

Abstract
Sensitive, inexpensive and rapid protease activity assays are of great merit for clinical diagnostics. Detection of protease-based toxins produced by Clostridium botulinum and Bacillus anthracis represents a particularly challenging task as exceptional sensitivity is a prerequisite due to the extreme potency of the toxins. Here, we present an inexpensive and sensitive assay platform for activity-based protease quantification utilizing filamentous bacteriophage as an exponentially amplifiable reporter and its application to detection of these bacterial toxins. The assay is based on specific cleavage of bacteriophage from a solid support and its subsequent quantification by means of infectivity or quantitative PCR. Detection of botulinum neurotoxin (BoNT) serotypes A and B and anthrax lethal factor in the pM range was demonstrated with a limit of detection of BoNT/A under optimized conditions of 2 pM.
The action of proteases has been implicated in a large number of crucial physiological and pathological processes, and consequently, sensitive, rapid and inexpensive protease assays are of great value for both clinical diagnostics and drug development. Timely and sensitive detection is particularly important in the case of protease-based bacterial toxins when fast intervention is needed to prevent or ease potential intoxication, such as with the toxins produced by Clostridium botulinum and Bacillus anthracis. Botulism and anthrax are acute lethal diseases caused by these toxins, with both being listed among the six highest risk bioterrorism threat agents by the US Centers for Disease Control and Prevention (CDC). At the molecular level, a zinc-dependent protease component of either toxin cleaves key host proteins, leading to the associated morbidity and mortality. C. botulinum produces botulinum neurotoxins (BoNTs) that cleave proteins critical for the release of acetylcholine from neuronal cells, inhibiting neuromuscular signal transduction.1 Similarly, the zinc protease of B. anthracis toxin, termed Lethal Factor (LF), cleaves mitogen-activated protein kinase kinases (MAPKK), effectively disturbing macrophage signaling.2
Detection of these toxins represents a significant analytical challenge.3 Due to their extreme potency (median lethal dose of BoNT serotype A is about 1 ng/kg), extreme sensitivity is a prerequisite of any viable diagnostic. Along with the sensitivity requirements, rapid detection and the ability to discriminate between active and inactive forms of the toxin is crucial in most scenarios, including a potential response to the abuse of these toxins as bio-weapons.3 Currently, BoNT detection is accomplished via a widely used mouse bioassay that is very sensitive (up to 20 pg/mL or 0.13 pM), but exceedingly slow.4 Alternatively, ELISA diagnostics are significantly faster and can have sensitivity approaching that of mouse bioassay, but are unable to discriminate between active and inactive forms of the toxin. While a number of alternative activity assays have been developed, none have seen adoption in clinical laboratories, presumably due to the advanced instrumentation required for the signal readout (e.g., fluorescence or MALDI-TOF MS).3 An ideal diagnostic would incorporate aspects of both bioassays and immunoassays, that is, be capable of detecting functional toxin and employ standard procedures and instrumentation utilized in current clinical diagnostics.
Here, we present an inexpensive and sensitive activity-based platform for protease quantification that utilizes filamentous bacteriophage as an exponentially amplifiable reporter and its application to the detection of bioterrorism agents. While phage has been extensively exploited in the investigation and engineering of protease substrate specificity5 as well as the protease inhibitor discovery process,6 a phage-based quantitative protease assay has not been reported. Like in other protease activity assays where the cleavage product is the analyte, inherent amplification of the output signal is achieved by the catalytic nature of substrate cleavage. However, this amplification alone is generally not sufficient for trace detection. To further increase signal gain, a second stage amplification was introduced utilizing post-cleavage propagation of filamentous phage. The exponential nature of phage amplification theoretically allows for the detection of as little as a single phage particle resulting from a single cleavage event.
