

Abstract
Although most agree that 17β-estradiol is neuroprotective via a variety of mechanisms, less is known about the role that biological sex plays in receptor-mediated estradiol neuroprotection. To address this issue we isolated primary cortical neurons from rat pups sorted by sex and assessed the ability of estradiol to protect the neurons from death induced by glutamate. Five minute pretreatment with 10 to 50nM 17β-estradiol protected female but not male neurons from glutamate toxicity 24 hours later. Both estrogen receptor alpha (ERα) and estrogen receptor beta (ERβ) are expressed in these cultures. Experiments using an ERα selective agonist or antagonist indicate that this receptor is important for neuroprotection in female cortical neurons. The ERβ selective agonist conveys a small degree of neuroprotection to both male and female cortical neurons. Interestingly, we found that 17α estradiol and the novel membrane estrogen receptor (mER) agonist STX, but not bovine serum albumin conjugated estradiol or the GPR30 agonist G1 were neuroprotective in both male and female neurons. Taken together these data highlight a role for ERα in sexually dimorphic neuroprotection.
Keywords: neuroprotection, sexually dimorphic, estrogen receptor alpha, estrogen receptor beta, primary cortical neuron, glutamate
Introduction
It is well known that 17β- estradiol has protective effects in the brain. Early work demonstrated that estradiol increased viability in neuronal and glial cell lines (Bishop and Simpkins, 1994). Estradiol neuroprotection was subsequently shown to occur via estrogen receptor (ER) independent antioxidant (Behl et al., 1995) or ER-dependent mechanisms (Singer et al., 1996; Singer et al., 1999). Work from this laboratory further demonstrated that ER-mediated neuroprotection in primary cortical neurons involves the rapid stimulation of the src-protein tyrosine kinase (PTK)-mitogen activated protein kinase (MAPK) pathway (Singer et al., 1999). More recently, it has been shown that ERα and ERβ can mediate neuroprotection against beta amyloid toxicity and glutamate mediated oxidative stress (Fitzpatrick et al., 2002; Mize et al., 2003). In the intervening years, ER-mediated cellular responses to estradiol have been loosely grouped into two interconnected categories: genomic and rapid non-genomic signaling (Bjornstrom and Sjoberg, 2005; McEwen, 2001). Presently, it appears that both rapid non-genomic and genomic responses to estrogen are required for estrogen’s trophic and protective effects.
Despite this progress in understanding the cellular mechanisms of estradiol neuroprotection (Behl et al., 1995; Cordey and Pike, 2005; Fitzpatrick et al., 2002; Green et al., 1996; Heyer et al., 2005; Marin et al., 2003; McEwen, 2001; Mize et al., 2003; Nilsen et al., 2002; Singer et al., 1999; Singer et al., 1996; Wu et al., 2005; Xia et al., 2009; Zhao and Brinton, 2007; Zhao et al., 2005; Zhao et al., 2004), one issue has been largely ignored until recent years. What role does biological sex play in estrogen neuroprotection? Accumulating in vitro evidence suggests that the vulnerability to neurological insult is sexually dimorphic. Male cortical neurons are reported to be more susceptible to nitrosative stress and excitotoxicity while female cortical neurons are more susceptible to agents that induce apoptosis (Du et al., 2004). Male hippocampal neurons have been reported to be more susceptible to hypoxia than female hippocampal neurons (Heyer et al., 2005). Male organotypic hippocampal cultures are more susceptible to NMDA excitotoxicity or oxygen glucose deprivation than female counterparts (Li et al., 2005). Finally, intact female rats sustain less ischemic injury that ovariectomized female or male counterparts (Alkayed et al., 1998). In light of these data it is reasonable to hypothesize that the protective effects of estradiol are sexually dimorphic.
In the present study we used primary cortical neurons sorted by biological sex to determine if estradiol neuroprotection against glutamate toxicity is sexually dimorphic. Our data indicate that estradiol protects neurons isolated from female but not male rat pups from glutamate toxicity. We provide evidence that the ERα selective ligand PPT provides dose dependent neuroprotection in female but not male cortical neurons. In contrast, the ERβ selective agonist DPN conveys a small degree of neuroprotection in females and male neurons. Similarly, 17α- estradiol and the estrogenic agent STX are neuroprotective in both male and female neurons. Taken together these data highlight a role for ERα in sexually dimorphic neuroprotection.
Methods
Primary Neuron Culture
The experiments described in this report conformed to guidelines established by the National Institutes of Health and Oregon Health & Science University for the humane treatment of animals. Primary cultures of cortical neurons were prepared from Sprague Dawley rat pups on embryonic day 19 (E19) as described in Brewer et al. (Brewer and Price, 1996; Brewer et al., 1993). Briefly, brains were sorted by biological sex (presence of testes) and dissected in Hibernate-E media (Brain Bits Inc, Springfield, Ill) containing B27 supplement. Hippoccampi and olfactory bulbs were removed from cortices and discarded. Cortices were then incubated in Papain (20units/ml) in Hibernate-E minus calcium for 30 minutes at room temperature. Neurons were subsequently dissociated by manual trituration in Hibernate/B27. Neurons were counted and plated (approximately 330,000-360,000 per well) on poly-d-lysine coated glass cover slips in 12 well plates in “complete” Neurobasal media containing 2% B27, 1% Glutamax and penicillin-streptomycin (Invitrogen, Carlsbad California). Cultures were maintained in a 5% CO2 atmosphere for 10 days in vitro (DIV) in a humidified incubator. Unless otherwise indicated, experiments were performed on 10-11 DIV.
