood JM, Schlievert PM. Toxic shock syndrome and bacterial superantigens: an update. Annu Rev Microbiol. 2001;55:77–104. doi: 10.1146/annurev.micro.55.1.77. [DOI] [PubMed] [Google Scholar]
- 2.Sundberg EJ, Li Y, Mariuzza RA. So many ways of getting in the way: diversity in the molecular architecture of superantigen-dependent T-cell signaling complexes. Curr Opin Immunol. 2002;14:36–44. doi: 10.1016/s0952-7915(01)00296-5. [DOI] [PubMed] [Google Scholar]
- 3.Kaul R, McGeer A, Norrby-Teglund A, Kotb M, Schwartz B, O'Rourke K, Talbot J, Low DE. Intravenous immunoglobulin therapy for streptococcal toxic shock syndrome--a comparative observational study. The Canadian Streptococcal Study Group. Clin Infect Dis. 1999;28:800–7. doi: 10.1086/515199. [DOI] [PubMed] [Google Scholar]
- 4.LeClaire RD, Bavari S. Human antibodies to bacterial superantigens and their ability to inhibit T-cell activation and lethality. Antimicrob Agents Chemother. 2001;45:460–3. doi: 10.1128/AAC.45.2.460-463.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Li Y, Li H, Dimasi N, McCormick JK, Martin R, Schuck P, Schlievert PM, Mariuzza RA. Crystal structure of a superantigen bound to the high-affinity, zinc-dependent site on MHC class II. Immunity. 2001;14:93–104. doi: 10.1016/s1074-7613(01)00092-9. [DOI] [PubMed] [Google Scholar]
- 6.Jardetzky TS, Brown JH, Gorga JC, Stern LJ, Urban RG, Chi YI, Stauffacher C, Strominger JL, Wiley DC. Three-dimensional structure of a human class II histocompatibility molecule complexed with superantigen. Nature. 1994;368:711–8. doi: 10.1038/368711a0. [DOI] [PubMed] [Google Scholar]
- 7.Kim J, Urban RG, Strominger JL, Wiley DC. Toxic shock syndrome toxin-1 complexed with a class II major histocompatibility molecule HLA-DR1. Science. 1994;266:1870–4. doi: 10.1126/science.7997880. [DOI] [PubMed] [Google Scholar]
- 8.Sundberg EJ, Li H, Llera AS, McCormick JK, Tormo J, Schlievert PM, Karjalainen K, Mariuzza RA. Structures of two streptococcal superantigens bound to TCR beta chains reveal diversity in the architecture of T cell signaling complexes. Structure. 2002;10:687–99. doi: 10.1016/s0969-2126(02)00759-1. [DOI] [PubMed] [Google Scholar]
- 9.Moza B, Varma AK, Zhu P, Buonpane RA, Herfst CA, Nicholson MJ, Wilbuer A-K, Seth NP, Wucherpfennig KW, McCormick JK, Kranz DM, Sundberg EJ. Structural basis of T cell receptor specificity and activation by the bacterial superantigen TSST-1. EMBO J. 2007;26:1187–1197. doi: 10.1038/sj.emboj.7601531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li H, Llera A, Tsuchiya D, Leder L, Ysern X, Schlievert PM, Karjalainen K, Mariuzza RA. Three-dimensional structure of the complex between a T cell receptor beta chain and the superantigen staphylococcal enterotoxin B. Immunity. 1998;9:807–16. doi: 10.1016/s1074-7613(00)80646-9. [DOI] [PubMed] [Google Scholar]
- 11.Proft T, Fraser JD. Bacterial superantigens. Clin Exp Immunol. 2003;133:299–306. doi: 10.1046/j.1365-2249.2003.02203.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brouillard J-NP, Gunther S, Varma AK, Gryski I, Herfst CA, Rahman AK, Leung DY, Schlievert PM, Madrenas J, Sundberg EJ, McCormick JK. Crystal structure of the streptococcal superantigen SpeI and functional role of a novel loop domain in T cell activation by Group V superantigens. J Mol Biol. 2007 doi: 10.1016/j.jmb.2007.01.024. in press. [DOI] [PubMed] [Google Scholar]
