

Summary
Major histocompatibility complex (MHC) class II molecules display peptides to the T cell receptor (TCR). The ability of the TCR to discriminate foreign from self peptides presented by MHC molecules is a requirement of an effective adaptive immune response. Dysregulation of this molecular recognition event often leads to a disease state. Recently, a number of structural studies have provided significant insight into several such dysregulated interactions between peptide/MHC complexes and TCR molecules. These include TCR recognition of self peptides, which results in autoimmune reactions, and of mutant self-peptides, common in the immunosurveillance of tumors, as well as the engagement of TCRs by superantigens, a family of bacterial toxins responsible for toxic shock syndrome.
Keywords: T cell receptor, major histocompatibility complex, superantigen, peptide antigen, T cell activity
1. TCR RECOGNITION OF PEPTIDE/MHC CLASS II COMPLEXES
The immune system has evolved through the requirement to distinguish non-self pathogens from self tissues. Whereas T cell recognition of foreign peptides is essential for immune defense against invading microorganisms, recognition of self peptides may cause autoimmune disease. A third category of T cell epitopes involves self peptides resulting from mutations accumulated during aging or disease [1, 2]. However, in terms of T cell recognition, the boundaries separating foreign, self, and altered self epitopes are not necessarily absolute. For example, immunity to cancer can arise from mutations in self proteins that render them visible to T cells [1, 2], a process that may also induce autoimmunity [3]. While much is known about TCR recognition of foreign antigens [4], only very recently have the structural and biophysical principles governing TCR recognition of self and mutant self begun to be elucidated. This portion of the review will focus on the latter.
1.1. Origins of autoimmunity
Central tolerance mechanisms known as negative selection preferentially delete autoreactive T cells during maturation of the immune system, thereby avoiding immune responses to self. However, in autoimmune diseases such as multiple sclerosis (MS) and type I diabetes, the presence of T cells in the periphery reactive with autoantigens demonstrates that such selection is imperfect. In MS, CD4+ T cells specific for central nervous system antigens, including myelin basic protein (MBP) and proteolipid protein, are believed to be a central factor in disease pathogenesis [5]. Likewise, CD4+ T cells reactive with various pancreatic islet proteins, such as insulin and glutamic acid decarboxylase, have been isolated from type I diabetics [6, 7].
Several mechanisms have been proposed to explain why some autoreactive T cells escape thymic deletion [8]. In some cases, escape from negative selection may simply result from lack of self-antigen expression in the thymus. However, most self antigens are expressed in medullary thymic epithelial cells [9]. A different mechanism for escaping negative selection involves expression of splice variants of self-proteins in the thymus that do not contain the relevant T cell epitope [10]. However, these two mechanisms do not account for most autoreactive T cells in the peripheral lymphoid compartment. Rather, thymic selection appears to be based on recognition of self peptide/MHC (pMHC) complexes, whereby weak interactions with self pMHC permit T cell survival (positive selection), whereas strong interactions induce apoptosis (negative selection) [11, 12]. Failure of negative selection could result from reduced TCR affinity for self pMHC ligands, such that the complex with TCR is too short-lived to permit negative selection [8]. Alternatively, unusually weak binding of the self peptide to MHC could destabilize the complex with TCR [13].
It has also been proposed that autoimmunity may result from mutations in self-proteins that render them immunogenic [2, 3]. Although animals possess sophisticated machinery to protect their genetic integrity, this machinery (like negative selection) is imperfect and errors accumulate during certain disease processes, most notably cancer. Tumor-specific antigens resulting from mutations in autologous gene products are biologically important examples of mutant self proteins, belonging to a category between truly self and foreign antigens. Such mutations may become visible to the immune system if they are incorporated into the recognized peptide epitope, or if they lead to aberrant processing of a normally cryptic wild-type epitope. An example of the former mechanism is a unique HLA-DR1-restricted human melanoma antigen derived from the glycolytic enzyme triosephosphate isomerase (TPI), in which a naturally-occurring point mutation replaced a threonine residue by isoleucine within the recognized epitope (TPI 23–37, Thr28Ile) [14]. Presentation of wild-type and mutant TPI (mutTPI) peptides by HLA-DR1 to melanoma-specific CD4+ tumor-infiltrating lymphocytes results in dramatically different T cell responses, such that recognition is enhanced 100,000-fold for the mutant relative to the wild-type peptide.
In the following sections, we review recent structural studies of autoimmune TCRs bound to self pMHC ligands [15-18], and of a tumor-specific TCR in complex with mutant and wild-type TPI peptides presented by HLA-DR1 [19].
1.2. Recognition of self peptide/MHC by autoimmune TCRs
Until 2005, structural studies of TCR/pMHC complexes had been restricted to TCRs specific for microbial and other foreign epitopes, or displaying alloreactivity [4]. These studies demonstrated remarkable similarities in the overall topology of TCR binding to pMHC, irrespective of MHC class I or class II restriction. In general, the TCR is positioned diagonally across the compound surface created by the peptide and the MHC α-helices that flank the peptide-binding groove, although some class I-restricted TCRs adopt a more orthogonal binding mode [20]. The diagonal orientation is exemplified by the structure of human TCR HA1.7 bound to an influenza virus hemagglutinin (HA) peptide and HLA-DR1 (Fig. 1a) [21]. The most structurally diverse CDR loops, CDR3α and CDR3β, are generally located over the central peptide residue at position P5, and form a pocket that accommodates the P5 side chain (Fig. 2a,e). This docking mode maximizes interactions between the CDR3 loops and the MHC-bound peptide.
Fig. 1.

Structures of human TCR/peptide/MHC class II complexes. (a) Ribbon diagram showing a top view of the anti-microbial HA1.7/HA/DR1 complex (PDB accession code 1FYT). TCR α-chain is blue and β-chain is gray; MHC α-chain is gold and β-chain is red; peptide is green. (b) The anti-tumor E8/mutTPI/DR1 complex (2IAM). (c) The autoimmune 3A6/MBP/DR2a complex (1ZGL). (d) The autoimmune Ob.1A12/MBP/DR2b complex (1YMM). In a–d, the central P5 residue of the peptide is represented as a green sphere.
Fig. 2.