Western Blot
Cultured neurons were scraped from 150 mm culture dishes (Corning) into ice cold Immunoprecipitation Buffer containing 1% triton and protease inhibitors (Bryant et al., 2005). Protein concentration was determined for each whole cell lysate using a commercially available BCA Assay (Pierce). After adding sample loading buffer, lysates were resolved using SDS-Page. Membranes were immunoblotted as described (Bryant et al., 2005) using antibodies against ERα (6F11, Novacastra), ERβ (PA1-310B, Thermo) and beta actin (Sigma). Densitometry was performed using software on a UVP EpiChemi Darkroom machine as described in (Bryant et al., 2005).
Chemicals
17β- Estradiol and 17α- estradiol were purchased from Steraloids. ICI 182,780(ICI), 1,3-Bis(4-hydroxyphenyl)-4-methyl-5-[4-(2-piperidinylet hoxy)phenol]-1H-pyrazole dihydrochloride (MPP dihydrochloride or MPP), Propylpyrazole-triol (PPT), Diarylpropionitrile (DPN), R, R-Tetrahydrochrysene (R, R-THC) and (±)-1-[(3aR*,4S*,9bS*)-4-(6-Bromo-1,3-benzodioxol-5-yl) - 3a,4,5,9b-tetrahydro-3H-cyclopenta[c]quinolin-8-yl]- ethanone) (G1) were purchased from Tocris Bioscience (Ellisville, Mo). Bovine serum albumin (BSA) conjugated estradiol was purchased from Steraloids (Newport RI). STX was kindly provided by Drs. Tom Scanlan and Martin J. Kelly, Department of Physiology and Pharmacology, Oregon Health & Science University.
Live Dead Assay
All stock solutions (except glutamate and STX, G1 and BSA conjugated estradiol) were made in 95 % ethanol. STX and G1 were dissolved in DMSO. BSA conjugated estradiol was dissolved in 50mM Tris pH 8.5. Free estradiol was washed away using a Microcon 3kd centrifugal filter device (Millipore, Bedford MA) as described in (Stevis et al., 1999). Stock solutions were diluted in buffer “G” containing 2 mM KCl, 1mM MgSO4, 2.5mM CaCl2, 1mM NaH2PO4, 4.2 mM NaHCO3, 12.5mM Hepes, 10mM glucose, 0.1 M NaCl (Singer et al., 1996) prior to treatment. L-Glutamate was dissolved in buffer G and diluted further in the same buffer. All treatments were performed on at 10DIV. In experiments using ER ligands (17β-estradiol, 17α- estradiol, DPN, PPT, R, R-THC, STX G1, BSA-estradiol), neurons were exposed to vehicle or ER ligand for 5 minutes followed by a 5 min exposure to glutamate. After a total of 10 minutes, neurons received one wash of complete neurobasal media. Fresh media was placed on neurons and they were returned to the incubator. In experiments using ER “antagonists” (ICI or MPP), neurons were pretreated for 15 minutes with the antagonist or vehicle followed by a 5 minute ER ligand or vehicle exposure followed by a 5 minute glutamate exposure. Neurons were thus exposed to the antagonist for a total of 25 minutes. The neurons received one media wash and then were returned to the incubator in fresh complete media. Twenty four hours after treatment (11DIV), neurons were stained using the commercially available Live/Dead (Invitrogen) assay using manufacturer’s instructions. Live cells were stained fluorescent green by Calcein AM, which stains esterases in living cells. Dead cells are stained fluorescent red by the ethidium bromide homodimer. Cover slips were placed on microscope slides and examined using FITC and TRITC filters on a Zeiss Axioskop FS microscope. Three images (TIFFs) were captured from each cover slip. Three cover slips were sampled for each experimental condition. A minimum of three platings (biological replicates) was used for each experiment. Percent survival (death) was calculated by dividing the number of live (dead) cells by the total number of cells. All data was normalized to the mean of vehicle (or glutamate, in the case of percent death) treated cells. A two way (Sex by Treatment) ANOVA was calculated followed by post-hoc Bonferroni t-tests for each experiment. ANOVAs were always statistically significant due to the glutamate mediated cell death. Unless otherwise indicated, we only report post-hoc t-tests comparing glutamate to ER ligand + glutamate (i.e. neuroprotection).
Results
Five minute pre-exposure to estradiol (50nM) protected female but not male neurons from glutamate (5mM) toxicity (***Bonferroni post hoc t (female glutamate vs. female estradiol+ glutamate) =3.911,p<0.001 Figure 1). Since susceptibility to glutamate/glycine toxicity has been reported to be sexually dimorphic (Du et al., 2004), we wanted to determine if there was a sex difference in neuronal survival 24 hours after a 5 minute glutamate exposure. We did not observe sexually dimorphic susceptibility to glutamate toxicity in the absence (Figure 2) or presence (data not shown) of glycine in our experimental paradigm. In order to determine whether the observed sexually dimorphic neuroprotection involved ER activation, male and female neurons were pre-exposed to vehicle (ethanol) or ICI 182,780 (1uM), for 15 minutes, followed by a 5 minute exposure to estradiol (10nM) and a subsequent 5 minute glutamate (5mM) exposure. We observed neuroprotection in estradiol treated female neurons but not male neurons (Figure 3a) (***Bonferroni post hoc t (glutamate vs. estradiol+ glutamate) =4.977, p<0.001) in vehicle pretreated neurons. Pre-exposure to ICI, blocked neuroprotection in female neurons and had no effect on male neuron survival (Figure 3b). Although ICI can have SERM-like effects, it has historically been used as an ER antagonist that implicates an ERα/β mediated response. Immunoblots of whole cell lysates (Figure 4) demonstrated that at 10DIV, ERα content of female neurons was greater than that male neurons (*t=4.773, p=0.0412). In contrast to ERα, there was no sex difference in ERβ expression.