- 13.Wen R, Cole GA, Surman S, Blackman MA, Woodland DL. Major histocompatibility complex class II-associated peptides control the presentation of bacterial superantigens to T cells. J Exp Med. 1996;183:1083–92. doi: 10.1084/jem.183.3.1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Moza B, Buonpane RA, Zhu P, Herfst CA, Rahman AK, McCormick JK, Kranz DM, Sundberg EJ. Long-range cooperative binding effects in a T cell receptor variable domain. Proc Natl Acad Sci U S A. 2006;103:9867–72. doi: 10.1073/pnas.0600220103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fields BA, Malchiodi EL, Li H, Ysern X, Stauffacher CV, Schlievert PM, Karjalainen K, Mariuzza RA. Crystal structure of a T-cell receptor beta-chain complexed with a superantigen. Nature. 1996;384:188–92. doi: 10.1038/384188a0. [DOI] [PubMed] [Google Scholar]
- 16.Abrahmsen L, Dohlsten M, Segren S, Bjork P, Jonsson E, Kalland T. Characterization of two distinct MHC class II binding sites in the superantigen staphylococcal enterotoxin A. EMBO J. 1995;14:2978–86. doi: 10.1002/j.1460-2075.1995.tb07300.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hudson KR, Tiedemann RE, Urban RG, Lowe SC, Strominger JL, Fraser JD. Staphylococcal enterotoxin A has two cooperative binding sites on major histocompatibility complex class II. J Exp Med. 1995;182:711–20. doi: 10.1084/jem.182.3.711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Petersson K, Thunnissen M, Forsberg G, Walse B. Crystal structure of a SEA variant in complex with MHC class II reveals the ability of SEA to crosslink MHC molecules. Structure. 2002;10:1619–26. doi: 10.1016/s0969-2126(02)00895-x. [DOI] [PubMed] [Google Scholar]
- 19.Petersson K, Hakansson M, Nilsson H, Forsberg G, Svensson LA, Liljas A, Walse B. Crystal structure of a superantigen bound to MHC class II displays zinc and peptide dependence. EMBO J. 2001;20:3306–12. doi: 10.1093/emboj/20.13.3306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li Y, Li H, Dimasi N, McCormick JK, Martin R, Schuck P, Schlievert PM, Mariuzza RA. Crystal structure of a superantigen bound to the high-affinity, zinc- dependent site on MHC class II. Immunity. 2001;14:93–104. doi: 10.1016/s1074-7613(01)00092-9. [DOI] [PubMed] [Google Scholar]
- 21.Sundberg EJ, Li H, Llera AS, McCormick JK, Tormo J, Schlievert PM, Karjalainen K, Mariuzza RA. Structures of two streptococcal superantigens bound to TCR beta chains reveal diversity in the architecture of T cell signaling complexes. Structure (Camb) 2002;10:687–99. doi: 10.1016/s0969-2126(02)00759-1. [DOI] [PubMed] [Google Scholar]
- 22.Fernandez MM, Guan R, Swaminathan CP, Malchiodi EL, Mariuzza RA. Crystal structure of staphylococcal enterotoxin I (SEI) in complex with a human major histocompatibility complex class II molecule. J Biol Chem. 2006;281:25356–64. doi: 10.1074/jbc.M603969200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li Y, Huang Y, Lue J, Quandt JA, Martin R, Mariuzza RA. Structure of a human autoimmune TCR bound to a myelin basic protein self-peptide and a multiple sclerosis-associated MHC class II molecule. Embo J. 2005;24:2968–79. doi: 10.1038/sj.emboj.7600771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Buonpane RA, Moza B, Sundberg EJ, Kranz DM. Characterization of T cell receptors engineered for high affinity against toxic shock syndrome toxin-1. J Mol Biol. 2005;353:308–21. doi: 10.1016/j.jmb.2005.08.041. [DOI] [PubMed] [Google Scholar]