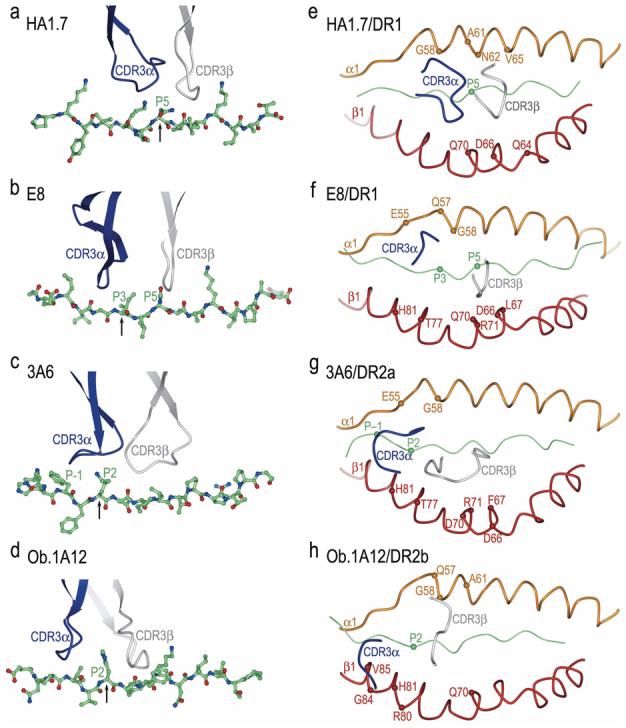
Position of TCR CDR3 loops over foreign, self, or mutant self peptide antigens in human TCR/peptide/MHC class II complexes. Color codes for TCR and MHC molecules are the same as Fig. 1. (a) In the HA1.7/HA/DR1 complex, CDR3α and CDR3β are positioned above the central P5 residue (arrow) of the influenza HA peptide. The peptide is drawn in ball-and-stick representation with carbon atoms in green, nitrogen atoms in blue, and oxygen atoms in red. (b) In the E8/mutTPI/DR1 complex, the CDR3 loops are centered over the P3 residue (arrow) of the mutant TPI self peptide, while maintaining contacts with P5. In the 3A6/MBP/DR2a (c) and Ob.1A12/MBP/DR2b (d) complexes, the two CDR3s converge over the P2 residue (arrow) of MBP, farther still toward the N-terminus of the peptide. The HA, mutTPI, and MBP peptides are aligned according to the P5 residue. (e-h) Positions of the CDR3 loops of TCRs HA1.7, E8, 3A6, and Ob.1A12 on the peptide/MHC surface. Peptide residues located within the pocket formed by CDR3α and CDR3β in the four complexes are indicated by spheres. For each complex, MHC residues contacted by the CDR3 loops are labeled.
The overall similarities among the initial structures of TCRs bound to MHC class I and II created the expectation that all TCRs bind pMHC complexes in similar fashion, and that pMHC recognition by autoreactive TCRs would be qualitatively indistinguishable from that by anti-foreign TCRs. In 2005, however, the first structures of autoimmune TCR-pMHC complexes were reported, including: (1) the complex between mouse TCR 172.10 and MBP 1–11 presented by I-Au [16]; (2) the complex between human TCR Ob.1A12 and MBP 85–99 presented by HLA-DR2b [18]; and (3) the complex between human TCR 3A6 and MBP 89–101 presented by HLA-DR2a [17]. TCR 172.10 is derived from a T cell clone that causes experimental autoimmune encephalomyelitis (EAE), an animal model of MS. TCRs Ob.1A12 and 3A6 were isolated from MS patients, and humanized mice transgenic for these TCRs and DR2b or DR2a develop symptoms typical of EAE. Remarkably, each of the three autoimmune TCRs engage pMHC with a distinct unconventional binding topology compared to TCRs specific for foreign antigens.
In the Ob.1A12/MBP/DR2b complex [18], the TCR is not centered over pMHC and only contacts the N-terminal portion of the MBP self peptide (Fig. 1d). Moreover, Ob.1A12 exhibits a counterclockwise rotation relative to pMHC compared to HA1.7 and other anti-foreign TCRs, resulting in a highly asymmetrical interaction with MHC. Thus, the orientation angle of Ob.1A12 to pMHC, defined as the angle between the line formed by the peptide direction and a line between the centers of mass of the Vα and Vβ domains, is 110° compared to 70° for HA1.7 (Fig. 1a,d) [21]. Significantly, this orientation angle lies far outside the range for all reported MHC class I- or class II-restricted TCRs (45–80°) [4], including autoimmune TCRs 172.10 and 3A6 (see below). Because of the overall shift in the Ob.1A12 footprint on MBP/DR2b (Fig. 1d), the TCR is tilted toward the DR2b β-chain, with which it makes many more contacts than the α-chain. In addition, the two CDR3 loops of Ob.1A12 form a broad pocket that accommodates the P2 side chain of MBP, as well as a side chain from the MHC molecule (His81β) (Fig. 2d,h). By contrast, this pocket accommodates only a single peptide residue (P5) in class II-restricted anti-foreign TCRs (Fig. 2a,e) [21, 22]. The focus of Ob.1A12 on the N-terminal, rather than central, portion of the self-peptide may be broadly characteristic of autoimmune TCRs (see below).
Importantly, the unusual binding topology found in the Ob.1A12/MBP/DR2b structure is supported by experiments with peptide analogs showing that MBP residues P2 and P3 are important TCR contacts, and that substitutions in the C-terminal half of the peptide do not affect TCR recognition (unless binding to MHC is decreased) [23]. Moreover, other MBP-reactive T cell clones derived from the same MS patient exhibited fine specificities very similar to Ob.1A12, implying that the corresponding TCRs engage MBP/DR2b with similar overall topologies.
For the 3A6/MBP/HLA-DR2a complex (Fig. 1c), the orientation angle of TCR to peptide/MHC is 65°, compared to 70° and 110° for the HA1.7/HA/HLA-DR1 and Ob.1A12/MBP/DR2b complexes, respectively [17]. Thus, 3A6 does not exhibit the asymmetrical interaction with MHC seen with Ob.1A12 [18]. In common with Ob.1A12 (Fig. 1d), however, the CDR footprint of 3A6 on MBP/DR2a (Fig. 1c) is shifted towards the N-terminus of the bound peptide, and towards the MHC β1 α-helix, compared to the CDR footprint of HA1.7 on its class II ligand (Fig. 1a).
In the 3A6/MBP/DR2a complex, CDR3α interacts with the N-terminal portion of the peptide, whose central and C-terminal portions engage all three CDR loops of Vβ. Compared to HA1.7 (Fig. 2a,e), large differences are observed in the position of both CDR3 loops along the MBP peptide, such that residue P–1 is enveloped by the CDR3α loop (Fig. 2c,g). Indeed, the pocket formed by CDR3α and CDR3β, which accommodates a single peptide side chain in other TCRs, including Ob.1A12 (Fig. 2d,h), accommodates residues P–1 and P2 in 3A6 (Fig. 2c,g).
Remarkably, no hydrogen bonds or salt bridges are observed between the CDR loops of 3A6 and MBP, involving either main-chain or side-chain atoms of the TCR or peptide, in contrast to all other TCR/pMHC complexes [4]. Interactions between TCR and peptide are restricted to van der Waals contacts, with poor shape complementarity. Therefore, 3A6 appears structurally degenerate in its recognition of the MBP self peptide. Functional degeneracy of the 3A6 interface is demonstrated by the isolation of superagonist peptides with multiple substitutions at TCR-contacting positions [24]. Some of these mimics stimulate 3A6 T cells up to 10,000-fold more efficiently than MBP itself. This degeneracy most likely results from the imperfect fit and lack of hydrogen bonds between 3A6 and MBP observed in the crystal structure, which offer ample opportunities for optimizing the interface. It is also possible that TCRs like 3A6 and Ob.1A12, which mainly recognize the N-terminal portion of peptides, are intrinsically more cross-reactive than TCRs recognizing the central portion, since the overall conformation of peptides bound to MHC class II molecules is far more conserved for residues P–1 to P4 than P5 to P9 [17]. As cross-reactivity would increase the probability of self pMHC recognition, the pathogenic potential of T cells expressing such TCRs would be enhanced, resulting in autoimmunity.