Figure 1.


50nM estradiol protects female but not male neurons from glutamate toxicity. At 10DIV, male and female primary cortical neurons were exposed to 0.005% ethanol or 50nM estradiol for 5 minutes prior to a five minute exposure to 5mM glutamate. After 1 wash in complete Neurobasal media, cells received fresh media and were returned to the incubator for 24 hours. A. Representative images from the Live/Dead assay (Invitrogen). Living neurons are stained green by Calcein AM. Dead cells are stained red by the ethidium bromide homodimer. B. 5 minute pre-exposure to 50nM estradiol protects female neurons (***Bonferroni t (glutamate vs. estradiol+ glutamate) =3.911, p<0.001) but not male neurons from glutamate toxicity (n=5).
Figure 2.

Glutamate toxicity is not sexually dimorphic in our experimental protocol. Male and female neurons were exposed to varying doses (.5mM, 1mM and 5mM) for 5 minutes. Neurons were handled as described in figure 1 and the methods section. There was no sex difference in percent survival (n=3).
Figure 3.


ICI 182, 780 blocks estrogen neuroprotection in female neurons. Male and female neurons were exposed to (A) 0.01% ethanol or (B) 1uM ICI for 15 minutes prior to a 5 minute exposure to 0.001% ethanol or 10nM estradiol. Neurons we subsequently exposed to 5mM glutamate for 5 minutes. (A) Estradiol (10 nM) protected female neurons but not male neurons from glutamate toxicity in ethanol pretreated neurons (***Bonferroni post hoc t (glutamate vs. estradiol+ glutamate) =4.977, p<0.001, n=4). (B) ICI pretreatment blocked estradiol protection in female neurons (n=4).
Figure 4.
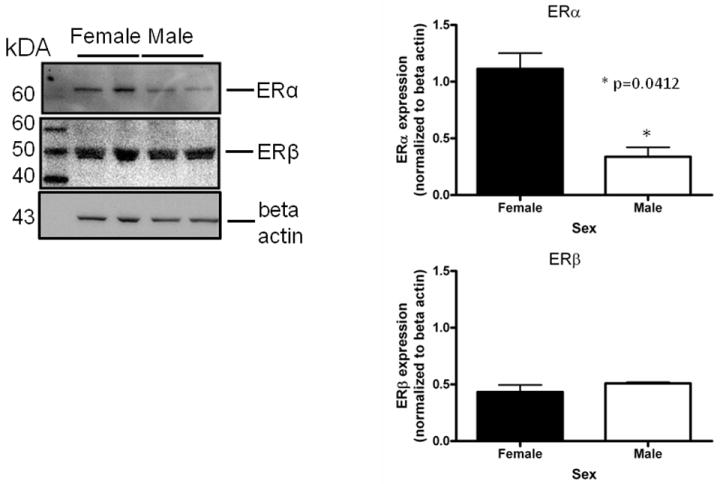
Female neurons contain more immunoreactive ERα than male neurons at 10DIV. Whole cell lysates from female and male neurons were immunoblotted using antibodies against ERα (6F11, Novacastra) ERβ (PA1-310B, Thermo) and beta actin (Sigma). Densitometric analysis revealed that there was a statistically significant difference in the ratio of ERα to beta actin (*t=4.773, p=0.0412, n=2) between female and male neurons.
Given the presence of both ERs in our cultures, pharmacological probes were used to assess the role of each receptor in neuroprotection. Neurons were pre-exposed to the ERα agonist propylpyrazole-triol (PPT) or the ERβ agonist diarylpropiolnitrile (DPN) for 5 minutes prior to a 5 minute glutamate exposure. The initial doses of PPT (10nM) and DPN (1nM) were chosen based on demonstrated transcriptional activation of estrogen response elements (EREs) via the target receptor but not the counterpart (Harrington et al., 2003). Female neurons exposed to PPT exhibited statistically significant neuroprotection (t=2.610, p<0.05 Figure 5a). PPT had no effect on male survival. In order to further evaluate the role of PPT in sexually dimorphic neuroprotection, we tested a range of doses, including 10nM, 1nM and .1nM. A two way (Sex X Treatment) analysis of variance indicated that there was an effect of Sex on PPT neuroprotection (F=6.11, p=0.02, Figure 5b). Bonferroni post-hoc tests indicate that PPT was neuroprotective at 10nM (t=2.42, p<0.05) and 1nM (t=6.96, p<0.05) but not 0.01nM in female neurons. At a 1nM concentration, DPN did not protect female or male neurons (Figure 5c). However, a follow up experiment examining a range of doses from 1nM to 0.01nM indicated that there was a modest overall effect of treatment in DPN exposed neurons (F =4.02, p=0.01). Post-hoc comparisons using the Bonferroni post test were not statistically significant. In order to further investigate the role of ERα in neuroprotection in female neurons we pretreated male and female neurons with the ERα antagonist MPP dihydrochloride (MPP, 1 ump) for 15 minutes prior to exposure to E2 and glutamate. MPP blocked E2 neuroprotection in female neurons (Figure 6). Thus, although our data suggests that the sex difference in estrogen neuroprotection is primarily ERα dependent, ERβ has a role in neuroprotection.
Figure 5.




