

Abstract
The extracellular matrix (ECM), once thought to solely provide physical support to a tissue, is a key component of a cell’s microenvironment responsible for directing cell fate and maintaining tissue specificity. It stands to reason, then, that changes in the ECM itself or in how signals from the ECM are presented to or interpreted by cells can disrupt tissue organization; the latter is a necessary step for malignant progression. In this review, we elaborate on this concept using the mammary gland as an example. We describe how the ECM directs mammary gland formation and function, and discuss how a cell’s inability to interpret these signals—whether as a result of genetic insults or physicochemical alterations in the ECM—disorganizes the gland and promotes malignancy. By restoring context and forcing cells to properly interpret these native signals, aberrant behavior can be quelled and organization re-established. Traditional imaging approaches have been a key complement to the standard biochemical, molecular, and cell biology approaches used in these studies. Utilizing imaging modalities with enhanced spatial resolution in live tissues may uncover additional means by which the ECM regulates tissue structure, on different length scales, through its pericellular organization (short-scale) and by biasing morphogenic and morphostatic gradients (long-scale).
Keywords: Extracellular matrix (ECM), Imaging, Microenvironment, Morphogenesis, Tissue polarity, Tumorigenesis
Introduction
The central goal of developmental biology is to understand how the genetic information within a single cell ultimately results in the formation of tissues, organs, and whole organisms. This involves sequential definition of boundary layers, initially through creation of germ layers. Germ layers give rise to specific tissues, which in turn organize themselves into layered organs. The end result is that, despite containing the same genetic information, cells acquire unique functions depending on where they reside (e.g., hepatocytes secrete bile acids while, thankfully, tongue epithelial cells do not). For humans, once growth has ceased, approximately 10 trillion of these cells must somehow retain their specificity for decades. These observations led us to question how gene expression is modulated in order to achieve specificity, and ask what prevents one cell from trans-differentiating and acquiring the unique properties of another—or from losing its specificity altogether. The answers lie, we believe, in the cell’s local environment. Specifically, we believe that the constant interactions of a cell with its microenvironment (i.e., the extracellular matrix (ECM), other cell types, and soluble factors produced as a result of interactions amongst these constituents) help dictate how specific genes will be expressed. If one takes this a step further, and believes that the manner in which these cues are presented, in addition to the cues themselves, is important, then one can argue that the architecture of a tissue is responsible for directing and maintaining the phenotype of its cells in a space- and time-specific fashion.
Amongst the most forceful evidence for this theory is that malignancy, an ingredient of which is a loss of cellular specificity, is characterized by the progressive degradation of tissue architecture as a tumor develops. In fact, melanomas and breast cancers are effectively staged according to how much the native architecture is disrupted (Burstein et al. 2004; Miller and Mihm 2006). What is still unclear is whether this is a cause or an effect: do changes in tissue architecture preceed and/or select for malignant cells or do genomic alterations occur first, creating “agents of destruction” that progressively destroy tissue architecture in their quest for more space? The answer is that both occur but that the tipping point, we believe, is the loss of tissue and organ architecture. This argument could be enhanced by applying more sensitive techniques than those traditionally utilized in our field to analyze cell–ECM interactions and ECM structure prior to, during, and after the onset of malignancy.
The purpose of this review is to summarize where we stand on these questions. We will focus on the mammary gland, as it provides a unique landscape to probe for answers: in mammals, it is the only organ that undergoes the majority of its development during puberty and cycles through stages of growth and differentiation (during pregnancy and lactation) and cell death and reorganization (during involution) in the adult. Using standard imaging approaches, our group and others have uncovered some of the critical cues conferred by the ECM and its remodeling enzymes to these processes in vivo as well as in culture by utilizing organotypic models which recapitulate some of the critical microenvironmental interactions and many of the discreet behaviors that occur in the native mammary gland (summarized in Fig. 1). Similar approaches have been applied to unravel how these signals are disrupted at the onset of malignancy. Before delving into these details, however, it is important to understand how it came to be recognized that the ECM is an active participant, rather than a passive bystander, in determining a cell’s phenotype.
Fig. 1.

The intricate structure of the mammary gland can be recapitulated in 3D laminin-rich gels and is context-dependent. a The human mammary gland is composed, in part, of a bilayered epithelium consisting of luminal epithelial cells lining the duct and myoepithelial cells lining the basal surface. This tree-like structure is separated from the surrounding stroma by a basement membrane (BM) rich in laminins and collagens other than type I. Three-dimensional culture of murine mammary gland explants allows us to study primary branching or the formation of alveoli (alveologenesis), depending on the context (e.g., the type of ECM present). In a laminin-rich ECM (such as Matrigel), organoids undergo, b alveologenesis or c form acini depending on the soluble factors present. Keratin (K) staining reveals that proper polarity is achieved in this culture model: K8 (green), an epithelial marker, stains throughout these structures while K14 (red), indicative of myoepithelial cells in vivo, is confined to the basal surface (* denotes lumen). On the other hand, culturing organoids within a type I collagen matrix (d), upon stimulation by any of a number of growth factors, yields branched, primary duct-like structures with hollow lumens (inset). b and c were reproduced, with permission from Elsevier, from Fata et al. (2007). d was reproduced, with permission from The Company of Biologists Ltd., from Simian et al. (2001)
The ECM: more than just a scaffold
The cell-secreted ECM, composed of large macromolecules (e.g., collagens, fibronectin, laminins) and polysaccharides (e.g., glycosaminoglycans such as hyaluronan) was initially thought to serve as nothing more than physical scaffolding meant to provide mechanical support to a tissue’s cellular constituents (Alberts et al. 2002). However, as investigators became suspicious that the components of the ECM served greater purposes, they uncovered that the ECM profoundly influences cell behavior. Doing so required intelligent experimental design and relatively basic imaging approaches to assess cell morphology. For instance, cell–cell interactions were found to be dictated by fragments of salt-insoluble collagen, which directed the formation of fused multi-nucleated muscle fibers from individual myoblasts (Hauschka and Konigsberg 1966). Collagen, present in the basal lamina of the lens capsule, was also found to influence the shape and growth of corneal epithelial cells. While corneal epithelial cells fail to synthesize native ECM, assume a flat morphology, and are unresponsive to epidermal growth factor (EGF) when cultured upon tissue culture plastic, they secrete ECM, assume their characteristic cuboidal morphology, and proliferate in response to EGF when cultured upon a collagen substratum (Meier and Hay 1974). Similarly, mammary epithelial cells (MECs) derived from pregnant or lactating mice fail to respond to lactogenic hormones and produce milk when cultured on tissue culture plastic, but do so when cultured in a manner that better approximates their in vivo environment; in this case, floating collagen gels (Emerman et al. 1977). Perhaps more striking, DNA synthesis of endothelial cells was found to be related to cell attachment and spreading on a culture surface (Folkman and Moscona 1978), and followup studies showed that modulation of cell shape influenced mRNA and protein synthesis as well (Ben-Ze’ev et al. 1979, 1980).
Thus, in a relatively short period of time, the concept of the ECM as mainly a physical support was debunked. Instead, it was clear that the ECM also regulated cell shape, proliferation, polarity, differentiation, transcription, synthesis, and secretion for a variety of cell types. This led us to hypothesize that a bidirectional crosstalk exists between the nucleus and chromatin of a cell and its surrounding ECM (i.e., a “dynamic reciprocity”), whether secreted from endogenously made molecules or supplied from the outside, where the ECM influences gene expression and the cell, in turn, can remodel the ECM, which then further acts on the cell, creating a feedback loop (Bissell et al. 1982). The concomitant identification of a family of transmembrane heterodimeric glycoproteins that connect the ECM with the cytoskeleton [subsequently classified as integrins (Hynes 1987)], of feedback elicited by cell-mediated proteolysis of ECM (Sternlicht and Werb 2001), of ECM-response elements in the promoter region of tissue-specific genes (Schmidhauser et al. 1992; Myers et al. 1998; Spencer et al. 2007), and of ECM-derived signals necessary to maintain cell-specific functions (discussed below) are amongst a growing body of evidence supporting the now widely accepted notion that the microenvironment in general and ECM in particular play active roles in determining and maintaining cell specificity. How this is accomplished in the mammary gland during development, pregnancy, and involution is discussed in the following section.
ECM regulation of mammary morphogenesis
The branched mammary gland is a product of interactions between the ectoderm-derived epithelium and mesoderm-derived mesenchyme. In the spirit of seminal work by Grobstein (1953), the specificity of these interactions was demonstrated by heterotypic recombination of epithelium and mesenchyme from mammary and salivary glands, demonstrating that factors from the salivary or mammary mesenchyme direct the branching phenotype and protein expression of the co-cultured epithelium, regardless of its tissue of origin (Sakakura et al. 1976). Many of the soluble factors mediating these interactions have since been identified, and are reviewed elsewhere (Nelson and Bissell 2006). These soluble cues synergize with insoluble cues from the ECM to dictate morphogenesis of the ductal tree. Because the mammary gland undergoes the majority of its growth and remodeling during puberty and pregnancy, it serves as a phenomenal case study of an organ where many of its developmental properties (e.g., invasion), in the adult, are also characteristic of tumor cells (Hanahan and Weinberg 2000). How this profound growth and remodeling is controlled is of great interest, and the ECM, its degrading enzymes (e.g., matrix metalloproteinases, MMPs) and their inhibitors (e.g., tissue inhibitors of metalloproteinases, TIMPs) play significant roles in this regard.