In the 172.10/MBP/I-Au complex [16], the CDR3 loops overlay the central region of the peptide-binding groove in the conventional manner. However, the MBP/I-Au ligand is unusual in that the N-terminal one-third of the binding groove is empty [25]. As a consequence, 172.10 recognizes only six peptide residues (P3 to P8), compared to nine (P–1 to P8) in the case of HA1.7. Furthermore, only two CDRs of 172.10, CDR3α and CDR3β, contact MBP, whereas HA1.7 uses four CDRs to engage HA. As for 3A6/MBP/DR2a, the interface of the 172.10/MBP/I-Au complex is characterized by a scarcity of hydrogen bonds between TCR and peptide, suggesting degeneracy. Thus, all three autoimmune TCRs engage pMHC with suboptimal topologies compared to TCRs specific for microbial and other foreign antigens.
The different ways in which anti-foreign and autoimmune TCRs recognize pMHC may reflect the distinct selection pressures exerted on anti-microbial versus autoreactive T cells [15, 17, 18]. In this view, the central diagonal orientation commonly observed for TCRs recognizing microbial epitopes represents an optimal binding mode for maximizing interactions between TCR and the MHC-bound peptide, resulting in high affinity for pMHC (KD ∼1–100 μM) [26]. As such, this docking mode confers a selective advantage during the intense competition among anti-microbial T cells following an infection [27]. By contrast, autoreactive T cells face different selection pressures, whereby cells expressing TCRs with too high affinity for self pMHC are deleted or inactivated by central and peripheral tolerance mechanisms [8]. The suboptimal binding mode of autoimmune TCRs [15-18] enables certain autoreactive T cells to escape thymic deletion, without necessarily precluding their activation in the periphery under appropriate conditions [28-30]. Indeed, both Ob.1A12 and 3A6 bind self pMHC with much lower affinities (KD >200 μM) than do TCRs recognizing foreign pMHC. Although TCR 172.2 binds MBP/I-Au relatively tightly (KD ∼5 μM), the very short half-life of the MBP/I-Au complex probably explains the escape of 172.2 T cells from negative selection [25].
1.3. Recognition of altered self by a tumor-specific TCR
The structure of a human tumor-specific TCR (E8) bound to the melanoma epitope mutTPI and HLA-DR1 has provided insights into T cell recognition of a naturally mutated self-antigen compared to recognition of native self or foreign antigens [19]. The E8/mutTPI/DR1 complex reveals a number of features intermediate between those of anti-foreign and autoimmune TCR-pMHC class II complexes that may reflect the hybrid nature of altered self. These include a shift of E8 toward the N-terminus of the bound peptide compared to anti-foreign TCRs (Fig. 2b), though not as extreme as for autoimmune TCRs, while maintaining the diagonal binding orientation of anti-foreign TCRs and autoimmune TCR 3A6 (Fig. 1b). As a consequence of this shift, the CDR3 loops of E8 are positioned directly over the substituted P3 residue of mutTPI (Fig. 2b,f), whereas in MBP-specific TCRs 3A6 and Ob.1A12 the CDR3 loops converge on residue P2 (Fig. 2c,d). This focus on the N-terminal half of self-peptides, which may be prevalent among TCRs like E8 and 3A6 that have escaped negative selection, implies that the N-terminal site is intrinsically less favorable for TCR binding than the central site typically utilized by TCRs recognizing foreign epitopes [4, 21, 22]. Consistent with this idea, E8 resembles autoimmune TCRs in binding TPI/DR1 (wild-type or mutant peptide) with very low affinity, although affinity is increased by the Thr-to-Ile mutation at TCR-contacting position P3 of TPI [19].
Also in common with autoimmune TCRs 3A6 and Ob.1A12, the CDR3 loops of E8 form a broad pocket that accommodates two ligand residues (P3 and P5 in the case of E8) (Fig. 2b,f), whereas the corresponding, but narrower, pocket of anti-foreign TCRs generally contains only a single residue (P5). On the other hand, as for anti-foreign complexes, the E8/mutTPI/DR1 structure indicates that residue P5 is crucial for TCR recognition, whereas the P5 position is relatively tolerant of substitutions in the 3A6/MBP/DR2a and Ob.1A12/MBP/DR2b complexes, in which P5 lies outside the CDR3 pocket (Fig. 2c,d) [16,17]. Finally, E8 is tilted toward the DR1 β-chain, with which it makes many more contacts (80% of the total) than does the α-chain, a feature that also distinguishes the autoimmune 3A6/MBP/DR2a and Ob.1A12/MBP/DR2b complexes from the anti-microbial HA1.7/HA/DR1 complex. This tilt precludes formation of a conserved salt bridge between CDR2β Asp/Glu56 and invariant class II residue Lys39α, which is believed to be important for complex stabilization [21, 22]. Whether these features generally distinguish class II-restricted TCRs recognizing altered self from ones recognizing self or non-self, however, must await determination of additional TCR/pMHC class II structures, of which there are currently few [4].
2. TCR RECOGNITION OF SUPERANTIGEN/MHC CLASS II COMPLEXES
Bacterial superantigens (SAGs) comprise a large family of disease-associated proteins that are produced predominantly by Staphylococcus aureus and Streptococcus pyogenes [31], on which this portion of the review will focus, as well as by a number of other bacteria and viruses. SAGs function by simultaneously interacting with class II MHC and TCR molecules on antigen presenting cells and T lymphocytes, respectively [32]. Contrary to the processed antigenic peptides discussed above, SAGs bind to MHC molecules outside of their peptide binding grooves and interact predominantly with only the Vβ domains of TCRs, resulting in the stimulation of up to 20 percent of the entire T cell population. In this way, SAGs initiate a systemic release of inflammatory cytokines that results in various immune-mediated diseases including a condition known as toxic shock syndrome (TSS) that can ultimately lead to multi-organ failure and death. SAGs have also been implicated in the pathogeneses of arthritis, asthma and inflammatory bowel syndrome, and are classified as Category B Select Agents by the U.S. Centers for Disease Control and Prevention.
2.1. Bacterial superantigens can be grouped evolutionarily
More than 30 distinct SAG serotypes from both staphylococci and streptococci belong to the pyrogenic toxin SAG family [33]. Although they are all believed to share a conserved tertiary structure, five distinct evolutionary Groups (I through V) have been proposed for these toxins due to their phylogenetic relationships [31] and there exist key differences in how the characterized representatives for each SAG Group engage their host receptors [34].
