

Abstract
cRNA microarray and real-time PCR (qPCR) studies from our lab identified five Cell Cycle Pathway (CCP) genes (CCNA2, CCNE2, CDC25A, CDKN1B, PLK-1) as targets for luteolin in PC-3 prostate cancer cells (Shoulars et. al, J Steroid Biochem Mol Biol, 118: 41–50, 2010). In this paper, Ingenuity Pathway Analysis of the microarray data identified 7 luteolin-regulated genes (EGFR, c-Fos, SOS, GRB2, JNK1, MKK4 and RasGAP) in the Epidermal Growth Factor Signaling Pathway (EGFSP) potentially involved in luteolin regulation of CCP genes and cell proliferation. To address these possibilities, we compared the response profiles (RNA and protein) of these EGFSP and CCP genes to luteolin and gefitinib by real-time PCR (qPCR) and Western blot analyses. Luteolin and gefitinib are known antagonists of EGFR-associated tyrosine protein kinase. Thus, the response profiles of EGFR regulated EGFSP or CCP genes should be very similar if genes in both pathways are controlled through this common mechanism of action. Treatment of PC-3 cell with luteolin for 24 hours caused a 4-fold stimulation of c-Fos gene expression, significant inhibition (p<0.001) of the CCP genes and G2/M arrest. Treatment of PC-3 cells with gefitinib also inhibited most of the CCP genes in a fashion similar to that of luteolin, however, the EGFR antagonist inhibited c-Fos gene expression, stimulated CDKN1B (p27) and arrested the cells in G0/G1. Thus, although the response patterns of most of the CCP genes to luteolin or gefitinib were similar, the effects of the two compounds on EGFSP gene expression and cell cycle arrest were clearly different. Combination studies revealed that the response of EGFSP genes to luteolin was not affected by gefitinib, even though the two compounds were additive with respect to their abilities to inhibit CCNA2, CCNE2, CDC25A and PCNA. These findings suggest that luteolin and gefitinib regulate CCP gene expression through a common mechanism involving EGFR-associated tyrosine kinase. Conversely, luteolin regulates PC-3 cell proliferation through an EGFR-tyrosine kinase independent mechanism(s), likely involving the epigenetic control of gene EGFSP gene expression through histone H4 binding interactions resulting in the upregulation of c-FOS and p21 gene expression.
Keywords: Bioflavonoid, Luteolin, Nuclear Type II [3H]Estradiol Binding Site Cell Cycle, Epidermal Growth Factor, Prostate Cancer
Introduction
Previous reports from our laboratory designated two classes of [3H]estradiol binding sites present in the rat uterus, rat, mouse and human mammary tumors and breast and prostate cancer cells as type I or type II [1–4]. Type I sites represent the classical ER (ERα or ERβ) which binds estrogens and antiestrogens with high affinity and function as transcription factors regulating gene expression [5]. Nuclear type II sites bind [3H]estradiol with a lower affinity and higher capacity than ER’s and are present in all mammalian tissues and cells. Very low levels (< 3000 sites/cell) of type II sites are present in non-proliferating cells and this binding site is rapidly stimulated (5–30-fold) by estrogenic hormones only under conditions that induce cellular DNA synthesis and proliferation [6–8]. Thus, it is not surprising that estrogen antagonists, including progesterone and dexamethasone, that block estradiol stimulation of type II sites also block cellular proliferation without measurable affects on ER function ([8]. These findings suggest that type II sites are components of the cellular growth response to estrogenic hormones. Similarly, malignant tissues contain high concentrations of type II sites, which is consistent with the uncontrolled DNA synthesis and cellular proliferation [4, 9].