Mammary gland development
At the onset of puberty, the rudimentary ductal tree present at birth (Hogg et al. 1983) undergoes a dramatic expansion to fill the surrounding fat pad. In rodents, terminal end buds residing at the ends of primary ducts and comprised of an outer layer of cap cells and a multilayered core of body cells rapidly penetrate the surrounding fat pad, periodically bifurcating at the leading edge to fill the vacant pad (Hinck and Silberstein 2005) while shedding myoepithelial and luminal epithelial cells in their wake to facilitate ductal elongation (Williams and Daniel 1983). Extensive sprouting from primary ducts to form secondary side-branches also takes place (Hinck and Silberstein 2005). Ducts are characterized by a bilayered epithelium consisting of luminal epithelial cells lining the apical/ductal surface and basally-located myoepithelial cells (Sternlicht 2006), and are ensheathed by a complex of ECM molecules collectively called the basement membrane (BM) (Fig. 1a). The BM is composed primarily of laminins, collagens other than collagen I, and various proteoglycans and calcium-binding proteins (Timpl 1996).
Early in genetic engineering, it was not simple to ablate genes encoding many of the critical ECM proteins or their cognate receptors because these deletions proved to be lethal at an early embryonic stage (Fassler and Meyer 1995; Stephens et al. 1995). Instead, the roles of various ECM proteins were probed via traditional imaging and functional assays. Case in point, the transcript expression of the interstitial matrix protein collagen I and BM proteins collagen IV and laminin-5 were examined via in situ hybridization in the mouse mammary gland before, during, and after puberty. Collagen I expression was shown to reach a maximum just before the peak growth period of the gland (weeks 4–7), and was absent thereafter, while the expression of BM proteins essentially displayed the opposite trend (Keely et al. 1995). These data suggested: (1) that collagen I plays a role in primary branching, (2) that interaction with type I collagen may be necessary for BM expression, and (3) that BM expression is necessary for functional differentiation of the mammary gland.
The first of these conclusions is supported by a three-dimensional (3D) culture explant model in which mammary glands are isolated from mice and minced before polymerization within a type I collagen gel. Though the minced product can come from any portion of the ductal tree (e.g., a primary duct, bifurcation, etc.), once embedded in collagen I and stimulated by any of several growth factors, these organoids form branched “spikes” within the gel (Simian et al. 2001). While the resulting structures do not necessarily recapitulate a functional unit of the mammary epithelium (i.e., they do not form alveoli), fixing and cross-sectioning these gels reveal the formation of duct-like structures characterized by hollow lumens and a correctly polarized, bilayered epithelium (Fig. 1d) (Simian et al. 2001). This result suggests that regardless of its point of origin within the gland, interactions with type I collagen induce ductal branching of the mammary epithelium. This assay can also be conducted with clusters of MECs, and has been utilized to demonstrate the dependence of ductal branching on the collagen I binding integrin α2β1 (Alford et al. 1998; Berdichevsky et al. 1994) and on matrix degrading enzymes, specifically of the MMP family (Simian et al. 2001). These results have since been confirmed in vivo (Keely et al. 1995; Wiseman et al. 2003).
While these data suggest that type I collagen mediates ductal morphogenesis, its expression also precedes that of BM proteins, implying that ligation of collagen I is necessary to produce the self-assembling BM which ultimately ensheaths these ducts. Providing MECs with a malleable collagen I substratum is indeed critical to induce de novo deposition of BM proteins (Streuli and Bissell 1990). In turn, the expression of BM, and its ligation by MECs, is critical to the structure, function, and survival of the gland. For instance, during menstruation, tertiary branches form along primary ducts (Sternlicht 2006), and the cyclic development and regression of these side branches coincide with remodeling of BM (Ferguson et al. 1992). MMP-2 mediated cleavage of the BM, specifically of laminin-5, generates fragments that enhance MEC motility, and likely help mediate branch invasion (Giannelli et al. 1999). Thus, while collagen I-derived signals are critical for primary branching and for BM production, BM remodeling is a common occurrence that stimulates the formation of temporary side branches during the estrous cycle. Signals from the BM are also critical during pregnancy and involution, as described below.
Pregnancy and involution
Upon pregnancy, the mammary gland undergoes a massive expansion marked by a dramatic increase in tertiary branches and lobulo-alveolar density, protagonized by the stromally secreted protease MMP-3 (Wiseman et al. 2003). Alveoli are characterized by their spherical nature and a unique cellular architecture; the luminal epithelium is enveloped by a “weave” of myoepithelial cells, enabling direct contact with the BM by both epithelial cell types (Oakes et al. 2006). Contact with the BM is critical to the formation of alveoli, as organoids embedded in matrices composed of Matrigel, a laminin-rich form of ECM derived from the BM of the murine Engleberth–Holm–Swarm tumor (Kleinman et al. 1986), recapitulate this bulbous architecture, either branching to form numerous alveolar-like structures upon stimulation by TGF-α (Fig. 1b) or growing to form a single cyst-like acinus upon stimulation by FGF-7 (Fig. 1c) (Fata et al. 2007) (please also see the accompanying movie, reproduced with permission).
Just before birth, functional differentiation is induced by the lactogenic switch. Luminal epithelial cells adapt a secretory phenotype and eject milk proteins into the adjoining duct upon stimulation by lactogenic hormones (Oakes et al. 2006). But, signals derived from the BM are also critical to functional differentiation. MECs cultured on tissue culture plastic do not express milk proteins such as β-casein when stimulated by lactogenic hormones (Li et al. 1987), nor do they express milk proteins when allowed to round or form multi-cellular clusters on non-adhesive substrata (Roskelley et al. 1994). Instead, it is clear that either endogenous or exogenous laminin, specifically laminin-1, is required to stimulate milk protein expression (Streuli et al. 1995). We now understand the underlying mechanism more clearly. Laminin-1 induces MEC polarization, which results in proper spatial expression of the prolactin receptor. Sustained ligation of this receptor is necessary for phosphorylation of the transcription factor STAT5 and chromatin reorganization, which in turn promotes transcription of the milk protein β-casein (Xu et al. 2009). While cell-cell contact is not a requisite for milk protein expression (Streuli et al. 1991), cell clustering induces more rapid expression of milk proteins in the presence of laminin (Roskelley et al. 1994), indicating that biochemical and biophysical cues converge to stimulate functional differentiation by regulating gene expression. Accordingly, the ability to interpret these cues is absolutely critical, and laminin-binding receptors such as α6 and β1 integrins and non-integrin receptors such as dystroglycan facilitate their interpretation (Miner and Yurchenco 2004). Function blocking antibodies against β1 and α6 attenuate the expression of β-casein (Muschler et al. 1999); the latter is completely inhibited by deletion of dystroglycan (Weir et al. 2006).
The unique double layered architecture of alveoli raises the question of whether integrin β1-mediated laminin signaling is critical in the luminal epithelial population, myoepithelial population, or both. Using genetic approaches, targeted and temporal deletion of β1 in the luminal and myoepithelial compartments has been achieved. Histological analysis of whole mounted or sectioned glands from mutant mice reveals that β1 deletion within the luminal population prior to pregnancy impairs formation of alveoli, whereas deletion during mid-pregnancy compromises the integrity of formed alveoli, despite a contiguous and intact BM produced by the still-normal basal compartment (Li et al. 2005; Naylor et al. 2005). In both cases, β-casein production is inhibited, impairing the ability of these mice to nurse (Naylor et al. 2005). Deletion of β1 in the basal (primarily myoepithelial) population of the mammary epithelium results in a similar fate; secondary and tertiary branching is significantly inhibited, formation of alveoli is restricted, and production of milk protein is suppressed (Taddei et al. 2008). Since mammary stem cells are thought to reside in the basal compartment as well (see next subsection), it is not clear how much stem cell impairment and how much myoepithelial cell impairment contribute to the observed effects. While the consequence of deleting only myoepithelial β1 with respect to laminin production is unclear at this juncture, it is apparent that β1- and dystroglycan-mediated laminin signaling is not only necessary for functional differentiation of luminal epithelial cells, but that the presence and likely the binding of β1 is necessary in the basal compartment to confer some critical signal(s) that support functional differentiation of the adjacent luminal epithelium.
The importance of the β1-laminin axis to the formation of alveoli and subsequent milk production suggests its continuous involvement also in loss of these functions once nursing has ceased. Involution is marked by a massive apoptotic event where regression of the mammary gland results in termination of milk production and removal of approximately 80% of the epithelium in the matter of days (Watson and Khaled 2008). This is accompanied by increased MMP activity, and transgenic models have shown that exogenous over-expression of MMP-3 results in BM fragmentation (Sympson et al. 1994; Witty et al. 1995) and disrupted production of β-casein (Sympson et al. 1994). One would assume then that BM degradation also contributes to apoptosis by causing cellular detachment from the BM and subsequent anoikis. Remarkably, however, involution is not driven simply by an inability of luminal epithelial cells to attach to the BM. Instead, the specific ligation of β1 integrin to the BM is critical. Evidence for this phenomenon was first provided in culture, where caspase-mediated apoptosis of MECs occurred on tissue culture plastic but was not observed on a laminin-rich (lr) ECM. Function blocking antibodies against integrin β1 or induced digestion of the lrECM via MMP-3 overexpression restored caspase activity and enhanced apoptosis (Boudreau et al. 1995). Ultrastructural analysis of the mammary gland prior to and during involution reveals that the BM does not undergo obvious structural or compositional remodeling during involution, nor is the expression of β1 on the basal surface of alveoli significantly altered (Prince et al. 2002). Instead, β1 ligation is suddenly diminished upon the onset of involution (Prince et al. 2002). This suggests that survival signals conferred by laminin are interpreted specifically by integrin β1, and loss of β1-mediated signaling is responsible for the wide-scale apoptosis observed during mammary gland involution. Accordingly, ECM-derived signals, especially those provided by laminin-1, are responsible for inducing tissue-specific function in the mammary gland. It is this step of signaling that is eliminated abruptly (i.e., when suckling is terminated) to induce involution of the gland.