Within this classification, toxic shock syndrome toxin-1 (TSST-1) from S. aureus is the only Group I SAG and is also unique in that it binds MHC through an N-terminal, low-affinity binding domain that is peptide-dependent [35, 36]. TSST-1 engages the TCR Vβ domain primarily through intermolecular contacts with residues from the second complementary determining region (CDR2) loop and the third framework region (FR3) [37] (Fig. 3a). No contacts with residues from the CDR1, CDR3 or HV4 loops are made with TSST-1. Hot spot residues located from the CDR2 and FR3 on opposite sides of the binding interface act synergistically to bind TSST-1 in an energetically cooperative manner [38].
Fig. 3.
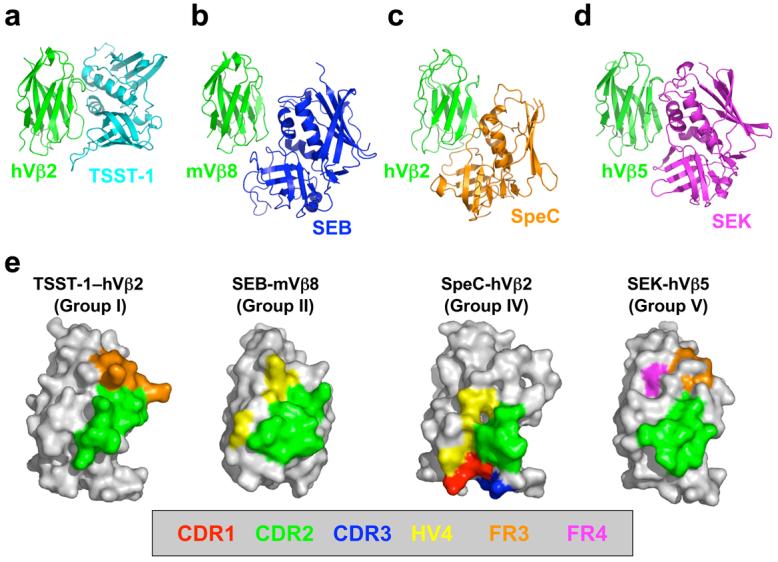
Superantigen engagement of the T cell receptor Vβ domain. Structures of the (a) TSST-1/hVβ2, (b) SEB/mVβ8, (c) SpeC/hVβ2, and (d) SEK/hVβ5 complexes. The Vβ domains in panels a-d are aligned to one another to highlight the distinct orientations by which these SAGs engage their TCR ligands. (e) TCR Vβ domain molecular surface buried by various SAGs. Hypervariable and framework region surface residues buried in the interface formed by TSST-1, SEB, SpeC and SEK are color-coded as follows: CDR1 (red); CDR2 (green); CDR3 (blue); HV4 (yellow); FR3 (orange); and FR4 (magenta). The Vβ domains in panel e are rotated counter-clockwise approximately 90° about the vertical axis of the page relative to their positions in panels a-d.
Group II contains both staphylococcal and streptococcal SAGs (including SEB, SEC and SpeA) that also bind the MHC α-chain through an N-terminal, low-affinity binding domain; however, in contrast to Group I, this binding is peptide-independent [39]. Group II SAGs engage the TCR Vβ through mostly conformationally-dependent mechanisms that are largely independent of specific Vβ amino acid side chains [40-42] (Fig. 3b). Engagement of TCR hypervariable regions by Group II SAGs is generally restricted to the CDR2 and HV4, although a shortened and conformationally-constrained disulphide loop in SpeA makes a single hydrogen bond with the CDR1 loop [42].
Group III SAGs contain only staphylococcal SAGs (such as SEA), and these toxins are able to crosslink MHC molecules [43, 44] through a low-affinity site similar to that used by Group II [45], as well as a high-affinity, zinc-dependent MHC binding interface located within the β-grasp domain of the SAG [46]. There is currently little available information regarding how Group III SAGs engage the TCR.
Group IV SAGs are restricted to only streptococcal members (such as SpeC), and these toxins contain a high-affinity MHC binding domain similar to that of Group III [47]. The structure of SpeC in complex with human Vβ2.1 (Fig. 3c) revealed that this SAG engages all TCR Vβ hypervariable loops, including each of CDR loops 1 through 3 and HV4 [48]. There exists a preponderance of side chain-to-side chain hydrogen bonds suggestive of a highly specific interaction. Furthermore, the Vβ2.1 domain contains non-canonical single residue insertions in both the CDR1 and CDR2 loops, that are involved in extensive networks of intermolecular contacts [42] that are energetically and functionally important [49].
Group V SAGs (including SpeI, SEI and SEK) are the most recently characterized of these toxins. A crystal structure of SEI in complex with HLA-DR1 showed that this group of SAGs binds to class II pMHC molecules in a similar fashion as do Group IV SAGs [50]. A key feature of Group V SAgs is the presence of a loop extension between the third α-helix and the eighth β-sheet (the α3-β8 loop). This ∼15-amino acid extension is not found in the other SAG Groups and is not involved in pMHC interactions. Instead, it has been shown recently that the α3-β8 loop of SpeI is functionally important for the activation of T cells [34]. The recently determined crystal structure of the SEK/hVβ5.1 complex (Fig. 3d) has revealed that residues from the α3-β8 loop of SEK make specific contacts with the TCR Vβ domain that are necessary for binding, are required functionally for the activation of hVβ5.1+ T cells, and extend the known TCR recognition site into the apical loop of FR4 [51].
2.2. SAG-TCR specificity and cross-reactivity
Although studies have shown that some SAGs expand T cells in a Vα-specific manner [52] or bind directly to the TCR Vα domain [53], TCR recognition by SAGs is primarily dictated by SAG-TCR Vβ interactions. The recently expanded database of SAG-TCR Vβ domain crystal structures allows the construction of a paradigm for how SAGs confer specificity and cross-reactivity in TCR recognition.
The least specific SAGs (including SEB and SEC3) depend primarily on a common conformation adopted by the CDR2 and HV4 loops in many Vβ domains [40, 41]. In these complexes, hydrogen bonds are made only to Vβ main chain atoms, such that numerous combinations of amino acid sequences in CDR2 and HV4 can satisfy the binding requirements for these SAGs, as long as they do not change the lengths of these hypervariable loops nor disrupt the common structural conformation adopted.
As TCR specificity increases (e.g., SpeA), the number of hypervariable loops with which the SAG interacts increases beyond CDR2 and HV4. Additionally, the interface becomes increasingly populated by hydrogen bonds formed directly between side chain atoms from both SAG and TCR [42].
As TCR Vβ domain binding partners become restricted even further (e.g., SpeC), the engagement of the entire repertoire of TCR hypervariable elements is observed. The CDR loops with which the SAG interacts also have incorporated non-canonical residue insertions that alter both their length and conformation to provide highly unique binding sites [42].