The assay is based on release of phage from a solid support upon specific cleavage of the displayed substrate domain in the phage-solid support linker (Figure 1). The linker is displayed on the pIII phage coat protein and consists of a substrate domain and N-terminus reactive attachment domain derived from O6-alkylguanine-DNA alkyltransferase (AGT), the latter forming a covalent bond with a benzylguanine modified support.7 We chose to examine BoNT/A for our initial testing given its extreme potency and the urgent need for trace level detection. Filamentous phage was prepared displaying a fusion of a 66 amino acid fragment of synaptosomal associated protein of 25 kDa (SNAP-25 (141 – 206))1 followed by the attachment domain. In the presence of toxin (BoNT/A light chain (LC) or holotoxin), the SNAP-25 substrate is cleaved, releasing the phage into the supernatant, and the number of phage particles released is then quantified simply by counting bacterial colonies in the presence of selection drug (Figures 2 and S1). The limits of detection of the method (LOD), defined as three standard deviations of the blank signal, were in pM range with best sensitivity achieved being 2 pM in BoNT/A-optimized reaction buffer (Table 1). Critically, this sensitivity is approaching that of the mouse bioassay standard for BoNT/A detection. It is particularly noteworthy that effective washing of untethered and/or nonspecifically bound phage from the magnetic beads prior to cleavage using high detergent concentrations (0.5% Tween 20) and elevated temperature (55 °C), was critical for enhancement of the signal to noise ratio (S/N).
Figure 1.
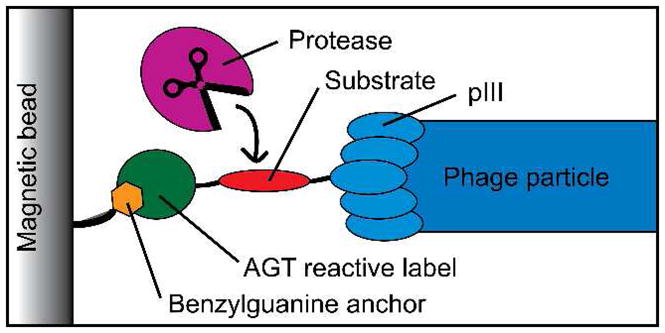
Phage reporter-based protease activity assay.
Figure 2.
Relative signal intensity (signal to noise ratio) at variable concentrations of BoNT/A. (a) HZTD buffer: 40 mM HEPES pH 7.4, 20 μM ZnCl2, 0.1% Tween 20, 1 mM DTT.
Table 1.
Limits of detection (LODs) determined for individual toxins.
| Toxin | Cleavage conditionsa | LOD [pM] |
|---|---|---|
| BoNT/A LC | HZTD bufferb | 10 |
| BoNT/A LC | 10% FBS in PBS | 4 |
| BoNT/A | HZTD bufferb | 2 |
| BoNT/A | PBS, 1 mM DTT | 120 |
| BoNT/A | 10% FBS in PBS | 600 |
| BoNT/B | HZTD bufferb | 850 |
| LF | 0.1 M NaCl in 40 mM HEPES pH 7.4 | 690 |
| LF | HZTD bufferb | 2000 |
Reactions were incubated for 3 h at 25 °C in the specified reaction buffer;
HZTD buffer: 40 mM HEPES pH 7.4, 20 μM ZnCl2, 0.1% Tween 20, 1 mM DTT.
To demonstrate the utility of this system for the detection of other toxins, synaptobrevin-phage (Syb-phage) and MAPKK-phage were constructed.8 In a similar fashion to SNAP25-phage, Syb-phage and MAPKK-phage were attached to magnetic beads and used as sensors for BoNT/B and LF, respectively. Both proteases performed well in the assay and the extent of cleavage readily quantified using the colony counting method (Figure S2). The LOD achieved with both proteases was greater than that observed with BoNT/A, however, both were within an acceptable range for detection of the respective toxin in biological samples (Table 1). In particular, the LOD of LF (690 pM) is notable since alternative non-MS activity based detection of LF are rare and less sensitive.9–10
In the case of a family of toxins such as BoNTs, the ability to quickly discriminate between serotypes is crucial, particularly as most biologic therapeutics (e.g., antibodies) are tailored to the specific serotype of toxin. Importantly, this assay can readily discriminate between BoNT/A and /B well, as demonstrated by lack of cross-reactivity when toxin was mismatched with phage-substrate (Figure S3), confirming that cleavage occurs specifically at the substrate domain.
Using colony counting as the analytical output, the assay can be completed within 16 hours starting from pre-attached phage, which can be stored at 4 °C for several weeks (Figure S4, Table S1). While simple colony counting is certainly advantageous from an economic perspective and allows for diagnostic measurements in the absence of specialized equipment, we envisioned that by using quantitative PCR (qPCR), the assay time could be shortened, while still retaining exponential amplification of the analytical signal. Amplification of filamentous phage DNA has been used for the genotyping of combinatorial library members,11 but we are unaware of any use of qPCR for the determination of filamentous phage titer. After brief thermal denaturation of phage particles (2 min at 95 °C), phage DNA can be precisely quantified by qPCR, showing linear dependency of the logarithm of the phage input on Ct values (Figure S5). Using this method, output phage could be quantified with comparable LOD and standard errors to colony counting, while significantly reducing the time needed to perform the assay to ~5 h (Figure 3).