The ERα selective ligand PPT is neuroprotective in female neurons only. Male and female neurons treated with the ERα selective ligand PPT (A, B) or the ERβ selective ligand DPN (C, D) for 5 minutes prior to a 5 minute glutamate exposure. (A) PPT was neuroprotective in female neurons Bonferroni t (glutamate vs. ppt+ glutamate)=2.610, p<0.05, n=3) but not male neurons. (B) PPT was neuroprotective in female neurons at 10nM (t=2.42, p<0.05) and 1nM (t=6.96, p<0.05) but not 0.01nM concentrations. (C) At a 1 nM concentration, DPN did not protect male or female neurons (n=4). (D) Neurons were exposed to three concentrations of (1nM, 0.1nM and 0.01nM) of dPN. A two way (Sex X Treatment) ANOVA indicated that there was an overall effect of treatment in neurons exposed to DPN (F=4.02, p=0.01). Post hoc tests were not statistically significant, however.
Figure 6.

The selective ERα antagonist MPP blocks estradiol neuroprotection.
(A) Male and female neurons were pretreated with 1uM MPP dihydrochloride for 15 minutes prior to exposure to estradiol and glutamate. Estradiol was not protective in female neurons under these conditions (n=4).
Since other ER candidates have been postulated in recent years, we assayed neuroprotection using ligands reported to be selective for these ER candidates. Neurons were exposed to STX, a novel diphenylacrylamide compound that has 100,000 fold reduced affinity for ERα and ERβ (Tobias et al., 2006). Five minute STX (10nM) pretreatment protected both male (*Bonferroni post hoc t (glutamate vs. STX+ glutamate) =2.557, p<0.05) and female (*Bonferroni post hoc t (glutamate vs. STX+ glutamate) =3.076, p<0.05) neurons from glutamate toxicity (Figure 7a). Since GPR30 has been reported to be a membrane estrogen receptor (Filardo et al., 2000; Revankar et al., 2005; Thomas et al., 2005), we evaluated the neuroprotective effect of the selective non-steroidal GPR30 agonist G1 on neuroprotection in female and male neurons. G1 was not protective in male or female neurons (Figure 7b). We also evaluated the neuroprotective effects of 17α-estradiol and bovine serum conjugated estradiol. 17α-estradiol was neuroprotective in both male (post hoc tglutamate vs 17α-estradiol +glutamate =3.507, p<0.01) and female (post hoc tglutamate+ 17α-estradiol +glutamate=3.18, p<0.05) neurons (Figure 7c). Finally, a 5 minute pre-exposure to bsa conjugated estradiol did not protect against glutamate toxicity (Figure 7d).
Figure 7.




(A) STX and 17α-estradiol protect male and female neurons. Neurons were exposed to 10 nM STX for 5 minutes prior to a 5 minute exposure to 5mM glutamate. STX protected both male (*Bonferroni post hoc t (glutamate vs. STX+ glutamate) =2.557,p<0.05, n=3) and female (*Bonferroni post hoc t (glutamate vs. STX+ glutamate) =3.076,p<0.05, n=3) neurons from glutamate toxicity.(7b) Neurons were exposed to 10nM of the non-steroidal GPR30 agonist G1for 5 minutes prior to exposure to glutamate. G1 was not neuroprotective (n=3) (7c) Neurons were exposed to 10 nM of 17α estradiol for 5 minutes prior to a 5 minute exposure to 5mM glutamate. 17α- estradiol is neuroprotective in male (post hoc tglutamate vs 17α- estradiol+glutamate=3.507,p<0.01) and female (post hoc tglutamate vs 17α-estradiol+glutamate=3.18, p<0.05) neurons (n=3). (7d) Neurons pre-exposed to 10nM bovine serum albumin conjugated estradiol for 5 minutes were not protected from glutamate toxicity (n=3).
Discussion
ERα activation is important for neuroprotection in female cortical neurons
Taken together, the data from this study indicate that in E19 isolated primary cortical neurons, estradiol neuroprotection against glutamate toxicity is sexually dimorphic. Furthermore, this sexual dimorphism is mostly mediated by ERα in rat female cortical neurons. The ERβ selective agonist, DPN provides some degree of neuroprotection in both male and female neurons. Similarly, 17α-estradiol and STX are also neuroprotective in both sexes. Sex segregation of primary cortical neuron cultures has allowed us to dissociate the neuroprotective effectiveness of PPT and DPN. The implication of these data is that there is a “sex by receptor” interaction underlying ER mediated neuroprotection in cortical neurons. Our findings are consistent with the literature, which highlight a role for ERα in estradiol neuroprotection in female mice and rats. Dubal et al., (Dubal et al., 2001) demonstrated that ERα is required for neuroprotection against delayed cell death after permanent middle cerebral artery occlusion (MCAO). In their studies, estradiol protected ERβKO mice but not ERαKO mice 24 hours after the onset of MCAO. They also observed an induction of ERα mRNA and protein in the cortex of MCAO treated estradiol replaced female rats (Dubal et al., 2006). We also observe more ERα expression in whole cell lysates from rat female neurons than male neurons at 10DIV. Estradiol is also cardioprotective in female rats against myocardial ischemia via an ERα-mediated mechanism (Jeanes et al., 2008). The ERα agonist ERA-45 was cardioprotective while the ERβ antagonist failed to block estrogen cardioprotection, further highlighting a role for ERα in estradiol protection against insult. In contrast to these findings, data from other studies did not support the idea that ERα is protective against transient MCAO (Sampei et al., 2000). This may be due to methodological differences such as permanent (Dubal et al., 2006; Dubal et al., 2001) versus transient MCAO (Sampei et al., 2000) and the use of intact animals in the later study compared to ovariectomized and/or estradiol replaced animals in the prior studies.