SAG-TCR specificity is thus accomplished with increased side chain-to-side chain hydrogen bond interactions, an expanded set of hypervariable elements engaged and an accumulation of non-canonical CDR loop structures, which is effectively exhausted at this point. In order to exhibit even greater specificity than SpeC, TSST-1 appears to target a structural element, the FR3 loop connecting the c” and d β-strands, that adopts a common conformation in all but a few Vβ domains, at the expense of interacting with each of the hypervariable structures. The fine specificity of TSST-1 for TCR Vβ domains is enhanced by requiring a particular residue (Lys) at a particular position (62) in FR3 in order to bind and efficiently activate T cells.
This targeting of rarely variable regions, at the expense of canonical hypervariable regions, in Vβ domains as a means for TCR specificity may constitute a general mechanism for enhancing SAG-TCR specificity, as the structural analysis of SEK in complex with one of its Vβ ligands, hVβ5.1, shows similar characteristics [51]. SEK appears to derive its specificity, at least in part, through interactions with relatively uncommon residues in FR3 and FR4, namely at positions 63 and 75, with which a residue in the SEK α3-β8 loop forms side chain-to-side chain hydrogen bonds [51].
The distinct orientations with which each of these representative SAGs from Groups I, II, IV and V engage the TCR Vβ domain result in unique patterns of hypervariable and framework region surfaces that are buried (Fig. 3e). Binding to the TCR Vβ CDR2 loop is a requirement for all bacterial SAGs, and the proportion of the SAG-TCR interface that is contributed by the CDR2 loop is invariably the greatest in any SAG-TCR complex, relative to any other single hypervariable or framework region. Involvement of Vβ domain regions beyond the CDR2 loop, however, plays a significant role in the TCR Vβ domain specificity and cross-reactivity of a SAG [38, 49, 54]. SEK and TSST-1 engage one or more framework region apical loops, at the expense of contacting the hypervariable elements. SEK buries significant molecular surface belonging to both the FR3 and FR4, while TSST-1 contacts only residues from FR3. The lower relative positions of SEB and SpeC on the Vβ domain result in their engagement of hypervariable elements at the expense of binding the apical loops of the framework regions. SEB buries molecular surface belonging to HV4, while SpeC contacts residues from CDR1, CDR3 and HV4.
2.3. Superantigen-mediated T cell signaling complexes
There exist three known binding modes for SAGs to interact with pMHC complexes. These binding modes are exemplified by the following SAGs: the Group I SAG TSST-1, which binds predominantly to the MHC α subunit at a site that overlaps with that of SEB but also extends over the surface of the peptide to make contacts with the β subunit [35]. Group II SAGs (i.e., SEB), which bind MHC exclusively to its α subunit with no contacts made with the antigenic peptide [39]; Group IV and V SAGs (i.e., SpeC and SEK, respectively) bind the MHC β subunit through coordination of a zinc ion and makes numerous contacts with the displayed peptide [50, 51, 55]. Crystal structures of TSST-1 [37], SEB [41], SpeC [42] and SEK [51] in complex with their TCR β chain ligands have allowed the construction of models of those MHC/SAG/TCR ternary complexes that are necessary for efficient T cell activation by Group I, II, IV and V SAGs, respectively, and are distinct from pMHC-TCR complexes.
TSST-1 (Group I) bridges the pMHC and TCR molecules such that two protein-protein interfaces, SAG/MHC and SAG/TCR, are formed (Fig. 4a). No direct MHC-TCR contacts are made. The relative orientation of the TCR and pMHC is such that a plane that passes through both the TCR α and β chains and one that is aligned with the MHC-displayed peptide are approximately perpendicular to one another.
Fig. 4.
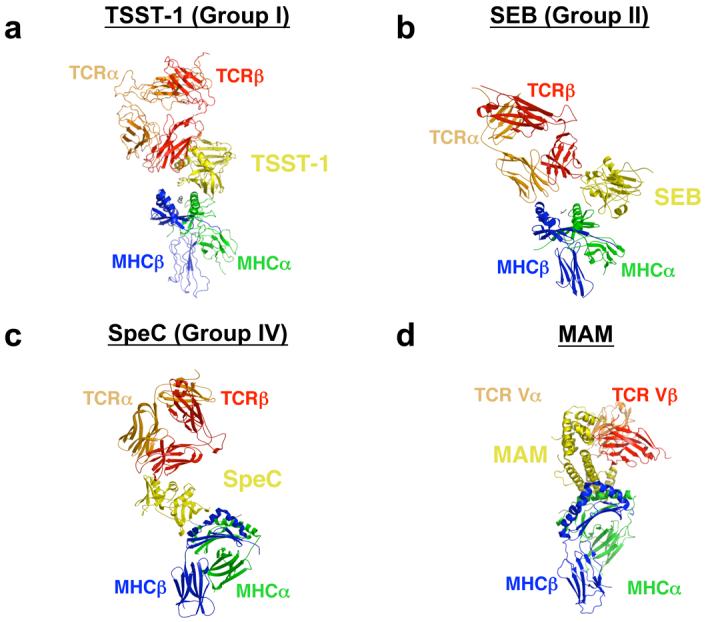
MHC/SAG/TCR ternary signaling complexes mediated by (a) TSST-1, (b) SEB, (c) SpeC, and (d) MAM. Colors are as follows: MHC α subunit, green; MHC β subunit, blue; antigenic peptide, gray; TCR α chain, orange; TCR β chain, red; SAGs, yellow. For clarity, the MHC/SAG/TCR complexes mediated by SpeC (panel c) and MAM (panel d) are rotated approximately 90° clockwise about the vertical axis of the page relative to those mediated by TSST-1 (panel a) and SEB (panel b).
In the SEB (Group II)-dependent T cell signaling complex (Fig. 4b), SEB acts as a wedge between the pMHC and TCR molecules, effectively rotating the TCR about a contact point between the MHC β subunit and the TCR α chain. This removes the antigenic peptide from any possible contacts with the TCR. The relative orientation of pMHC and TCR is otherwise akin to that observed in the TSST-1–mediated T cell signaling complex model. In this supramolecular complex there exist three protein-protein interfaces: SEB/MHC, SEB/TCR and MHC/TCR. The presence of the direct MHC/TCR interaction (as indicated by the arrow in Fig. 4b) has been verified biochemically [56].
SpeC (Group IV), in contrast to SEB but similar to TSST-1, bridges the MHC and TCR molecules (Fig. 4c). There exists no direct interaction between MHC and TCR, and thus only two distinct protein-protein interfaces (i.e., SAG/MHC and SAG/TCR) comprise this complex. However, the TCR and pMHC are oriented such that planes passing through the TCR α and β chains and the antigenic peptide are approximately parallel to one another. Because SEK (Group V) engages pMHC almost identically to SpeC (Group IV) [50], the MHC-SEK-TCR complex is structurally similar to that formed by SpeC. However, since SEK engages the TCR Vβ domain such that it can bind to the FR apical loops, while SpeC engages the Vβ domain such that it binds all of the CDR loops, the angle formed between the axes of the MHC-displayed peptide and the interface between the TCR Vα and Vβ domains is more acute in the SEK- versus SpeC-dependent complexes.