The role of ECM in guiding stem cell fate
The extensive remodeling that must occur within the mammary gland during pregnancy and involution suggests that a population of precursor cells exist within the gland (Taylor-Papadimitriou et al. 1983). Further evidence suggesting that mammary epithelial-specific stem cells exist and persist throughout the gland’s lifetime was drawn from observations that portions of the gland from mice of any age were capable of regenerating a complete and functional epithelial tree upon transplantation into a cleared mammary fat pad (Smith and Medina 1988). Definitive proof has come when cells isolated from the mouse mammary gland were shown to have the capacity to regenerate all functional cell types of the mammary gland (Shackleton et al. 2006; Stingl et al. 2006). In humans, mammary stem cells were shown to reside in terminal ducts adjacent to fully differentiated cell types (i.e., committed luminal- and myo-epithelial cells) (Villadsen et al. 2007), bringing into question how mammary stem cells reside next to committed cells yet maintain their progenitor status; that is, what defines their niche?
Integrin β1 is also involved here, as its selective deletion from the luminal epithelium prevents alveolar expansion (Li et al. 2005) while its deletion from the basal epithelium impairs the ability of these cells to repopulate cleared fat pads of recipient glands (Taddei et al. 2008). Beta-1 deletion also causes asymmetrical cell division of putative mammary stem cells (Taddei et al. 2008), which implies that β1-mediated signaling may be crucial to the maintenance of the stem cell niche. Little is known, however, about the role the ECM plays in maintaining this niche, and going a step further, how signaling molecules and cell-cell interactions bias cues from the ECM to either maintain quiescence or drive the cells down a trajectory that leads to a particular lineage. Our group has recently utilized microarray patterning technology to lay down complex yet precise patterns of a large number of ECM proteins and signaling molecules to create dozens of unique microenvironments on which mammary stem cells can be cultured. Expression profiles derived from these Microenvironment Arrays (MEArrays™) suggests that cues from the ECM are crucial to guiding cell fate decision, and are biased by signaling proteins to either maintain stemness (in this case, laminin in combination with the notch ligand jagged-1) or move towards a differentiated state (several ECM types in combination with cadherin-mediated cell-cell interactions) (LaBarge et al. 2009).
In summary, signals from the ECM synergize with soluble cues to guide mammary gland development, functional differentiation, and involution, and contribute to the maintenance of the gland’s stem cell niche. Thus it should not come as a surprise that disruption of ECM structure or a misinterpretation of ECM-derived signals could cause or promote malignancy.
Interaction with the ECM influences malignant progression
It is clear that tumors contain many genetic alterations and that there are single mutations that increase susceptibility. However, it is also apparent that cancer is not simply a disease that manifests from a single cell acquiring the same shopping list of genetic alterations. The abnormalities possessed by transformed cells vary not only from tumor to tumor, but within a tumor itself (Folkman et al. 2000). Irrespective of these genetic changes, there are clearly traits that can be ascribed to a “successful” tumor (Hanahan and Weinberg 2000), and the majority of these traits coincide with the sequential destruction of a tissue’s architecture. What is not yet obvious is which comes first.
Evidence suggests that disrupted ECM composition or architecture can in fact at times preceed tumor formation, or even trigger cancer-causing genomic alterations. Earlier studies demonstrated that wounding to create an inflammatory environment, characterized not only by increased ECM deposition, but by increased angiogenesis, inflammatory cell invasion, and an elevated concentration of inflammatory mediators (Coussens and Werb 2002) caused malignancy in prone animal models (i.e., those possessing oncogenic mutations or inoculated with cancer causing viruses) (Dolberg et al. 1985; Lacey et al. 1986; Nerenberg et al. 1987; Schuh et al. 1990). More recently, it has been demonstrated that a specific ECM molecule, collagen VII, is necessary for tumorigenesis of Ras-transformed keratinocytes in a model of squamous cell carcinoma (Ortiz-Urda et al. 2005). Even in the absence of oncogenic transformation, however, overexpression of ECM remodeling enzymes such as MMP-3 or MMP-14 can cause malignancy in the murine mammary gland (Sternlicht et al. 1999; Ha et al. 2001). For the former, these mechanisms have been elucidated. MMP-3 overexpression stimulates the formation of a reactive stroma characterized by increased collagen I deposition prior to tumor formation (Thomasset et al. 1998), and its overexpression or even addition to the culture medium causes genomic instability within MECs and stimulates epithelial to mesenchymal transition via the generation of intracellular reactive oxygen species (Radisky et al. 2005). So the sustained and inappropriate overexpression of a MMP normally expressed during mammary gland development and differentiation can directly cause tumorigenesis, in part by disrupting ECM structure, and also by cleavage of a cell-cell adhesion molecule, E-cadherin (Lochter et al. 1997). In turn, changes in ECM structure and composition can disrupt cellular organization within tissues on its own.
Changes in ECM composition and structure disrupt tissue organization
The BM serves as the principle barrier that must be compromised as a breast tumor proceeds from an in situ malignancy to an invasive one (Vargo-Gogola and Rosen 2007). Even in untransformed cells, the loss of BM-derived signals yields an unmistakable change in function and appearance: unlike cells in lrECM (Fig. 2a), luminal epithelial cells cultured in a type I collagen matrix become “reverse-polarized,” even if they growth arrest (Fig. 2b) (Gudjonsson et al. 2002). Incorporating myoepithelial cells results in structures with proper polarity (Fig. 2c) because these cells in vivo are the ones that secrete laminin. Laminin, in turn, restores appropriate signaling to the luminal epithelial cells (Gudjonsson et al. 2002). This is not only a culture phenomenon: incorporating normal myoepithelial cells with a ductal carcinoma in situ (DCIS)-like cell line, which otherwise adopts an invasive phenotype in vivo in the presence of normal or tumor-associated fibroblasts or tumor myoepithelial cells, inhibits conversion to invasive carcinoma, allowing them to remain as DCIS (Hu et al. 2008). This property appears to be lost in breast cancer, as myoepithelial cells isolated from breast cancers lose their ability to secrete suYcient amounts of laminin-1 (Fig. 2d) (Gudjonsson et al. 2002) or begin to secrete cytokines that are detrimental to organization of luminal epithelial cells (Allinen et al. 2004). Indeed, there are likely roles for other types of laminins in maintaining tissue organization: analysis of clinical samples revealed that certain ECM molecule expression signatures, including genes which encode some of the subunit chains that comprise laminin-1, -2, and -8, amongst others, correlate with high aggressiveness and poor clinical outcome in breast tumors (Bergamaschi et al. 2008).
Fig. 2.

Laminin α1 chain derived from a normal myoepithelium is necessary to confer polarity to luminal epithelial cells in a type I collagen gel. a While luminal epithelial cells (LEC) display proper organization and a basally-secreted basement membrane (BM) in a laminin-rich ECM (lrECM), b culture of LEC within a type I collagen (Coll I) gel results in disorganized structures which growth arrest but fail to deposit a BM. c Addition of myoepithelial cells (MEP) results in acini with proper polarity and restores formation of endogenous BM. d However, human breast cancer-derived MEP fail to confer polarity to LEC, as evidenced by a complete lack of lumen-containing structures within these cultures and disorganized staining of the apical marker sialomucin (green). This figure was reproduced with minor modifications from Gudjonsson et al. (2002) with permission from The Company of Biologists Ltd
In addition to the biochemical signals provided by the BM, the physical properties of the stromal-derived ECM influence malignant progression. A long-appreciated feature of breast tumors is that they can be identified via physical palpation—they have a rigidity distinct from that of the surrounding tissue (Huang and Ingber 2005). The precise origin of this enhanced tissue stiffness, or desmoplasia, has not been elucidated, but conversion of stromal fibroblasts to an activated phenotype (i.e., myofibroblast) which is capable of depositing large amounts of collagen and collagen cross-linkers into the stroma is certainly one component (Paszek and Weaver 2004; Walker 2001). Mechanotransducing elements such as integrins, focal adhesion complexes and cytoskeletal proteins convert these physical forces into chemical signals, which in turn have been shown to influence the behaviors of many discreet cell types (for reviews, see Discher et al. 2005; Peyton et al. 2007). In the mammary gland, elevating tissue stiffness out of the physiological range widely impacts MEC function by increasing intracellular elasticity (Alcaraz et al. 2009), altering cell shape (Emerman et al. 1977), steering MECs away from a tubulogenic phenotype (Wozniak et al. 2003), enhancing fibronectin deposition (Wolf et al. 2007), promoting MEC proliferation (in concert with fibronectin) (Williams et al. 2008), and disrupting the milk protein secretion profile of MECs (Parry et al. 1982). Mechanistically, matrix stiffness acts through β1 integrin clustering and sustained activation of the GTPase Rho to disrupt MEC differentiation and induce a DCIS-like phenotype; upregulating integrin β1 clustering or RhoA activity on a soft substratum disrupts MEC organization and causes MECs to behave as if they were cultured on a stiff substratum (Paszek et al. 2005). These effects of increased ECM density have been confirmed in vivo in a bi-transgenic murine model that spontaneously forms mammary tumors and displays increased stromal collagen density in the mammary gland (Provenzano et al. 2008a). Tumor formation and metastasis was drastically increased in the collagen-dense mutant (Provenzano et al. 2008a), again implicating ECM-derived signals in promoting malignancy.