ERβ conveys some degree of neuroprotection in male and female neurons
Although our data indicate that ERα has a prominent role in sexually dimorphic neuroprotection, it appears that ERβ is modestly neuroprotective in both sexes. Our findings contrast with Xia et al., (Xia et al., 2009) who did not observe neuroprotection in mixed primary cortical neurons exposed to 0.1 nM PPT or 0.1nM DPN (co-administration or 24 hour pretreatment) 18 hours after glutamate exposure. The lack of protection may be due to ligand-dependent ER down-regulation. For example, ERα expression is down-regulated in human aortic smooth muscle cells after 48 hour exposure to Estradiol, PPT, and DPN (Barchiesi et al., 2004). This data, along with our own highlight the interaction of ligand exposure time and ER expression levels on ER mediated responses. Consistent with our findings, ERβ has been shown to be protective in males under other experimental conditions. Estradiol and the ERβ selective agonist DPN were neuroprotective against transient global ischemia resultant from cardiac arrest/cardiopulmonary resuscitation (CA/CPR) in the hippocampal CA1, caudate putamen of male mice (Noppens et al., 2009). Conversely, the selective ERα agonist, PPT was not protective in the caudate putamen or the CA1 field of the hippocampus. Taken together these data indicate that ER mediated sexually dimorphic neuroprotection can vary from one brain structure to the next.
Despite differences between our experimental protocol and that of published reports, consistent themes do emerge. Cordey and Pike (Cordey and Pike, 2005) used mixed (male and female) cortical neurons to determine which ER was important for neuroprotection against beta amyloid (Aβ) toxicity. Neurons were exposed to ER ligands for 1 hour prior to a 24 hour Aβ exposure. They observed ICI sensitive neuroprotection with estradiol, 17α-estradiol, PPT and DPN. Furthermore, PKC activation was required for neuroprotection via all of these ligands except DPN (Cordey and Pike, 2005). Our data is consistent with these findings in that17α-estradiol is neuroprotective and the response to DPN is not identical to that of via estradiol or PPT. Our data is somewhat consistent with that of Zhao et al., (Zhao et al., 2004), who demonstrated that ERα and ERβ activation is important for neuroprotection against glutamate toxicity in mixed primary hippocampal neurons. Neuroprotection was most robust in the picomolar range which minimally activates several transcription factors including ERE, sodium – hydrogen exchanger regulatory factor ezrin-radixin-moesin binding protein 50 (NHE-RF/EBP50), and TGFβ after 24 hours (Harrington et al., 2003). Although our current findings are generally consistent with (Zhao et al., 2004) and prior work in this laboratory (Fitzpatrick et al., 2002; Mize et al., 2003), we find that sexually dimorphic neuroprotection appears to be due solely to ERα.
Sexually dimorphic neuroprotection does not involve novel ER candidates
Our data suggest that sexually dimorphic neuroprotection against glutamate toxicity is a consequence of ERα and not other ER candidates. Several candidate estrogen receptors have been postulated, including the STX-responsive membrane estrogen receptor (Qiu et al., 2003; Qiu et al., 2006), ER”X” (Toran-Allerand et al., 2002) and GPR30 (Filardo et al., 2000; Revankar et al., 2005; Thomas et al., 2005). We find that STX is neuroprotective in both male and female neurons. Our data is consistent with previously reported STX neuroprotection, in vivo. It protects neurons in the CA1 field of the female rat hippocampus from transient global ischemia (Lebesgue et al., 2010). STX is a diphenylacrylamide compound (Tobias et al., 2006) that is known to stimulate rapid, estradiol-sensitive, non-genomic responses such as, phospholipase C and protein kinase A and protein kinase C, MAPK and PI3K (Qiu et al., 2003; Qiu et al., 2006; Lin et al., 2009). STX has a 100,000-fold reduced affinity for ERα and ERβ compared to estradiol (Tobias et al., 2006) and alters gene expression (Roepke et al., 2008) but it is not uterotrophic in vivo (Qiu et al., 2003). Furthermore, STX does not stimulate or block ERE transcription via ERα or ERβ (Lin et al., 2009). Although direct STX binding to GPR30 has not been demonstrated, recently published data suggests that GPR30 could be involved in STX stimulated non-genomic and genomic signaling. STX stimulates the phosphorylation of the estrogen steroidogenic factor 1(SF-1) via MAPK and PI3K. (Lin et al., 2009). SiRNA knock down of GPR30 blocks estradiol and STX stimulated aromatase (Cyp19a1) transcription (Lin et al., 2009). The idea that GPR30 is a bona fide estrogen receptor is controversial as the evidence for estradiol binding for GPR30 has been called into question (Langer et al., 2009). Finally, our data are consistent with the idea that GPR30 and the STX responsive membrane estrogen receptor are not the same vis-à-vis neuroprotection given the lack of G1 neuroprotection in this experimental paradigm.
Although 17α- estradiol has historically been considered an inactive isomer, there is evidence that it is neuroprotective in HT22 cells (Behl et al., 1997) and SK-N-SH cells (Green et al 1997). At the time it was postulated that 17α- estradiol worked via an estrogen receptor independent mechanism. In contrast to this early data, 17α-estradiol has more recently been postulated as a selective ligand for the ER candidate ERX. Evidence has been published supporting the idea that17α-estradiol activates MAPK signaling via developmentally regulated, caveolae localized ERX (Toran-Allerand et al., 2002). Our data are consistent with early work which indicates that 17α-estradiol is a neuroprotective steroid. Our data also imply that this ligand might operate via a mechanism that is independent of ERα, at least in male neurons. Finally, we did not observe neuroprotection in neurons exposed to bovine serum albumin conjugated estradiol. This data implies that sexually dimorphic neuroprotection involves primarily an ERα mediated transcriptional response.