The crystal structure of a complete MHC/SAG/TCR ternary complex has been determined recently for the SAG Mycoplasma arthritidis mitogen (MAM; Fig. 4d) [53]. There are two distinguishing features of this T cell signaling complex. First, MAM makes extensive intermolecular contacts not only with the TCR Vβ domain, but also with the Vα domain. Second, the orientation of the TCR is such that the axis of the interface between the TCR α and β chains is nearly parallel to the axis of the antigenic peptide.
An approximate affinity range of 10−7 – 10−5 M is required of pMHC/TCR interactions for the initiation of T cell activation [57]. The structurally diverse MHC/SAG/TCR signaling complexes are able to achieve affinities within this range in a variety of ways. Although the respective affinities (KDs) of the SEB/MHC and SEB/TCR interactions are only 54 and 150 μM [56, 58], and thus insufficient for efficient T cell activation, the direct MHC/TCR interface acts in a cooperative energetic manner in order to increase the affinity of the entire MHC/SEB/TCR ternary complex to 1.4 μM [59], sufficient for T cell signaling. The energetics of the SpeC-dependent T cell signaling complex are markedly different from that of the SEB-dependent complex. SpeC binds pMHC through a high affinity (KD ≥ 0.1 μM) site on the polymorphic β subunit concomitant with the coordination of a zinc ion. The interaction of SpeC with Vβ has an affinity of 13 μM [60]. Together, these affinities allow the overall MHC/SpeC/TCR complex to achieve an affinity within the range for efficient T cell activation. The affinities of TSST-1 for pMHC and TCR are 1 μM [37] and 0.6 μM [54], respectively. The overall sub-μM affinity of the MHC/TSST-1/TCR complex is thus within the range exhibited by most pMHC/TCR interactions [57].
2.4. Anti-superantigen therapeutic development
Despite the intense research efforts that have been directed toward the characterization of SAGs, therapeutics capable of neutralizing SAG-mediated T cell activation in humans are clinically unavailable. Intravenously-administered pooled human immunoglobulin (IVIG) has been used with some success, but its supply is limited and its effectiveness is variable [61, 62]. Mouse monoclonal antibodies have been generated against SEB [63, 64], but have not been humanized for clinical use. A potentially more general anti-inflammatory agent, a recombinant cell-penetrating form of the suppressor of cytokine signaling 3 (SOCS3) has exhibited some efficacy in protecting mice challenged with lethal doses of SEB [65].
A strategy of using affinity-matured forms of TCR Vβ domains, the natural receptors of these toxins, as potential therapeutics has been developed recently. Vβ domain-derived SAG antagonists that bind to their SAG targets, including SEC3, SEB and TSST, have been engineered with affinities up to a million-fold higher than the wild type SAG/Vβ interactions [54, 66, 67]. One of these Vβ variants completely neutralizes the lethal activity of SEB in animal models [66]. Beyond engineering anti-SAG therapeutics, the affinity maturation of a drug target's natural ligand to create a competitive inhibitor may constitute a generally applicable approach to therapeutic development.
Because SAGs bring together TCR and pMHC molecules resulting in cytokine production and cell division, they had always been presumed to activate T cells through the well-documented signaling cascade induced by TCR engagement by pMHC or anti-CD3 anitbody [68]. Indeed, this pathway is utilized by SAGs, but an alternative signal transduction pathway that is SAG-specific has been discovered recently [69]. This pathway is dependent on Gα11, a member of the pertussin toxin-insensitive Gq family of Gα proteins that regulate PLC-β activity, suggesting that SAGs may use a G protein-coupled receptor as a co-receptor on T cells. Inhibiting this novel SAG-specific pathway either by protein therapeutics that prevent the engagement of SAGs with this as yet unknown co-receptor or by small molecules that block the associated downstream signal transduction events present viable drug development strategies for antagonizing SAG-mediated disease that would not be globally immuno-suppressive.
Acknowledgements
RAM is supported by grants from the National Multiple Sclerosis Society (RG2747) and the National Institutes of Health (AI36900). LD is a Cancer Research Institute Postdoctoral Fellow.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Rosenberg SA. Progress in human tumour immunology and immunotherapy. Nature. 2001;411(6835):380–4. doi: 10.1038/35077246. [DOI] [PubMed] [Google Scholar]
- 2.Houghton AN, et al. Immune recognition of self in immunity against cancer. J Clin Invest. 2004;114(4):468–71. doi: 10.1172/JCI22685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Engelhorn ME, et al. Autoimmunity and tumor immunity induced by immune responses to mutations in self. Nat Med. 2006;12(2):198–206. doi: 10.1038/nm1363. [DOI] [PubMed] [Google Scholar]
- 4.Rudolph MG, et al. How TCRs bind MHCs, peptides, and coreceptors. Annu Rev Immunol. 2006;24:419–66. doi: 10.1146/annurev.immunol.23.021704.115658. [DOI] [PubMed] [Google Scholar]
- 5.Sospedra M, et al. Immunology of multiple sclerosis. Annu Rev Immunol. 2005;23:683–747. doi: 10.1146/annurev.immunol.23.021704.115707. [DOI] [PubMed] [Google Scholar]
- 6.Kent SC, et al. Expanded T cells from pancreatic lymph nodes of type 1 diabetic subjects recognize an insulin epitope. Nature. 2005;435(7039):224–8. doi: 10.1038/nature03625. [DOI] [PubMed] [Google Scholar]
- 7.Nepom GT, et al. Identification and modulation of a naturally processed T cell epitope from the diabetes-associated autoantigen human glutamic acid decarboxylase 65 (hGAD65) Proc Natl Acad Sci U S A. 2001;98(4):1763–8. doi: 10.1073/pnas.98.4.1763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ohashi PS. Negative selection and autoimmunity. Curr Opin Immunol. 2003;15(6):668–76. doi: 10.1016/j.coi.2003.09.013. [DOI] [PubMed] [Google Scholar]
- 9.Derbinski J, et al. Promiscuous gene expression in medullary thymic epithelial cells mirrors the peripheral self. Nat Immunol. 2001;2(11):1032–9. doi: 10.1038/ni723. [DOI] [PubMed] [Google Scholar]
- 10.Klein L, et al. Shaping of the autoreactive T-cell repertoire by a splice variant of self protein expressed in thymic epithelial cells. Nat Med. 2000;6(1):56–61. doi: 10.1038/71540. [DOI] [PubMed] [Google Scholar]
- 11.Kappler JW, et al. T cell tolerance by clonal elimination in the thymus. Cell. 1987;49(2):273–80. doi: 10.1016/0092-8674(87)90568-x. [DOI] [PubMed] [Google Scholar]