Attenuation of ECM- and growth factor-derived signals restores tissue organization
In addition to compositional changes within the ECM, a cell’s failure to properly interpret its extracellular environment contributes to malignant behavior. To study this phenomenon, we utilized a unique progression series of MECs with identical genetic backgrounds isolated from reduction mammoplasty of a fibrocystic breast tissue (Briand et al. 1996). The epithelial component of this tissue was passaged repeatedly in defined medium and produced a spontaneously immortalized, EGF-dependent nontumorigenic cell line (referred to as HMT-3522-S1) (Briand et al. 1987). Continued culture in the absence of EGF yielded the S2 population, which eventually produced a malignant tumor upon repeated injection into mice (Briand et al. 1996). Re-explantation of one of these tumors in culture followed by a second round of injection/isolation yielded the tumorigenic T4-2 subline (Briand et al. 1996).
When cultured two-dimensionally on tissue culture plastic, normal breast epithelial cells are difficult to distinguish from their transformed counterparts (Petersen et al. 1992). However, culturing these cells in a physiologically relevant environment, in this case a 3D lrECM, has allowed us and others to easily distinguish the normal and malignant cells based on the structures they form. Primary breast cells or non-malignant breast cell lines such as S1 differentiate into acini-like structures which are growth arrested and have cell–cell junctions containing E-cadherin, basal or baso-lateral integrin localization, and basal secretion of BM proteins laminin, collagen IV, and other BM components (Fig. 3) (Weaver et al. 1997, 2002). Despite identical microenvironments, primary tumor cells or cancer cell lines such as T4-2 form disorganized and proliferative colonies (Fig. 3) (Weaver et al. 1997). Cancer cells fail to properly interpret the signals provided by their endogenously produced ECM because the balance of their adhesion receptors such as integrin β1 and other surface receptors such as EGFR, as well as related signaling pathways, are skewed (reviewed in (Bissell et al. 2005)).
Fig. 3.

Treatment of malignant cells with reverting agents results in organized structures with proper polarity. Confocal microscopy of labeled nuclei, F-actin, β-catenin, β4 integrin, and laminin-5 reveals that while malignant cells form dense and disorganized clusters (middle column) marked by diffuse staining of F-actin, β-catenin, and integrin β4, and fail to deposit a basement membrane (BM), treating malignant cells with any of several reverting agents (β1 integrin targeting monoclonal antibody AIIB2 shown, right column) results in normalized clusters (compare to non-malignant cells, left column). These polarized clusters exhibit cortically organized F-actin, β-catenin concentrated at cell–cell junctions, basally localized β4 integrin, and basally secreted BM. This figure was reproduced, with permission from Elsevier, from Weaver et al. (2002)
Remarkably, targeting the aberrantly over-expressed cell-surface receptors or signaling proteins with specific antibodies or small molecule inhibitors not only restores polarity and growth-arrest to malignant cells (Fig. 3), it also normalizes expression levels of other deviant pathways in T4-2 cells (Weaver et al. 1997; Wang et al. 1998, 2002; Liu et al. 2004; Kenny and Bissell 2007; Itoh et al. 2007). As a result, possible new therapies targeting some of these molecules have emerged as a viable treatment for human xenografts in mice. For instance, growth of tumors derived from metastatic cell lines is significantly inhibited by treatment with an integrin β1 inhibitory antibody (Park et al. 2006), and the number of tumors in a murine breast cancer model is significantly reduced with β1 integrin deletion (White et al. 2004). In fact, the only tumors formed are in those cells where β1 integrin is not deleted, demonstrating that this mediator of cell–ECM interactions is critical for tumor induction and its growth. Accordingly, even in the absence of changes to ECM composition or architecture, a failure to correctly interpret ECM-derived signals can lead to a loss of cellular organization and inappropriate growth. Correcting this aberrant ECM–cell signaling can restore the proper structure to a community of cells which still contain the malignant genome; thus, even in cancer cells, phenotype can override genotype.
Imag(in)ing a crisper future to understand how the ECM contributes to morphogenesis and tumorigenesis
It is important to realize that the vast majority of the studies described thus far have utilized fairly basic imaging techniques to draw significant conclusions about how cell–ECM interactions ultimately influence cell behavior. Techniques such as immunohistochemistry (IHC), immunofluorescence (IF), and electron microscopy (EM) typically require fixing of the tissue sample and often involve sectioning, limiting our ability to image over longer time-scales in live samples to observe dynamic cell behavior. EM is further restricted because the fixing process may require dehydrating the tissue sample, so the images obtained do not necessarily represent the tissue’s native architecture. On the other hand, using traditional microscopes for IHC and IF limits spatial resolution and does not facilitate crisp 3D imaging. With all of the excellent research that has been conducted using these traditional approaches, imagine how far the envelope can be pushed by adopting more sophisticated modalities to image live samples over longer time periods and at higher spatial resolution. The functional promise of this course has been demonstrated through the utilization of live confocal microscopy to identify the collective migration dynamics that drive cancer cell invasion (Wolf et al. 2007) and mammary branching morphogenesis (Ewald et al. 2008). Authors from both studies have contributed to this issue and the reader is referred to those articles to learn more.
Multiphoton microscopy to image ECM structure in live tissue samples
To observe live cell–ECM interactions as well as other means by which the ECM can alter cell behavior in 3D cultures or in vivo requires the use of increasingly sophisticated modes of imaging that would allow high spatial resolution at enhanced tissue depths, and ideally would not oblige the use of exogenous fluorophores. Multiphoton microscopy (MPM) is one of the techniques developed for such purposes. MPM takes advantage of the principle that a fluorophore can be excited by the near-simultaneous absorption of two low energy photons (e.g., two photons each of a wavelength twice the fluorophore’s excitation wavelength) (Friedl et al. 2007). Because this is a highly unlikely event outside of the focus plane, photodamage and phototoxicity is not much of a concern with MPM. Further, the use of low energy wavelengths facilitates greater imaging depths, because these longer wavelengths are less scattered by a tissue sample, which inherently possesses countless changes in its refractive index (Sidani et al. 2006).
An additional advantage of MPM is the ability to take advantage of the noncentrosymmetric structure of collagen to generate harmonic signals via second harmonic generation (SHG) (Provenzano et al. 2008b). Here, pulsing low energy wavelengths at fibrillar collagen generates an emitted photon exactly half the wavelength of the incident beam. This technique is not only useful for drawing conclusions about a tissue’s mechanical properties non-invasively (Raub et al. 2008), but also has been applied to study the effects of collagen organization on terminal end bud structure (Ingman et al. 2006). In malignant tissues, SHG has been utilized to identify signature collagen alignments around pre-palpable tumors which change from a perpendicular alignment to a radial one which guides invasion (Provenzano et al. 2006). Applying SHG in vivo to facilitate earlier detection and staging of breast tumors based on these collagen signatures holds promise, as does utilizing SHG in live imaging of murine tumors to identify whether changes in collagen microstructure select for aberrant cell types and facilitate their growth.
Fluorescent fusion proteins to image how changes in ECM structure influence protein gradients
In conjunction with more sophisticated imaging techniques such as MPM, advancements in molecular cell biology, e.g., through the creation of functional fluorescent fusion proteins in Nobel Prize winning work (Giepmans et al. 2006), may eventually enable the imaging of live cytokine gradients. Not only would this allow us to monitor the interactions between tumor cells and activated stromal components, but it would also shed further light on how changes in the physicochemical properties of the ECM impact cell behavior in a less direct fashion by disrupting signaling fields to promote or support tumorigenesis. Current evidence substantiates the notion that simple changes in ECM composition can alter diffusion and permeability through the ECM (Helm et al. 2007; Leddy et al. 2004), and that increased ECM density (as in a desmoplastic response) decreases matrix porosity (Ryan et al. 1999; Ghajar et al. 2006) and significantly restricts macromolecular diffusion (Fig. 4a–c) (Netti et al. 2000; Ghajar et al. 2008). While these transport measurements were conducted using fluorescein-tagged dextran molecular weight markers, the effective diffusion of a protein in a dense ECM would likely be even more restricted due to the substantial interactions known to occur between signaling molecules and the ECM (Sahni and Francis 2000; Sahni et al. 1998). Diffusion restrictions could foster tumorigenesis over short and long length-scales by restricting the clearance of factors secreted by enveloped cells, causing an upregulation of receptors for these factors and self-sufficient signaling (short-scale) (Tschumperlin et al. 2004); and by disrupting putative morphostatic gradients meant to maintain tissue form (long-scale) (Potter 2007). Real-time monitoring of a fluorescent morphogenic fusion protein in the Drosophila embryo via MPM has facilitated a number of significant conclusions about the spatial and temporal nature of morphogenic gradients in this organism (Gregor et al. 2007a, b). Although imaging growth factor gradients in live tissue samples has thus far proven difficult, imaging growth factor distribution in fixed and stained samples has elucidated how vessel geometry can control tissue patterning by the controlling the direction in which a morphoregulator (in this case, TGF-β1) is secreted (Fig. 4d, e) (Nelson et al. 2006). Whether MPM can be utilized in combination with fusion proteins to determine if changes in ECM composition and microstructure disrupt critical signaling gradients to steer cells away from a differentiated phenotype and towards a tumorigenic phenotype remains to be seen.
Fig. 4.