We did not observe sexually dimorphic susceptibility to glutamate toxicity in the absence of presence (data not shown) of glycine in our experimental paradigm. Our findings contrast somewhat with Du et al, (Du et al., 2004), who reported that male neurons were more susceptible to glutamate/glycine excitotoxicity than female neurons. In their excitotoxicity experiments, neurons were exposed to glutamate/glycine for 24 hours prior to examining cell viability. However, our findings are consistent with Du et al (Du et al., 2004) in that we observe estradiol neuroprotection in female neurons but not male neurons using a 10 or 50nM dose..
Conclusions
Taken together, our data indicate that estradiol neuroprotection is sexually dimorphic in 10-11DIV neurons. This sex difference appears to involve ERα and not ERβ and is correlated with increased ERα expression in female neurons. Ligands selective for ERβ, the STX sensitive membrane receptor and ERX are also neuroprotective in both sexes, suggesting that (non) genomic responses downstream of these receptor candidates are not identical to that of ERα. Ongoing work is aimed at examining sexually dimorphic downstream signaling responses to estradiol.
Acknowledgments
We would like to acknowledge Drs. Robert Shapiro and Laird Sheldahl for helpful suggestions and discussions as well as Drs. Tom Scanlan and Martin Kelly for kindly providing the STX. This work was supported by NIH NS 20311 (DMD)
Footnotes
Disclosure Statement: The authors have nothing to disclose
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- Alkayed NJ, Harukuni I, Kimes AS, London ED, Traystman RJ, Hurn PD. Gender-linked brain injury in experimental stroke. Stroke. 1998;29:159–165. doi: 10.1161/01.str.29.1.159. [DOI] [PubMed] [Google Scholar]
- Barchiesi F, Jackson EK, Imthurn B, Fingerle J, Gillespie DG, Dubey RK. Differential regulation of estrogen receptor subtypes alpha and beta in human aortic smooth muscle cells by oligonucleotides and estradiol. J Clin Endocrinol Metab. 2004;89:2373–2381. doi: 10.1210/jc.2003-030821. [DOI] [PubMed] [Google Scholar]
- Behl C, Widmann M, Trapp T, Holsboer F. 17-beta estradiol protects neurons from oxidative stress-induced cell death in vitro. Biochem Biophys Res Commun. 1995;216:473–482. doi: 10.1006/bbrc.1995.2647. [DOI] [PubMed] [Google Scholar]
- Behl C, Skutella T, Lezoualc’h F, Post A, Widmann M, Newton CJ, Holsboer F. Neuroprotection against oxidative stress by estrogens: structure-activity relationship. Mol Parmacol. 1997;51:535–541. [PubMed] [Google Scholar]
- Bishop J, Simpkins JW. Estradiol treatment increases viability of glioma and neuroblastoma cells in vitro. Mol Cell Neurosci. 1994;5:303–308. doi: 10.1006/mcne.1994.1036. [DOI] [PubMed] [Google Scholar]
- Bjornstrom L, Sjoberg M. Mechanisms of estrogen receptor signaling: convergence of genomic and nongenomic actions on target genes. Mol Endocrinol. 2005;19:833–842. doi: 10.1210/me.2004-0486. [DOI] [PubMed] [Google Scholar]
- Brewer GJ, Price PJ. Viable cultured neurons in ambient carbon dioxide and hibernation storage for a month. Neuroreport. 1996;7:1509–1512. doi: 10.1097/00001756-199606170-00014. [DOI] [PubMed] [Google Scholar]
- Brewer GJ, Torricelli JR, Evege EK, Price PJ. Optimized survival of hippocampal neurons in B27-supplemented Neurobasal, a new serum-free medium combination. J Neurosci Res. 1993;35:567–576. doi: 10.1002/jnr.490350513. [DOI] [PubMed] [Google Scholar]
- Bryant DN, Bosch MA, Ronnekleiv OK, Dorsa DM. 17-Beta estradiol rapidly enhances extracellular signal-regulated kinase 2 phosphorylation in the rat brain. Neuroscience. 2005;133:343–352. doi: 10.1016/j.neuroscience.2005.02.024. [DOI] [PubMed] [Google Scholar]
- Cordey M, Pike CJ. Neuroprotective properties of selective estrogen receptor agonists in cultured neurons. Brain Res. 2005;1045:217–223. doi: 10.1016/j.brainres.2005.03.032. [DOI] [PubMed] [Google Scholar]
- Du L, Bayir H, Lai Y, Zhang X, Kochanek PM, Watkins SC, Graham SH, Clark RS. Innate gender-based proclivity in response to cytotoxicity and programmed cell death pathway. J Biol Chem. 2004;279:38563–38570. doi: 10.1074/jbc.M405461200. [DOI] [PubMed] [Google Scholar]
- Dubal DB, Rau SW, Shughrue PJ, Zhu H, Yu J, Cashion AB, Suzuki S, Gerhold LM, Bottner MB, Dubal SB, Merchanthaler I, Kindy MS, Wise PM. Differential modulation of estrogen receptors (ERs) in ischemic brain injury: a role for ERalpha in estradiol-mediated protection against delayed cell death. Endocrinology. 2006;147:3076–3084. doi: 10.1210/en.2005-1177. [DOI] [PubMed] [Google Scholar]