- 12.Alam SM, et al. T-cell-receptor affinity and thymocyte positive selection. Nature. 1996;381(6583):616–20. doi: 10.1038/381616a0. [DOI] [PubMed] [Google Scholar]
- 13.Anderton SM, et al. Negative selection during the peripheral immune response to antigen. J Exp Med. 2001;193(1):1–11. doi: 10.1084/jem.193.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pieper R, et al. Biochemical identification of a mutated human melanoma antigen recognized by CD4(+) T cells. J Exp Med. 1999;189(5):757–66. doi: 10.1084/jem.189.5.757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nicholson MJ, et al. Unusual features of self-peptide/MHC binding by autoimmune T cell receptors. Immunity. 2005;23(4):351–60. doi: 10.1016/j.immuni.2005.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Maynard J, et al. Structure of an autoimmune T cell receptor complexed with class II peptide-MHC: insights into MHC bias and antigen specificity. Immunity. 2005;22(1):81–92. doi: 10.1016/j.immuni.2004.11.015. [DOI] [PubMed] [Google Scholar]
- 17.Li Y, et al. Structure of a human autoimmune TCR bound to a myelin basic protein self-peptide and a multiple sclerosis-associated MHC class II molecule. Embo J. 2005;24(17):2968–79. doi: 10.1038/sj.emboj.7600771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hahn M, et al. Unconventional topology of self peptide-major histocompatibility complex binding by a human autoimmune T cell receptor. Nat Immunol. 2005;6(5):490–6. doi: 10.1038/ni1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Deng L, et al. Structural basis for the recognition of mutant self by a tumor-specific, MHC class II-restricted T cell receptor. Nat Immunol. 2007 doi: 10.1038/ni1447. [DOI] [PubMed] [Google Scholar]
- 20.Clements CS, et al. Specificity on a knife-edge: the alphabeta T cell receptor. Curr Opin Struct Biol. 2006;16(6):787–95. doi: 10.1016/j.sbi.2006.09.004. [DOI] [PubMed] [Google Scholar]
- 21.Hennecke J, et al. Structure of a covalently stabilized complex of a human alphabeta T-cell receptor, influenza HA peptide and MHC class II molecule, HLA-DR1. Embo J. 2000;19(21):5611–24. doi: 10.1093/emboj/19.21.5611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Reinherz EL, et al. The crystal structure of a T cell receptor in complex with peptide and MHC class II. Science. 1999;286(5446):1913–21. doi: 10.1126/science.286.5446.1913. [DOI] [PubMed] [Google Scholar]
- 23.Hausmann S, et al. Structural features of autoreactive TCR that determine the degree of degeneracy in peptide recognition. J Immunol. 1999;162(1):338–44. [PubMed] [Google Scholar]
- 24.Hemmer B, et al. Contribution of individual amino acids within MHC molecule or antigenic peptide to TCR ligand potency. J Immunol. 2000;164(2):861–71. doi: 10.4049/jimmunol.164.2.861. [DOI] [PubMed] [Google Scholar]
- 25.He XL, et al. Structural snapshot of aberrant antigen presentation linked to autoimmunity: the immunodominant epitope of MBP complexed with I-Au. Immunity. 2002;17(1):83–94. doi: 10.1016/s1074-7613(02)00340-0. [DOI] [PubMed] [Google Scholar]
- 26.van der Merwe PA, et al. Molecular interactions mediating T cell antigen recognition. Annu Rev Immunol. 2003;21:659–84. doi: 10.1146/annurev.immunol.21.120601.141036. [DOI] [PubMed] [Google Scholar]
- 27.Kedl RM, et al. Epitope dominance, competition and T cell affinity maturation. Curr Opin Immunol. 2003;15(1):120–7. doi: 10.1016/s0952-7915(02)00009-2. [DOI] [PubMed] [Google Scholar]
- 28.Goodnow CC, et al. Cellular and genetic mechanisms of self tolerance and autoimmunity. Nature. 2005;435(7042):590–7. doi: 10.1038/nature03724. [DOI] [PubMed] [Google Scholar]
- 29.Gronski MA, et al. TCR affinity and negative regulation limit autoimmunity. Nat Med. 2004;10(11):1234–9. doi: 10.1038/nm1114. [DOI] [PubMed] [Google Scholar]
- 30.Zehn D, et al. T cells with low avidity for a tissue-restricted antigen routinely evade central and peripheral tolerance and cause autoimmunity. Immunity. 2006;25(2):261–70. doi: 10.1016/j.immuni.2006.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.McCormick JK, et al. Toxic shock syndrome and bacterial superantigens: an update. Annu Rev Microbiol. 2001;55:77–104. doi: 10.1146/annurev.micro.55.1.77. [DOI] [PubMed] [Google Scholar]
- 32.Sundberg EJ, et al. So many ways of getting in the way: diversity in the molecular architecture of superantigen-dependent T-cell signaling complexes. Curr Opin Immunol. 2002;14(1):36–44. doi: 10.1016/s0952-7915(01)00296-5. [DOI] [PubMed] [Google Scholar]
- 33.Proft T, et al. Bacterial superantigens. Clin Exp Immunol. 2003;133(3):299–306. doi: 10.1046/j.1365-2249.2003.02203.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Brouillard J-NP, et al. Crystal structure of the streptococcal superantigen SpeI and functional role of a novel loop domain in T cell activation by Group V superantigens. J Mol Biol. 2007 doi: 10.1016/j.jmb.2007.01.024. in press. [DOI] [PubMed] [Google Scholar]
- 35.Kim J, et al. Toxic shock syndrome toxin-1 complexed with a class II major histocompatibility molecule HLA-DR1. Science. 1994;266(5192):1870–4. doi: 10.1126/science.7997880. [DOI] [PubMed] [Google Scholar]
- 36.Wen R, et al. Major histocompatibility complex class II-associated peptides control the presentation of bacterial superantigens to T cells. J Exp Med. 1996;183(3):1083–92. doi: 10.1084/jem.183.3.1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Moza B, et al. Structural basis of T cell receptor specificity and activation by the bacterial superantigen TSST-1. EMBO J. 2007;26(4):1187–1197. doi: 10.1038/sj.emboj.7601531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Moza B, et al. Long-range cooperative binding effects in a T cell receptor variable domain. Proc Natl Acad Sci U S A. 2006;103(26):9867–72. doi: 10.1073/pnas.0600220103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jardetzky TS, et al. Three-dimensional structure of a human class II histocompatibility molecule complexed with superantigen. Nature. 1994;368(6473):711–8. doi: 10.1038/368711a0. [DOI] [PubMed] [Google Scholar]
- 40.Fields BA, et al. Crystal structure of a T-cell receptor beta-chain complexed with a superantigen. Nature. 1996;384(6605):188–92. doi: 10.1038/384188a0. [DOI] [PubMed] [Google Scholar]
- 41.Li H, et al. Three-dimensional structure of the complex between a T cell receptor beta chain and the superantigen staphylococcal enterotoxin B. Immunity. 1998;9(6):807–16. doi: 10.1016/s1074-7613(00)80646-9. [DOI] [PubMed] [Google Scholar]