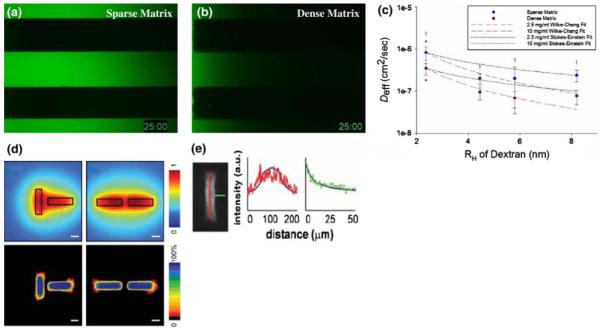
ECM composition and tissue architecture bias soluble factor gradients. To study the effects of ECM density on the passive diffusion of signaling molecules, the transport of fluorescein-tagged 10 kDa molecular weight dextran markers was monitored in microchannels containing a sparse (2.5 mg/ml fibrin) and b dense (10 mg/ml fibrin) tissues. After 25 min, it is clear that diffusion is greatly hindered in the dense matrix. c Effective diffusion coefficients (Deff) were extrapolated from these data for a range of molecular weight markers (presented as hydrodynamic radius, RH) to demonstrate the quantitative significance of this restriction († denotes P < 0.05 when comparing sparse to dense matrix conditions). These data are bounded by two well-known models of solute transport through fluid media (dotted and dashed lines). d The architecture of a tissue can also bias gradients of soluble factors. In this case, secretion of an inhibitory factor and its diffusion through a collagenous ECM was modeled computationally for mammary epithelial cell (MEC)-seeded tubules oriented perpendicular (left column) and parallel (right column) to each other. In both cases, inhibitor concentration was predicted to reach a maxim between the tubules (top row). Functionally, this predicted that MECs would branch in regions with reduced concentrations of the putative inhibitor. Heat maps (bottom row) generated from several images of tubules arranged in the described architectures illustrate that MECs do indeed branch in this fashion. Loss- and gain-of function studies demonstrated that the inhibitor in question was TGF-β1, and e fixing and staining these tissues for TGF-β1 demonstrates that the distribution of this factor matches that predicted by the computational model. Thus, knowing the distribution of an inhibitory gradient allows us to predict where branching will occur in a tissue. Developing techniques to image gradients within live tissues would allow the investigation of how the distribution of factors critical to maintaining homeostasis are disrupted by physicochemical changes within the matrix at the onset of malignancy. a–c of this figure were reproduced, with permission from The Biophysical Society, from Ghajar et al. (2008), while d and e were reproduced, with the permission from the American Association for the Advancement of Science, from Nelson et al. (2006)
Supplementary Material
Acknowledgments
We apologize to those whose work could not be cited due to space limitations. We have cited reviews where possible. We would like to thank members of the Bissell Laboratory, specially Hidetoshi Mori, Jamie Inman, Mark LaBarge, and Virginia Spencer for valuable discussions. The work from the authors’ laboratory is supported by grants from the OBER Office of Biological and Environmental Research; awards from the National Cancer Institute; Innovator awards from the DOD breast cancer program and a Distinguished Fellowship Award from the OBER, Department of Energy (M.J.B.). C.M.G. is supported by a Glenn T. Seaborg Postdoctoral Fellowship from Lawrence Berkeley National Laboratory.
Footnotes
Electronic supplementary material The online version of this article (doi:10.1007/s00418-008-0537-1) contains supplementary material, which is available to authorized users.
References
- Alberts B, Johnson A, Lewis J, Raff M, Roberts K, Walter P. Molecular biology of the cell. Garland Science; New York: 2002. [Google Scholar]
- Alcaraz J, Xu R, Mori H, Nelson CM, Mroue R, Spencer VA, Brown-field D, Radisky D, Bustamante C, Bissell MJ. Laminin and biomimetic extracellular elasticity enhance functional differentiation in mammary epithelia. EMBO J. 2009 doi: 10.1038/emboj.2008.206. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alford D, Baeckstrom D, Geyp M, Pitha P, Taylor-Papadimitriou J. Integrin-matrix interactions affect the form of the structures developing from human mammary epithelial cells in collagen or fibrin gels. J Cell Sci. 1998;111(Pt 4):521–532. doi: 10.1242/jcs.111.4.521. [DOI] [PubMed] [Google Scholar]
- Allinen M, Beroukhim R, Cai L, Brennan C, Lahti-Domenici J, Huang H, Porter D, Hu M, Chin L, Richardson A, Schnitt S, Sellers WR, Polyak K. Molecular characterization of the tumor microenvironment in breast cancer. Cancer Cell. 2004;6:17–32. doi: 10.1016/j.ccr.2004.06.010. [DOI] [PubMed] [Google Scholar]
- Ben-Ze’ev A, Farmer SR, Penman S. Mechanisms of regulating tubulin synthesis in cultured mammalian cells. Cell. 1979;17:319–325. doi: 10.1016/0092-8674(79)90157-0. [DOI] [PubMed] [Google Scholar]
- Ben-Ze’ev A, Farmer SR, Penman S. Protein synthesis requires cell-surface contact while nuclear events respond to cell shape in anchorage-dependent fibroblasts. Cell. 1980;21:365–372. doi: 10.1016/0092-8674(80)90473-0. [DOI] [PubMed] [Google Scholar]
- Berdichevsky F, Alford D, D’Souza B, Taylor-Papadimitriou J. Branching morphogenesis of human mammary epithelial cells in collagen gels. J Cell Sci. 1994;107(Pt 12):3557–3568. doi: 10.1242/jcs.107.12.3557. [DOI] [PubMed] [Google Scholar]
- Bergamaschi A, Tagliabue E, Sorlie T, Naume B, Triulzi T, Orlandi R, Russnes HG, Nesland JM, Tammi R, Auvinen P, Kosma VM, Menard S, Borresen-Dale AL. Extracellular matrix signature identifies breast cancer subgroups with different clinical outcome. J Pathol. 2008;214:357–367. doi: 10.1002/path.2278. [DOI] [PubMed] [Google Scholar]
- Bissell MJ, Hall HG, Parry G. How does the extracellular matrix direct gene expression? J Theor Biol. 1982;99:31–68. doi: 10.1016/0022-5193(82)90388-5. [DOI] [PubMed] [Google Scholar]
- Bissell MJ, Kenny PA, Radisky DC. Microenvironmental regulators of tissue structure and function also regulate tumor induction and progression: the role of extracellular matrix and its degrading enzymes. Cold Spring Harb Symp Quant Biol. 2005;70:343–356. doi: 10.1101/sqb.2005.70.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boudreau N, Sympson CJ, Werb Z, Bissell MJ. Suppression of ICE and apoptosis in mammary epithelial cells by extracellular matrix. Science. 1995;267:891–893. doi: 10.1126/science.7531366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briand P, Petersen OW, Van Deurs B. A new diploid nontumorigenic human breast epithelial cell line isolated and propagated in chemically defined medium. In Vitro Cell Dev Biol. 1987;23:181–188. doi: 10.1007/BF02623578. [DOI] [PubMed] [Google Scholar]
- Briand P, Nielsen KV, Madsen MW, Petersen OW. Trisomy 7p and malignant transformation of human breast epithelial cells following epidermal growth factor withdrawal. Cancer Res. 1996;56:2039–2044. [PubMed] [Google Scholar]
- Burstein HJ, Polyak K, Wong JS, Lester SC, Kaelin CM. Ductal carcinoma in situ of the breast. N Engl J Med. 2004;350:1430–1441. doi: 10.1056/NEJMra031301. [DOI] [PubMed] [Google Scholar]
- Coussens LM, Werb Z. Inflammation and cancer. Nature. 2002;420:860–867. doi: 10.1038/nature01322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Discher DE, Janmey P, Wang YL. Tissue cells feel and respond to the stiffness of their substrate. Science. 2005;310:1139–1143. doi: 10.1126/science.1116995. [DOI] [PubMed] [Google Scholar]
- Dolberg DS, Hollingsworth R, Hertle M, Bissell MJ. Wounding and its role in RSV-mediated tumor formation. Science. 1985;230:676–678. doi: 10.1126/science.2996144. [DOI] [PubMed] [Google Scholar]
- Emerman JT, Enami J, Pitelka DR, Nandi S. Hormonal effects on intracellular and secreted casein in cultures of mouse mammary epithelial cells on floating collagen membranes. Proc Natl Acad Sci USA. 1977;74:4466–4470. doi: 10.1073/pnas.74.10.4466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewald AJ, Brenot A, Duong M, Chan BS, Werb Z. Collective epithelial migration and cell rearrangements drive mammary branching morphogenesis. Dev Cell. 2008;14:570–581. doi: 10.1016/j.devcel.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fassler R, Meyer M. Consequences of lack of beta 1 integrin gene expression in mice. Genes Dev. 1995;9:1896–1908. doi: 10.1101/gad.9.15.1896. [DOI] [PubMed] [Google Scholar]
- Fata JE, Mori H, Ewald AJ, Zhang H, Yao E, Werb Z, Bissell MJ. The MAPK(ERK-1, 2) pathway integrates distinct and antagonistic signals from TGFalpha and FGF7 in morphogenesis of mouse mammary epithelium. Dev Biol. 2007;306:193–207. doi: 10.1016/j.ydbio.2007.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson JE, Schor AM, Howell A, Ferguson MW. Changes in the extracellular matrix of the normal human breast during the menstrual cycle. Cell Tissue Res. 1992;268:167–177. doi: 10.1007/BF00338066. [DOI] [PubMed] [Google Scholar]