- Dubal DB, Zhu H, Yu J, Rau SW, Shughrue PJ, Merchenthaler I, Kindy MS, Wise PM. Estrogen receptor alpha, not beta, is a critical link in estradiol-mediated protection against brain injury. Proc Natl Acad Sci U S A. 2001;98:1952–1957. doi: 10.1073/pnas.041483198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filardo EJ, Quinn JA, Bland KI, Frackelton AR., Jr Estrogen-induced activation of Erk-1 and Erk-2 requires the G protein-coupled receptor homolog, GPR30, and occurs via trans-activation of the epidermal growth factor receptor through release of HB-EGF. Mol Endocrinol. 2000;14:1649–1660. doi: 10.1210/mend.14.10.0532. [DOI] [PubMed] [Google Scholar]
- Fitzpatrick JL, Mize AL, Wade CB, Harris JA, Shapiro RA, Dorsa DM. Estrogen-mediated neuroprotection against beta-amyloid toxicity requires expression of estrogen receptor alpha or beta and activation of the MAPK pathway. J Neurochem. 2002;82:674–682. doi: 10.1046/j.1471-4159.2002.01000.x. [DOI] [PubMed] [Google Scholar]
- Green PS, Gridley KE, Simpkins JW. Estradiol protects against beta-amyloid (25-35)-induced toxicity in SK-N-SH human neuroblastoma cells. Neurosci Lett. 1996;218:165–168. doi: 10.1016/s0304-3940(96)13148-7. [DOI] [PubMed] [Google Scholar]
- Green PS, Bishop J, Simpkins JW. 17 alpha-estradiol exerts neuroprotective effects on SK-N-SH cells. J Neurosci. 1997;17:511–515. doi: 10.1523/JNEUROSCI.17-02-00511.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrington WR, Sheng S, Barnett DH, Petz LN, Katzenellenbogen JA, Katzenellenbogen BS. Activities of estrogen receptor alpha- and beta-selective ligands at diverse estrogen responsive gene sites mediating transactivation or transrepression. Mol Cell Endocrinol. 2003;206:13–22. doi: 10.1016/s0303-7207(03)00255-7. [DOI] [PubMed] [Google Scholar]
- Heyer A, Hasselblatt M, von AN, Hafner H, Siren AL, Ehrenreich H. In vitro gender differences in neuronal survival on hypoxia and in 17beta-estradiol-mediated neuroprotection. J Cereb Blood Flow Metab. 2005;25:427–430. doi: 10.1038/sj.jcbfm.9600056. [DOI] [PubMed] [Google Scholar]
- Jeanes HL, Tabor C, Black D, Ederveen A, Gray GA. Oestrogen-mediated cardioprotection following ischaemia and reperfusion is mimicked by an oestrogen receptor (ER)alpha agonist and unaffected by an ER beta antagonist. J Endocrinol. 2008;197:493–501. doi: 10.1677/JOE-08-0071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langer G, Bader B, Meoli L, Isensee J, Delbeck M, Noppinger PR, Otto C. A critical review of fundamental controversies in the field of GPR30 research. Steroids. 2010;75:603–610. doi: 10.1016/j.steroids.2009.12.006. [DOI] [PubMed] [Google Scholar]
- Lebesgue D, Traub M, De Butte-Smith M, Chen C, Zukin RS, Kelly MJ, Etgen AM. Acute administration of non-classical estrogen receptor agonists attenuates ischemia-induced hippocampal neuron loss in middle-aged female rats. PLoS One. 2010;5:e8642. doi: 10.1371/journal.pone.0008642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Pin S, Zeng Z, Wang MM, Andreasson KA, McCullough LD. Sex differences in cell death. Ann Neurol. 2005;58:317–321. doi: 10.1002/ana.20538. [DOI] [PubMed] [Google Scholar]
- Lin BC, Suzawa M, Blind RD, Tobias SC, Bulun SE, Scanlan TS, Ingraham HA. Stimulating the GPR30 estrogen receptor with a novel tamoxifen analogue activates SF-1 and promotes endometrial cell proliferation. Cancer Res. 2009;69:5415–5423. doi: 10.1158/0008-5472.CAN-08-1622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marin R, Guerra B, Hernandez-Jimenez JG, Kang XL, Fraser JD, Lopez FJ, Alonso R. Estradiol prevents amyloid-beta peptide-induced cell death in a cholinergic cell line via modulation of a classical estrogen receptor. Neuroscience. 2003;121:917–926. doi: 10.1016/s0306-4522(03)00464-0. [DOI] [PubMed] [Google Scholar]
- McEwen BS. Invited review: Estrogens effects on the brain: multiple sites and molecular mechanisms. J Appl Physiol. 2001;91:2785–2801. doi: 10.1152/jappl.2001.91.6.2785. [DOI] [PubMed] [Google Scholar]
- Mize AL, Shapiro RA, Dorsa DM. Estrogen receptor-mediated neuroprotection from oxidative stress requires activation of the mitogen-activated protein kinase pathway. Endocrinology. 2003;144:306–312. doi: 10.1210/en.2002-220698. [DOI] [PubMed] [Google Scholar]
- Nilsen J, Chen S, Brinton RD. Dual action of estrogen on glutamate-induced calcium signaling: mechanisms requiring interaction between estrogen receptors and src/mitogen activated protein kinase pathway. Brain Res. 2002;930:216–234. doi: 10.1016/s0006-8993(02)02254-0. [DOI] [PubMed] [Google Scholar]
- Noppens RR, Kofler J, Grafe MR, Hurn PD, Traystman RJ. Estradiol after cardiac arrest and cardiopulmonary resuscitation is neuroprotective and mediated through estrogen receptor-beta. J Cereb Blood Flow Metab. 2009;29:277–286. doi: 10.1038/jcbfm.2008.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orikasa C, Kondo Y, Sakuma Y. Transient transcription of the somatostatin gene at the time of estrogen-dependent organization of the sexually dimorphic nucleus of the rat preoptic area. Endocrinology. 2009;148:1144–1149. doi: 10.1210/en.2006-1214. [DOI] [PubMed] [Google Scholar]