Figure 3.
Relative qPCR signal intensity (signal to noise ratio) at variable concentrations of BoNT/A, BoNT/B and LF. (a) HZTD buffer: 40 mM HEPES pH 7.4, 20 μM ZnCl2, 0.1% Tween 20, 1 mM DTT; (b) HEPES/NaCl buffer: 40 mM HEPES pH 7.4, 100 mM NaCl.
Given that a number of diagnostics currently employ qPCR as a detection modality,12 a phage activity-based diagnostic can be implemented clinically without the need for new or specialized instrumentation. Economically, colony counting is also very efficient as it is estimated that the cost per reaction would be <$0.04 per assay. While the sensitivity does not entirely supplant the current mouse bioassay used for BoNT/A detection, it is critical to note that BoNT detection is a demanding example where immense sensitivity is required, particularly in the case of testing in complex matrices. If required, additional enhancement of the sensitivity and specificity can be achieved by incorporation of a pre-enrichment step, as has been previously performed.13 Furthermore, this assay does not require the use of animals, and is faster to perform, independent of a bacteria titer or qPCR readout.
The need for easily implemented rapid diagnostic technologies for agents of bioterrorism remains large, and a bacteriophage activity diagnostic provides a powerful and robust tool for trace detection of active toxins. However, the application of this technology is not limited solely to toxins. By taking advantage of the genotype-phenotype link inherent to display technologies, a mixture of phages can be treated with an unknown sample and the identity of the cleaved substrate, and consequently, the corresponding protease, deduced from the phage DNA. Using this premise, we anticipate further development will allow for application to other clinical scenarios, including the multiplexed profiling of panels of proteases.
Supplementary Material
Acknowledgments
This work was supported by Kythera Biopharmaceuticals and the National Institutes of Health (AI 082190 to T.J.D.).
Footnotes
Supporting Information Available: Experimental procedures and supporting figures S1–S4. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Schiavo G, Matteoli M, Montecucco C. Physiol Rev. 2000;80:717. doi: 10.1152/physrev.2000.80.2.717. [DOI] [PubMed] [Google Scholar]
- 2.Tonello F, Montecucco C. Mol Aspects Med. 2009;30:431. doi: 10.1016/j.mam.2009.07.006. [DOI] [PubMed] [Google Scholar]
- 3.Čapek P, Dickerson TJ. Toxins. 2010;2:24. doi: 10.3390/toxins2010024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Solomon HM, Lilly TJ. Bacteriological Analytical Manual. U.S. Food and Drug Administration; 2001. [Google Scholar]
- 5.Deperthes D. Biol Chem. 2002;383:1107. doi: 10.1515/BC.2002.119. [DOI] [PubMed] [Google Scholar]
- 6.Zani ML, Moreau T. Biochimie. 2010 doi: 10.1016/j.biochi.2010.05.003. [DOI] [PubMed] [Google Scholar]
- 7.Keppler A, Gendreizig S, Gronemeyer T, Pick H, Vogel H, Johnsson K. Nat Biotechnol. 2003;21:86. doi: 10.1038/nbt765. [DOI] [PubMed] [Google Scholar]
- 8.See Supporting Information for details.
- 9.Macrae MX, Blake S, Jiang XY, Capone R, Estes DJ, Mayer M, Yang J. ACS Nano. 2009;3:3567. doi: 10.1021/nn901231h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Guarise C, Pasquato L, De Filippis V, Scrimin P. Proc Natl Acad Sci USA. 2006;103:3978. doi: 10.1073/pnas.0509372103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yu L, Bloem LJ. Methods Mol Biol. 1996;58:335. doi: 10.1385/0-89603-402-X:335. [DOI] [PubMed] [Google Scholar]
- 12.Kaltenboeck B, Wang C. Adv Clin Chem. 2005;40:219. doi: 10.1016/S0065-2423(05)40006-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kalb SR, Moura H, Boyer AE, McWilliams LG, Pirkle JL, Barr JR. Anal Biochem. 2006;351:84. doi: 10.1016/j.ab.2006.01.027. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.