- 42.Sundberg EJ, et al. Structures of two streptococcal superantigens bound to TCR beta chains reveal diversity in the architecture of T cell signaling complexes. Structure. 2002;10(5):687–99. doi: 10.1016/s0969-2126(02)00759-1. [DOI] [PubMed] [Google Scholar]
- 43.Abrahmsen L, et al. Characterization of two distinct MHC class II binding sites in the superantigen staphylococcal enterotoxin A. EMBO J. 1995;14(13):2978–86. doi: 10.1002/j.1460-2075.1995.tb07300.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hudson KR, et al. Staphylococcal enterotoxin A has two cooperative binding sites on major histocompatibility complex class II. J Exp Med. 1995;182(3):711–20. doi: 10.1084/jem.182.3.711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Petersson K, et al. Crystal structure of a SEA variant in complex with MHC class II reveals the ability of SEA to crosslink MHC molecules. Structure. 2002;10(12):1619–26. doi: 10.1016/s0969-2126(02)00895-x. [DOI] [PubMed] [Google Scholar]
- 46.Petersson K, et al. Crystal structure of a superantigen bound to MHC class II displays zinc and peptide dependence. EMBO J. 2001;20(13):3306–12. doi: 10.1093/emboj/20.13.3306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li Y, et al. Crystal structure of a superantigen bound to the high-affinity, zinc-dependent site on MHC class II. Immunity. 2001;14(1):93–104. doi: 10.1016/s1074-7613(01)00092-9. [DOI] [PubMed] [Google Scholar]
- 48.Sundberg EJ, et al. Structures of two streptococcal superantigens bound to TCR beta chains reveal diversity in the architecture of T cell signaling complexes. Structure (Camb) 2002;10(5):687–99. doi: 10.1016/s0969-2126(02)00759-1. [DOI] [PubMed] [Google Scholar]
- 49.Rahman AK, et al. Molecular Basis of TCR Selectivity, Cross-Reactivity, and Allelic Discrimination by a Bacterial Superantigen: Integrative Functional and Energetic Mapping of the SpeC-Vbeta2.1 Molecular Interface. J Immunol. 2006;177(12):8595–603. doi: 10.4049/jimmunol.177.12.8595. [DOI] [PubMed] [Google Scholar]
- 50.Fernandez MM, et al. Crystal structure of staphylococcal enterotoxin I (SEI) in complex with a human major histocompatibility complex class II molecule. J Biol Chem. 2006;281(35):25356–64. doi: 10.1074/jbc.M603969200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gunther S, et al. A novel loop domain in superantigens extends their T cell receptor recognition site. doi: 10.1016/j.jmb.2007.05.038. Submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Petersson K, et al. Staphylococcal enterotoxin H induces V alpha-specific expansion of T cells. J Immunol. 2003;170(8):4148–54. doi: 10.4049/jimmunol.170.8.4148. [DOI] [PubMed] [Google Scholar]
- 53.Wang L, et al. Crystal structure of a complete ternary complex of TCR, superantigen and peptide-MHC. Nat Struct Mol Biol. 2007 doi: 10.1038/nsmb1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Buonpane RA, et al. Characterization of T cell receptors engineered for high affinity against toxic shock syndrome toxin-1. J Mol Biol. 2005;353(2):308–21. doi: 10.1016/j.jmb.2005.08.041. [DOI] [PubMed] [Google Scholar]
- 55.Li Y, et al. Crystal structure of a superantigen bound to the high-affinity, zinc-dependent site on MHC class II. Immunity. 2001;14(1):93–104. doi: 10.1016/s1074-7613(01)00092-9. [DOI] [PubMed] [Google Scholar]
- 56.Andersen PS, et al. Role of the T cell receptor alpha chain in stabilizing TCRsuperantigen- MHC class II complexes. Immunity. 1999;10(4):473–83. doi: 10.1016/s1074-7613(00)80047-3. [DOI] [PubMed] [Google Scholar]
- 57.Davis MM, et al. Ligand recognition by alpha beta T cell receptors. Annu Rev Immunol. 1998;16:523–44. doi: 10.1146/annurev.immunol.16.1.523. [DOI] [PubMed] [Google Scholar]
- 58.Malchiodi EL, et al. Superantigen binding to a T cell receptor beta chain of known three- dimensional structure. J Exp Med. 1995;182(6):1833–45. doi: 10.1084/jem.182.6.1833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Andersen PS, et al. Quantifying the energetics of cooperativity in a ternary protein complex. Biochemistry. 2002;41(16):5177–84. doi: 10.1021/bi0200209. [DOI] [PubMed] [Google Scholar]
- 60.Rahman AKMN, et al. Molecular basis of T cell receptor selectivity, cross-reactivity and allelic discrimination by a bacterial superantigen: integrative functional and energetic mapping of the SpeC-Vb2.1 molecular interface. J Immunol. 2006;17(12) doi: 10.4049/jimmunol.177.12.8595. [DOI] [PubMed] [Google Scholar]
- 61.Kaul R, et al. Intravenous immunoglobulin therapy for streptococcal toxic shock syndrome--a comparative observational study. The Canadian Streptococcal Study Group. Clin Infect Dis. 1999;28(4):800–7. doi: 10.1086/515199. [DOI] [PubMed] [Google Scholar]
- 62.LeClaire RD, et al. Human antibodies to bacterial superantigens and their ability to inhibit T-cell activation and lethality. Antimicrob Agents Chemother. 2001;45(2):460–3. doi: 10.1128/AAC.45.2.460-463.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hamad AR, et al. Monoclonal antibodies defining functional sites on the toxin superantigen staphylococcal enterotoxin B. J Exp Med. 1994;180(2):615–21. doi: 10.1084/jem.180.2.615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Pang LT, et al. Inhibition of staphylococcal enterotoxin B-induced lymphocyte proliferation and tumor necrosis factor alpha secretion by MAb5, an anti-toxic shock syndrome toxin 1 monoclonal antibody. Infect Immun. 2000;68(6):3261–8. doi: 10.1128/iai.68.6.3261-3268.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Jo D, et al. Intracellular protein therapy with SOCS3 inhibits inflammation and apoptosis. Nat Med. 2005;11(8):892–8. doi: 10.1038/nm1269. [DOI] [PubMed] [Google Scholar]
- 66.Buonpane RA, et al. Neutralization of staphylococcal enterotoxin B by soluble, high affinity receptor antagonists. Nat Med. 2007 doi: 10.1038/nm1584. in press. [DOI] [PubMed] [Google Scholar]
- 67.Kieke MC, et al. High affinity T cell receptors from yeast display libraries block T cell activation by superantigens. J Mol Biol. 2001;307(5):1305–15. doi: 10.1006/jmbi.2001.4560. [DOI] [PubMed] [Google Scholar]
- 68.Kane LP, et al. Signal transduction by the TCR for antigen. Curr Opin Immunol. 2000;12(3):242–9. doi: 10.1016/s0952-7915(00)00083-2. [DOI] [PubMed] [Google Scholar]
- 69.Bueno C, et al. Bacterial superantigens bypass Lck-dependent T cell receptor signaling by activating a Galpha11-dependent, PLC-beta-mediated pathway. Immunity. 2006;25(1):67–78. doi: 10.1016/j.immuni.2006.04.012. [DOI] [PubMed] [Google Scholar]