- Qiu J, Bosch MA, Tobias SC, Grandy DK, Scanlan TS, Ronnekleiv OK, Kelly MJ. Rapid signaling of estrogen in hypothalamic neurons involves a novel G-protein-coupled estrogen receptor that activates protein kinase C. J Neurosci. 2003;23:9529–9540. doi: 10.1523/JNEUROSCI.23-29-09529.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu J, Bosch MA, Tobias SC, Krust A, Graham SM, Murphy SJ, Korach KS, Chambon P, Scanlan TS, Ronnekleiv OK, Kelly MJ. A G-protein-coupled estrogen receptor is involved in hypothalamic control of energy homeostasis. J Neurosci. 2006;26:5649–5655. doi: 10.1523/JNEUROSCI.0327-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Revankar CM, Cimino DF, Sklar LA, Arterburn JB, Prossnitz ER. A transmembrane intracellular estrogen receptor mediates rapid cell signaling. Science. 2005;307:1625–1630. doi: 10.1126/science.1106943. [DOI] [PubMed] [Google Scholar]
- Roepke TA, Xue C, Bosch MA, Scanlan TS, Kelly MJ, Ronnekleiv OK. Genes associated with membrane initiated signaling of estrogen and energy homeostasis. Endocrinology. 2008;149:6113–6124. doi: 10.1210/en.2008-0769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sampei K, Goto S, Alkayed NJ, Crain BJ, Korach KS, Traystman RJ, Demas GE, Nelson RJ, Hurn PD. Stroke in estrogen receptor-alpha-deficient mice. Stroke. 2000;31:738–743. doi: 10.1161/01.str.31.3.738. [DOI] [PubMed] [Google Scholar]
- Singer CA, Figueroa-Masot XA, Batchelor RH, Dorsa DM. The mitogen-activated protein kinase pathway mediates estrogen neuroprotection after glutamate toxicity in primary cortical neurons. J Neurosci. 1999;19:2455–2463. doi: 10.1523/JNEUROSCI.19-07-02455.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singer CA, Rogers KL, Strickland TM, Dorsa DM. Estrogen protects primary cortical neurons from glutamate toxicity. Neurosci Lett. 1996;212:13–16. doi: 10.1016/0304-3940(96)12760-9. [DOI] [PubMed] [Google Scholar]
- Stevis PE, Deecher DC, Suhadolnik L, Mallis LM, Frail DE. Differential effects of estradiol and estradiol-BSA conjugates. Endocrinology. 1999;140:5455–5458. doi: 10.1210/endo.140.11.7247. [DOI] [PubMed] [Google Scholar]
- Thomas P, Pang Y, Filardo EJ, Dong J. Identity of an estrogen membrane receptor coupled to a G protein in human breast cancer cells. Endocrinology. 2005;146:624–632. doi: 10.1210/en.2004-1064. [DOI] [PubMed] [Google Scholar]
- Tobias SC, Qiu J, Kelly MJ, Scanlan TS. Synthesis and biological evaluation of SERMs with potent nongenomic estrogenic activity. ChemMedChem. 2006;1:565–571. doi: 10.1002/cmdc.200500098. [DOI] [PubMed] [Google Scholar]
- Toran-Allerand CD, Guan X, MacLusky NJ, Horvath TL, Diano S, Singh M, Connolly ES, Jr, Nethrapalli IS, Tinnikov AA. ER-X: a novel, plasma membrane-associated, putative estrogen receptor that is regulated during development and after ischemic brain injury. J Neurosci. 2002;22:8391–8401. doi: 10.1523/JNEUROSCI.22-19-08391.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu TW, Wang JM, Chen S, Brinton RD. 17Beta-estradiol induced Ca2+ influx via L-type calcium channels activates the Src/ERK/cyclic-AMP response element binding protein signal pathway and BCL-2 expression in rat hippocampal neurons: a potential initiation mechanism for estrogen-induced neuroprotection. Neuroscience. 2005;135:59–72. doi: 10.1016/j.neuroscience.2004.12.027. [DOI] [PubMed] [Google Scholar]
- Xia Y, Xing JZ, Krukoff TL. Neuroprotective effects of R,R-tetrahydrochrysene against glutamate-induced cell death through anti-excitotoxic and antioxidant actions involving estrogen receptor-dependent and -independent pathways. Neuroscience. 2009;162:292–306. doi: 10.1016/j.neuroscience.2009.04.068. [DOI] [PubMed] [Google Scholar]
- Zhao L, Brinton RD. Estrogen receptor alpha and beta differentially regulate intracellular Ca(2+) dynamics leading to ERK phosphorylation and estrogen neuroprotection in hippocampal neurons. Brain Res. 2007;1172:48–59. doi: 10.1016/j.brainres.2007.06.092. [DOI] [PubMed] [Google Scholar]
- Zhao L, Chen S, Ming WJ, Brinton RD. 17beta-estradiol induces Ca2+ influx, dendritic and nuclear Ca2+ rise and subsequent cyclic AMP response element-binding protein activation in hippocampal neurons: a potential initiation mechanism for estrogen neurotrophism. Neuroscience. 2005;132:299–311. doi: 10.1016/j.neuroscience.2004.11.054. [DOI] [PubMed] [Google Scholar]
- Zhao L, Wu TW, Brinton RD. Estrogen receptor subtypes alpha and beta contribute to neuroprotection and increased Bcl-2 expression in primary hippocampal neurons. Brain Res. 2004;1010:22–34. doi: 10.1016/j.brainres.2004.02.066. [DOI] [PubMed] [Google Scholar]