- Folkman J, Moscona A. Role of cell shape in growth control. Nature. 1978;273:345–349. doi: 10.1038/273345a0. [DOI] [PubMed] [Google Scholar]
- Folkman J, Hahnfeldt P, Hlatky L. Cancer: looking outside the genome. Nat Rev Mol Cell Biol. 2000;1:76–79. doi: 10.1038/35036100. [DOI] [PubMed] [Google Scholar]
- Friedl P, Wolf K, von Andrian UH, Harms G. Biological second and third harmonic generation microscopy. Curr Protoc Cell Biol. 2007;4(4):15. doi: 10.1002/0471143030.cb0415s34. [DOI] [PubMed] [Google Scholar]
- Ghajar CM, Blevins KS, Hughes CC, George SC, Putnam AJ. Mesenchymal stem cells enhance angiogenesis in mechanically viable prevascularized tissues via early matrix metalloproteinase upregulation. Tissue Eng. 2006;12:2875–2888. doi: 10.1089/ten.2006.12.2875. [DOI] [PubMed] [Google Scholar]
- Ghajar CM, Chen X, Harris JW, Suresh V, Hughes CC, Jeon NL, Putnam AJ, George SC. The effect of matrix density on the regulation of 3-D capillary morphogenesis. Biophys J. 2008;94:1930–1941. doi: 10.1529/biophysj.107.120774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giannelli G, Pozzi A, Stetler-Stevenson WG, Gardner HA, Quaranta V. Expression of matrix metalloprotease-2-cleaved laminin-5 in breast remodeling stimulated by sex steroids. Am J Pathol. 1999;154:1193–1201. doi: 10.1016/S0002-9440(10)65371-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giepmans BN, Adams SR, Ellisman MH, Tsien RY. The fluorescent toolbox for assessing protein location and function. Science. 2006;312:217–224. doi: 10.1126/science.1124618. [DOI] [PubMed] [Google Scholar]
- Gregor T, Tank DW, Wieschaus EF, Bialek W. Probing the limits to positional information. Cell. 2007a;130:153–164. doi: 10.1016/j.cell.2007.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregor T, Wieschaus EF, McGregor AP, Bialek W, Tank DW. Stability and nuclear dynamics of the bicoid morphogen gradient. Cell. 2007b;130:141–152. doi: 10.1016/j.cell.2007.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grobstein C. Inductive epitheliomesenchymal interaction in cultured organ rudiments of the mouse. Science. 1953;118:52–55. doi: 10.1126/science.118.3054.52. [DOI] [PubMed] [Google Scholar]
- Gudjonsson T, Ronnov-Jessen L, Villadsen R, Rank F, Bissell MJ, Petersen OW. Normal and tumor-derived myoepithelial cells differ in their ability to interact with luminal breast epithelial cells for polarity and basement membrane deposition. J Cell Sci. 2002;115:39–50. doi: 10.1242/jcs.115.1.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ha HY, Moon HB, Nam MS, Lee JW, Ryoo ZY, Lee TH, Lee KK, So BJ, Sato H, Seiki M, Yu DY. Overexpression of membrane-type matrix metalloproteinase–1 gene induces mammary gland abnormalities and adenocarcinoma in transgenic mice. Cancer Res. 2001;61:984–990. [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- Hauschka SD, Konigsberg IR. The influence of collagen on the development of muscle clones. Proc Natl Acad Sci USA. 1966;55:119–126. doi: 10.1073/pnas.55.1.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helm CL, Zisch A, Swartz MA. Engineered blood and lymphatic capillaries in 3-D VEGF-fibrin-collagen matrices with interstitial flow. Biotechnol Bioeng. 2007;96:167–176. doi: 10.1002/bit.21185. [DOI] [PubMed] [Google Scholar]
- Hinck L, Silberstein GB. Key stages in mammary gland development: the mammary end bud as a motile organ. Breast Cancer Res. 2005;7:245–251. doi: 10.1186/bcr1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogg NA, Harrison CJ, Tickle C. Lumen formation in the developing mouse mammary gland. J Embryol Exp Morphol. 1983;73:39–57. [PubMed] [Google Scholar]
- Hu M, Yao J, Carroll DK, Weremowicz S, Chen H, Carrasco D, Richardson A, Violette S, Nikolskaya T, Nikolsky Y, Bauerlein EL, Hahn WC, Gelman RS, Allred C, Bissell MJ, Schnitt S, Polyak K. Regulation of in situ to invasive breast carcinoma transition. Cancer Cell. 2008;13:394–406. doi: 10.1016/j.ccr.2008.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang S, Ingber DE. Cell tension, matrix mechanics, and cancer development. Cancer Cell. 2005;8:175–176. doi: 10.1016/j.ccr.2005.08.009. [DOI] [PubMed] [Google Scholar]
- Hynes RO. Integrins: a family of cell surface receptors. Cell. 1987;48:549–554. doi: 10.1016/0092-8674(87)90233-9. [DOI] [PubMed] [Google Scholar]
- Ingman WV, Wyckoff J, Gouon-Evans V, Condeelis J, Pollard JW. Macrophages promote collagen fibrillogenesis around terminal end buds of the developing mammary gland. Dev Dyn. 2006;235:3222–3229. doi: 10.1002/dvdy.20972. [DOI] [PubMed] [Google Scholar]
- Itoh M, Nelson CM, Myers CA, Bissell MJ. Rap1 integrates tissue polarity, lumen formation, and tumorigenic potential in human breast epithelial cells. Cancer Res. 2007;67:4759–4766. doi: 10.1158/0008-5472.CAN-06-4246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keely PJ, Wu JE, Santoro SA. The spatial and temporal expression of the alpha 2 beta 1 integrin and its ligands, collagen I, collagen IV, and laminin, suggest important roles in mouse mammary morphogenesis. Differentiation. 1995;59:1–13. doi: 10.1046/j.1432-0436.1995.5910001.x. [DOI] [PubMed] [Google Scholar]
- Kenny PA, Bissell MJ. Targeting TACE-dependent EGFR ligand shedding in breast cancer. J Clin Invest. 2007;117:337–345. doi: 10.1172/JCI29518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleinman HK, McGarvey ML, Hassell JR, Star VL, Cannon FB, Laurie GW, Martin GR. Basement membrane complexes with biological activity. Biochemistry. 1986;25:312–318. doi: 10.1021/bi00350a005. [DOI] [PubMed] [Google Scholar]
- LaBarge MA, Nelson CM, Villadsen R, Fridriksdottir AJ, Ruth JR, Stampfer MM, Petersen OW, Bissell MJ. Human mammary progenitor cell fate decisions are products of interactions with combinatorial microenvironments. Integrative Biol. 2009 doi: 10.1039/b816472j. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacey M, Alpert S, Hanahan D. Bovine papillomavirus genome elicits skin tumours in transgenic mice. Nature. 1986;322:609–612. doi: 10.1038/322609a0. [DOI] [PubMed] [Google Scholar]
- Leddy HA, Awad HA, Guilak F. Molecular diffusion in tissue-engineered cartilage constructs: effects of scaffold material, time, and culture conditions. J Biomed Mater Res B Appl Biomater. 2004;70:397–406. doi: 10.1002/jbm.b.30053. [DOI] [PubMed] [Google Scholar]
- Li ML, Aggeler J, Farson DA, Hatier C, Hassell J, Bissell MJ. Influence of a reconstituted basement membrane and its components on casein gene expression and secretion in mouse mammary epithelial cells. Proc Natl Acad Sci USA. 1987;84:136–140. doi: 10.1073/pnas.84.1.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li N, Zhang Y, Naylor MJ, Schatzmann F, Maurer F, Wintermantel T, Schuetz G, Mueller U, Streuli CH, Hynes NE. Beta1 integrins regulate mammary gland proliferation and maintain the integrity of mammary alveoli. Embo J. 2005;24:1942–1953. doi: 10.1038/sj.emboj.7600674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Radisky DC, Wang F, Bissell MJ. Polarity and proliferation are controlled by distinct signaling pathways downstream of PI3-kinase in breast epithelial tumor cells. J Cell Biol. 2004;164:603–612. doi: 10.1083/jcb.200306090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lochter A, Galosy S, Muschler J, Freedman N, Werb Z, Bissell MJ. Matrix metalloproteinase stromelysin-1 triggers a cascade of molecular alterations that leads to stable epithelial-to-mesenchymal conversion and a premalignant phenotype in mammary epithelial cells. J Cell Biol. 1997;139:1861–1872. doi: 10.1083/jcb.139.7.1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meier S, Hay ED. Control of corneal differentiation by extracellular materials. Collagen as a promoter and stabilizer of epithelial stroma production. Dev Biol. 1974;38:249–270. doi: 10.1016/0012-1606(74)90005-0. [DOI] [PubMed] [Google Scholar]
- Miller AJ, Mihm MC., Jr Melanoma. N Engl J Med. 2006;355:51–65. doi: 10.1056/NEJMra052166. [DOI] [PubMed] [Google Scholar]
- Miner JH, Yurchenco PD. Laminin functions in tissue morphogenesis. Annu Rev Cell Dev Biol. 2004;20:255–284. doi: 10.1146/annurev.cellbio.20.010403.094555. [DOI] [PubMed] [Google Scholar]