- 25.Rahman AK, Herfst CA, Moza B, Shames SR, Chau LA, Bueno C, Madrenas J, Sundberg EJ, McCormick JK. Molecular Basis of TCR Selectivity, Cross-Reactivity, and Allelic Discrimination by a Bacterial Superantigen: Integrative Functional and Energetic Mapping of the SpeC-Vbeta2.1 Molecular Interface. J Immunol. 2006;177:8595–603. doi: 10.4049/jimmunol.177.12.8595. [DOI] [PubMed] [Google Scholar]
- 26.Jarraud S, Peyrat MA, Lim A, Tristan A, Bes M, Mougel C, Etienne J, Vandenesch F, Bonneville M, Lina G. egc, a highly prevalent operon of enterotoxin gene, forms a putative nursery of superantigens in Staphylococcus aureus. J Immunol. 2001;166:669–77. doi: 10.4049/jimmunol.166.1.669. [DOI] [PubMed] [Google Scholar]
- 27.Orwin PM, Leung DY, Donahue HL, Novick RP, Schlievert PM. Biochemical and biological properties of Staphylococcal enterotoxin K. Infect Immun. 2001;69:360–6. doi: 10.1128/IAI.69.1.360-366.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Prindiville TP, Cantrell MC, Matsumoto T, Brown WR, Ansari AA, Kotzin BL, Gershwin ME. Analysis of function, specificity and T cell receptor expression of cloned mucosal T cell lines in Crohn's disease. J Autoimmun. 1996;9:193–204. doi: 10.1006/jaut.1996.0023. [DOI] [PubMed] [Google Scholar]
- 29.Nakajima T, Yamazaki K, Hara K. Biased T cell receptor V gene usage in tissues with periodontal disease. J Periodontal Res. 1996;31:2–10. doi: 10.1111/j.1600-0765.1996.tb00457.x. [DOI] [PubMed] [Google Scholar]
- 30.McDermott M, Kastner DL, Holloman JD, Schmidt-Wolf G, Lundberg AS, Sinha AA, Hsu C, Cashin P, Molloy MG, Mulcahy B, et al. The role of T cell receptor beta chain genes in susceptibility to rheumatoid arthritis. Arthritis Rheum. 1995;38:91–5. doi: 10.1002/art.1780380114. [DOI] [PubMed] [Google Scholar]
- 31.Bueno C, Lemke CD, Criado G, Baroja ML, Ferguson SS, Rahman AK, Tsoukas CD, McCormick JK, Madrenas J. Bacterial superantigens bypass Lck-dependent T cell receptor signaling by activating a Galpha11-dependent, PLC-beta-mediated pathway. Immunity. 2006;25:67–78. doi: 10.1016/j.immuni.2006.04.012. [DOI] [PubMed] [Google Scholar]
- 32.Otwinowski Z, Minor W. Processing X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 33.Storoni LC, McCoy AJ, Read RJ. Likelihood-enhanced fast rotation functions. Acta Crystallogr D Biol Crystallogr. 2004;60:432–8. doi: 10.1107/S0907444903028956. [DOI] [PubMed] [Google Scholar]
- 34.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr. 1997;53:240–55. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 35.Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, GrosseKunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography and NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr. 1998;D54:905–21. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 36.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–32. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 37.Weiss A, Stobo JD. Requirement for the coexpression of T3 and the T cell antigen receptor on a malignant human T cell line. J Exp Med. 1984;160:1284–99. doi: 10.1084/jem.160.5.1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ohashi P, Mak T, Elsen P. v. d., Yanagi Y, Yoshikai Y, Calan A, Terhorst C, Stobo J, Weiss A. Reconstitution of an active surface T3/T-cell antigen receptor by DNA transfer. Nature. 1985;316:606–609. doi: 10.1038/316606a0. [DOI] [PubMed] [Google Scholar]
- 39.Strathdee C, McLeod M, Hall J. Efficient control of tetracycline-responsive gene expression from an autoregulated bi-directional expression vector. Gene. 1999;229:21–29. doi: 10.1016/s0378-1119(99)00045-1. [DOI] [PubMed] [Google Scholar]