- Muschler J, Lochter A, Roskelley CD, Yurchenco P, Bissell MJ. Division of labor among the alpha6beta4 integrin, beta1 integrins, and an E3 laminin receptor to signal morphogenesis and betacasein expression in mammary epithelial cells. Mol Biol Cell. 1999;10:2817–2828. doi: 10.1091/mbc.10.9.2817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers CA, Schmidhauser C, Mellentin-Michelotti J, Fragoso G, Roskelley CD, Casperson G, Mossi R, Pujuguet P, Hager G, Bissell MJ. Characterization of BCE-1, a transcriptional enhancer regulated by prolactin and extracellular matrix and modulated by the state of histone acetylation. Mol Cell Biol. 1998;18:2184–2195. doi: 10.1128/mcb.18.4.2184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naylor MJ, Li N, Cheung J, Lowe ET, Lambert E, Marlow R, Wang P, Schatzmann F, Wintermantel T, Schuetz G, Clarke AR, Mueller U, Hynes NE, Streuli CH. Ablation of beta1 integrin in mammary epithelium reveals a key role for integrin in glandular morphogenesis and differentiation. J Cell Biol. 2005;171:717–728. doi: 10.1083/jcb.200503144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson CM, Bissell MJ. Of extracellular matrix, scaffolds, and signaling: tissue architecture regulates development, homeostasis, and cancer. Annu Rev Cell Dev Biol. 2006;22:287–309. doi: 10.1146/annurev.cellbio.22.010305.104315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson CM, Vanduijn MM, Inman JL, Fletcher DA, Bissell MJ. Tissue geometry determines sites of mammary branching morphogenesis in organotypic cultures. Science. 2006;314:298–300. doi: 10.1126/science.1131000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nerenberg M, Hinrichs SH, Reynolds RK, Khoury G, Jay G. The tat gene of human T-lymphotropic virus type 1 induces mesenchymal tumors in transgenic mice. Science. 1987;237:1324–1329. doi: 10.1126/science.2888190. [DOI] [PubMed] [Google Scholar]
- Netti PA, Berk DA, Swartz MA, Grodzinsky AJ, Jain RK. Role of extracellular matrix assembly in interstitial transport in solid tumors. Cancer Res. 2000;60:2497–2503. [PubMed] [Google Scholar]
- Oakes SR, Hilton HN, Ormandy CJ. The alveolar switch: coordinating the proliferative cues and cell fate decisions that drive the formation of lobuloalveoli from ductal epithelium. Breast Cancer Res. 2006;8:207. doi: 10.1186/bcr1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortiz-Urda S, Garcia J, Green CL, Chen L, Lin Q, Veitch DP, Sakai LY, Lee H, Marinkovich MP, Khavari PA. Type VII collagen is required for Ras-driven human epidermal tumorigenesis. Science. 2005;307:1773–1776. doi: 10.1126/science.1106209. [DOI] [PubMed] [Google Scholar]
- Park CC, Zhang H, Pallavicini M, Gray JW, Baehner F, Park CJ, Bissell MJ. Beta1 integrin inhibitory antibody induces apoptosis of breast cancer cells, inhibits growth, and distinguishes malignant from normal phenotype in three dimensional cultures and in vivo. Cancer Res. 2006;66:1526–1535. doi: 10.1158/0008-5472.CAN-05-3071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parry G, Lee E, Bissell MJ. Modulation of the differentiated phenotype of cultured mouse mammary epithelial cells by collagen substrata. In: Hawkes SP, Wang J, editors. The extracellular matrix. Academic Press; New York: 1982. pp. 303–308. [Google Scholar]
- Paszek MJ, Weaver VM. The tension mounts: mechanics meets morphogenesis and malignancy. J Mammary Gland Biol Neoplasia. 2004;9:325–342. doi: 10.1007/s10911-004-1404-x. [DOI] [PubMed] [Google Scholar]
- Paszek MJ, Zahir N, Johnson KR, Lakins JN, Rozenberg GI, Gefen A, Reinhart-King CA, Margulies SS, Dembo M, Boettiger D, Hammer DA, Weaver VM. Tensional homeostasis and the malignant phenotype. Cancer Cell. 2005;8:241–254. doi: 10.1016/j.ccr.2005.08.010. [DOI] [PubMed] [Google Scholar]
- Petersen OW, Ronnov-Jessen L, Howlett AR, Bissell MJ. Interaction with basement membrane serves to rapidly distinguish growth and differentiation pattern of normal and malignant human breast epithelial cells. Proc Natl Acad Sci USA. 1992;89:9064–9068. doi: 10.1073/pnas.89.19.9064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peyton SR, Ghajar CM, Khatifiala CB, Putnam AJ. The emergence of ECM mechanics and cytoskeletal tension as important regulators of cell function. Cell Biochem Biophys. 2007;47:300–320. doi: 10.1007/s12013-007-0004-y. [DOI] [PubMed] [Google Scholar]
- Potter JD. Morphogens, morphostats, microarchitecture and malignancy. Nat Rev Cancer. 2007;7:464–474. doi: 10.1038/nrc2146. [DOI] [PubMed] [Google Scholar]
- Prince JM, Klinowska TC, Marshman E, Lowe ET, Mayer U, Miner J, Aberdam D, Vestweber D, Gusterson B, Streuli CH. Cell–matrix interactions during development and apoptosis of the mouse mammary gland in vivo. Dev Dyn. 2002;223:497–516. doi: 10.1002/dvdy.10070. [DOI] [PubMed] [Google Scholar]
- Provenzano PP, Eliceiri KW, Campbell JM, Inman DR, White JG, Keely PJ. Collagen reorganization at the tumor-stromal interface facilitates local invasion. BMC Med. 2006;4:38. doi: 10.1186/1741-7015-4-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Provenzano PP, Inman DR, Eliceiri KW, Knittel JG, Yan L, Rueden CT, White JG, Keely PJ. Collagen density promotes mammary tumor initiation and progression. BMC Med. 2008a;6:11. doi: 10.1186/1741-7015-6-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Provenzano PP, Eliceiri KW, Keely PJ. Multiphoton microscopy and fluorescence lifetime imaging microscopy (FLIM) to monitor metastasis and the tumor microenvironment. Clin Exp Metastasis. 2008b doi: 10.1007/s10585-008-9204-0. [DOI] [PubMed] [Google Scholar]
- Radisky DC, Levy DD, Littlepage LE, Liu H, Nelson CM, Fata JE, Leake D, Godden EL, Albertson DG, Nieto MA, Werb Z, Bissell MJ. Rac1b and reactive oxygen species mediate MMP-3-induced EMT and genomic instability. Nature. 2005;436:123–127. doi: 10.1038/nature03688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raub CB, Unruh J, Suresh V, Krasieva T, Lindmo T, Gratton E, Tromberg BJ, George SC. Image correlation spectroscopy of multiphoton images correlates with collagen mechanical properties. Biophys J. 2008;94:2361–2373. doi: 10.1529/biophysj.107.120006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roskelley CD, Desprez PY, Bissell MJ. Extracellular matrix-dependent tissue-specific gene expression in mammary epithelial cells requires both physical and biochemical signal transduction. Proc Natl Acad Sci USA. 1994;91:12378–12382. doi: 10.1073/pnas.91.26.12378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan EA, Mockros LF, Weisel JW, Lorand L. Structural origins of fibrin clot rheology. Biophys J. 1999;77:2813–2826. doi: 10.1016/S0006-3495(99)77113-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahni A, Francis CW. Vascular endothelial growth factor binds to fibrinogen and fibrin and stimulates endothelial cell proliferation. Blood. 2000;96:3772–3778. [PubMed] [Google Scholar]
- Sahni A, Odrljin T, Francis CW. Binding of basic fibroblast growth factor to fibrinogen and fibrin. J Biol Chem. 1998;273:7554–7559. doi: 10.1074/jbc.273.13.7554. [DOI] [PubMed] [Google Scholar]
- Sakakura T, Nishizuka Y, Dawe CJ. Mesenchyme-dependent morphogenesis and epithelium-specific cytodifferentiation in mouse mammary gland. Science. 1976;194:1439–1441. doi: 10.1126/science.827022. [DOI] [PubMed] [Google Scholar]
- Schmidhauser C, Casperson GF, Myers CA, Sanzo KT, Bolten S, Bissell MJ. A novel transcriptional enhancer is involved in the prolactin- and extracellular matrix-dependent regulation of beta-casein gene expression. Mol Biol Cell. 1992;3:699–709. doi: 10.1091/mbc.3.6.699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuh AC, Keating SJ, Monteclaro FS, Vogt PK, Breitman ML. Obligatory wounding requirement for tumorigenesis in v-jun transgenic mice. Nature. 1990;346:756–760. doi: 10.1038/346756a0. [DOI] [PubMed] [Google Scholar]
- Shackleton M, Vaillant F, Simpson KJ, Stingl J, Smyth GK, Asselin-Labat ML, Wu L, Lindeman GJ, Visvader JE. Generation of a functional mammary gland from a single stem cell. Nature. 2006;439:84–88. doi: 10.1038/nature04372. [DOI] [PubMed] [Google Scholar]
- Sidani M, Wyckoff J, Xue C, Segall JE, Condeelis J. Probing the microenvironment of mammary tumors using multiphoton microscopy. J Mammary Gland Biol Neoplasia. 2006;11:151–163. doi: 10.1007/s10911-006-9021-5. [DOI] [PubMed] [Google Scholar]
- Simian M, Hirai Y, Navre M, Werb Z, Lochter A, Bissell MJ. The interplay of matrix metalloproteinases, morphogens and growth factors is necessary for branching of mammary epithelial cells. Development. 2001;128:3117–3131. doi: 10.1242/dev.128.16.3117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith GH, Medina D. A morphologically distinct candidate for an epithelial stem cell in mouse mammary gland. J Cell Sci. 1988;90(Pt 1):173–183. doi: 10.1242/jcs.90.1.173. [DOI] [PubMed] [Google Scholar]
- Spencer VA, Xu R, Bissell MJ. Extracellular matrix, nuclear and chromatin structure, and gene expression in normal tissues and malignant tumors: a work in progress. Adv Cancer Res. 2007;97:275–294. doi: 10.1016/S0065-230X(06)97012-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens LE, Sutherland AE, Klimanskaya IV, Andrieux A, Meneses J, Pedersen RA, Damsky CH. Deletion of beta 1 integrins in mice results in inner cell mass failure and peri-implantation lethality. Genes Dev. 1995;9:1883–1895. doi: 10.1101/gad.9.15.1883. [DOI] [PubMed] [Google Scholar]
- Sternlicht MD. Key stages in mammary gland development: the cues that regulate ductal branching morphogenesis. Breast Cancer Res. 2006;8:201. doi: 10.1186/bcr1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sternlicht MD, Werb Z. How matrix metalloproteinases regulate cell behavior. Annu Rev Cell Dev Biol. 2001;17:463–516. doi: 10.1146/annurev.cellbio.17.1.463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sternlicht MD, Lochter A, Sympson CJ, Huey B, Rougier JP, Gray JW, Pinkel D, Bissell MJ, Werb Z. The stromal proteinase MMP3/stromelysin–1 promotes mammary carcinogenesis. Cell. 1999;98:137–146. doi: 10.1016/s0092-8674(00)81009-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stingl J, Eirew P, Ricketson I, Shackleton M, Vaillant F, Choi D, Li HI, Eaves CJ. Purification and unique properties of mammary epithelial stem cells. Nature. 2006;439:993–997. doi: 10.1038/nature04496. [DOI] [PubMed] [Google Scholar]
- Streuli CH, Bissell MJ. Expression of extracellular matrix components is regulated by substratum. J Cell Biol. 1990;110:1405–1415. doi: 10.1083/jcb.110.4.1405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Streuli CH, Bailey N, Bissell MJ. Control of mammary epithelial differentiation: basement membrane induces tissue-specific gene expression in the absence of cell–cell interaction and morphological polarity. J Cell Biol. 1991;115:1383–1395. doi: 10.1083/jcb.115.5.1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Streuli CH, Schmidhauser C, Bailey N, Yurchenco P, Skubitz AP, Roskelley C, Bissell MJ. Laminin mediates tissue-specific gene expression in mammary epithelia. J Cell Biol. 1995;129:591–603. doi: 10.1083/jcb.129.3.591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sympson CJ, Talhouk RS, Alexander CM, Chin JR, Clift SM, Bissell MJ, Werb Z. Targeted expression of stromelysin-1 in mammary gland provides evidence for a role of proteinases in branching morphogenesis and the requirement for an intact basement membrane for tissue-specific gene expression. J Cell Biol. 1994;125:681–693. doi: 10.1083/jcb.125.3.681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taddei I, Deugnier MA, Faraldo MM, Petit V, Bouvard D, Medina D, Fassler R, Thiery JP, Glukhova MA. Beta1 integrin deletion from the basal compartment of the mammary epithelium affects stem cells. Nat Cell Biol. 2008;10:716–722. doi: 10.1038/ncb1734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor-Papadimitriou J, Lane EB, Chang SE. Cell lineages and interactions in neoplastic expression in the human breast. In: Rich MA, Hager JC, Furmanski P, editors. Understanding breast cancer: clinical and laboratory concepts. Dekker; New York: 1983. pp. 215–246. [Google Scholar]
- Thomasset N, Lochter A, Sympson CJ, Lund LR, Williams DR, Behrendtsen O, Werb Z, Bissell MJ. Expression of autoactivated stromelysin-1 in mammary glands of transgenic mice leads to a reactive stroma during early development. Am J Pathol. 1998;153:457–467. doi: 10.1016/S0002-9440(10)65589-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timpl R. Macromolecular organization of basement membranes. Curr Opin Cell Biol. 1996;8:618–624. doi: 10.1016/s0955-0674(96)80102-5. [DOI] [PubMed] [Google Scholar]
- Tschumperlin DJ, Dai G, Maly IV, Kikuchi T, Laiho LH, McVittie AK, Haley KJ, Lilly CM, So PT, Lauffenburger DA, Kamm RD, Drazen JM. Mechanotransduction through growth-factor shedding into the extracellular space. Nature. 2004;429:83–86. doi: 10.1038/nature02543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vargo-Gogola T, Rosen JM. Modelling breast cancer: one size does not fit all. Nat Rev Cancer. 2007;7:659–672. doi: 10.1038/nrc2193. [DOI] [PubMed] [Google Scholar]
- Villadsen R, Fridriksdottir AJ, Ronnov-Jessen L, Gudjonsson T, Rank F, LaBarge MA, Bissell MJ, Petersen OW. Evidence for a stem cell hierarchy in the adult human breast. J Cell Biol. 2007;177:87–101. doi: 10.1083/jcb.200611114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker RA. The complexities of breast cancer desmoplasia. Breast Cancer Res. 2001;3:143–145. doi: 10.1186/bcr287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F, Weaver VM, Petersen OW, Larabell CA, Dedhar S, Briand P, Lupu R, Bissell MJ. Reciprocal interactions between beta1-integrin and epidermal growth factor receptor in three-dimensional basement membrane breast cultures: a different perspective in epithelial biology. Proc Natl Acad Sci USA. 1998;95:14821–14826. doi: 10.1073/pnas.95.25.14821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F, Hansen RK, Radisky D, Yoneda T, Barcellos-Hoff MH, Petersen OW, Turley EA, Bissell MJ. Phenotypic reversion or death of cancer cells by altering signaling pathways in three-dimensional contexts. J Natl Cancer Inst. 2002;94:1494–1503. doi: 10.1093/jnci/94.19.1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson CJ, Khaled WT. Mammary development in the embryo and adult: a journey of morphogenesis and commitment. Development. 2008;135:995–1003. doi: 10.1242/dev.005439. [DOI] [PubMed] [Google Scholar]
- Weaver VM, Petersen OW, Wang F, Larabell CA, Briand P, Damsky C, Bissell MJ. Reversion of the malignant phenotype of human breast cells in three-dimensional culture and in vivo by integrin blocking antibodies. J Cell Biol. 1997;137:231–245. doi: 10.1083/jcb.137.1.231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaver VM, Lelievre S, Lakins JN, Chrenek MA, Jones JC, Giancotti F, Werb Z, Bissell MJ. beta4 integrin-dependent formation of polarized three-dimensional architecture confers resistance to apoptosis in normal and malignant mammary epithelium. Cancer Cell. 2002;2:205–216. doi: 10.1016/s1535-6108(02)00125-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weir ML, Oppizzi ML, Henry MD, Onishi A, Campbell KP, Bissell MJ, Muschler JL. Dystroglycan loss disrupts polarity and beta-casein induction in mammary epithelial cells by perturbing laminin anchoring. J Cell Sci. 2006;119:4047–4058. doi: 10.1242/jcs.03103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White DE, Kurpios NA, Zuo D, Hassell JA, Blaess S, Mueller U, Muller WJ. Targeted disruption of beta1-integrin in a transgenic mouse model of human breast cancer reveals an essential role in mammary tumor induction. Cancer Cell. 2004;6:159–170. doi: 10.1016/j.ccr.2004.06.025. [DOI] [PubMed] [Google Scholar]
- Williams JM, Daniel CW. Mammary ductal elongation: differentiation of myoepithelium and basal lamina during branching morphogenesis. Dev Biol. 1983;97:274–290. doi: 10.1016/0012-1606(83)90086-6. [DOI] [PubMed] [Google Scholar]
- Williams CM, Engler AJ, Slone RD, Galante LL, Schwarzbauer JE. Fibronectin expression modulates mammary epithelial cell proliferation during acinar differentiation. Cancer Res. 2008;68:3185–3192. doi: 10.1158/0008-5472.CAN-07-2673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiseman BS, Sternlicht MD, Lund LR, Alexander CM, Mott J, Bissell MJ, Soloway P, Itohara S, Werb Z. Site-specific inductive and inhibitory activities of MMP-2 and MMP-3 orchestrate mammary gland branching morphogenesis. J Cell Biol. 2003;162:1123–1133. doi: 10.1083/jcb.200302090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witty JP, Wright JH, Matrisian LM. Matrix metalloproteinases are expressed during ductal and alveolar mammary morphogenesis, and misregulation of stromelysin-1 in transgenic mice induces unscheduled alveolar development. Mol Biol Cell. 1995;6:1287–1303. doi: 10.1091/mbc.6.10.1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf K, Wu YI, Liu Y, Geiger J, Tam E, Overall C, Stack MS, Friedl P. Multi-step pericellular proteolysis controls the transition from individual to collective cancer cell invasion. Nat Cell Biol. 2007;9:893–904. doi: 10.1038/ncb1616. [DOI] [PubMed] [Google Scholar]
- Wozniak MA, Desai R, Solski PA, Der CJ, Keely PJ. ROCK-generated contractility regulates breast epithelial cell differentiation in response to the physical properties of a three-dimensional collagen matrix. J Cell Biol. 2003;163:583–595. doi: 10.1083/jcb.200305010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu R, Nelson CM, Muschler JL, Veiseh M, Vonderhaar BK, Bissell MJ. Continuous laminin signaling is required for sustained activation of STAT5 and maintenance of mammary-specific function. J Cell Biol. 2009 doi: 10.1083/jcb.200807021. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.