Type II sites were originally discovered and characterized on the basis of their ability to bind relatively low concentrations (4–40 nM) of [3H]estradiol. However, their function is to bind an endogenous ligand purified and identified from serum [10] as methyl-p-hydroxyphenyllactate (MeHPLA). That MeHPLA is a bioflavonoid or tyrosine metabolite is consistent with the fact that the compound is essentially ubiquitous in mammalian tissues and fluids, even though malignant tissues are devoid of this type II site ligand [10–12]. Synthetic preparations of MeHPLA bind to type II sites with a very high affinity (Kd < 5 nM), block estrogen stimulation of rat uterine growth and inhibit MCF-7 human breast cancer cell proliferation. Thus, MeHPLA is an important cell growth regulating agent in mammalian systems [10]. Further studies led to the identification of an enzyme (MeHPLA esterase) that is under estrogen regulation in the rat uterus, and constitutively expressed at high levels in malignant cells. MeHPLA esterse hydrolyzes MeHPLA to p-hydroxyphenyllactate (HPLA), the corresponded free acid [13–15]. HPLA binds to type II site with very low affinity (Kd > 200 nM) and does not block estrogen stimulation of rat uterine growth or inhibit breast cancer cell proliferation [10]. Thus, an esterase-induced deficiency of MeHPLA in malignant cells leads to a high level of unoccupied type II sites and the loss of regulatory control. Consequently, the methyl ester moiety of MeHPLA is critical for retaining high binding affinity for nuclear type II sites and cell inhibitory activity in vivo and in vitro. On the basis of these observations we developed a number of esterase stable ligands for nuclear type II sites including as 4-(3,4-dihydroxyphenyl)but-3-en-2-one (ZN-2) and 2,6-bis((3-methoxy-4-hydroxyphenyl)methylene)cyclohexanone (BMHPC), that bind to nuclear type II sites with high affinity also inhibit the proliferation of breast [10, 13, 16], pancreatic [17], prostatic [18], colorectal [19], ovarian cancer cells [19], lymphoblastoid cells [20], and leukemia [21] in vitro and in vivo strongly supporting our findings that type II sites are ubiquitous and MeHPLA is an important cell growth regulating agent in mammalian cells.
The identification of MeHPLA as a bioflavonoid metabolite represents one critical missing link between the consumption of fruits and vegetables and the lower incidence of cancer in man [22–24]. Studies in our laboratory and others have shown that bioflavonoids, such as luteolin and quercetin, inhibit estradiol stimulation of nuclear type II sites and uterine growth in the rat, and these compounds are also capable of occupying type II sites and inhibiting the growth and proliferation of malignant cells and tissues in vitro and in vivo [3, 14, 18, 25, 26]. These studies led to the delineation of a novel epigenetic mechanism for the regulation of normal and malignant breast and prostate cell growth by MeHPLA and related compounds including luteolin. The recent discovery that the nuclear type II site represent a binding component of histone H4 [27–29] suggests that ligands binding to this site are capable of modifying gene transcription through an epigenetic mechanism. This concept was recently extended by cRNA microarray analysis on luteolin treated PC-3 human prostate cancer cells which revealed that luteolin treatment significantly altered the expression of 3331 genes in these cells [30]. GenMapp analyses of the microarray data identified 22-downregulated genes and one upregulated gene in the cell cycle pathway (CCP), findings consistent with the inhibitory effects of luteolin on PC-3 cell proliferation in vitro and in vivo. The microarray studies were confirmed by real-time polymerase chain reactions (qPCR) and western blots for 6 selected CCP genes including cyclin A2 (CCNA2), cyclin E2 (CCNE2), cell division cycle 25A (CDC25A), cyclin-dependent kinase inhibitor 1B (CDKN1B), and polo-like kinase I (PLK1). Furthermore, chromatin immunoprecipitation studies (ChIP assays) indicated that luteolin altered the acetylation state of promoter-associated histone H4 associated with the PLK1 gene promoter in PC-3 cells [30]. This finding supports an epigenetic mechanism for the control of gene expression in prostate cancer cells by nuclear type II site ligands.
The studies described in the present manuscript have identified genes in the Epidermal Growth Factor Signaling Pathway (EGFSP) as key regulatory sites for luteolin in PC-3 and DU-145 prostate cancer cells. EGFSP genes encode a number of transcription factors which regulate CCP genes (including the cyclins, and cyclin-dependent kinases) suggesting that luteolin regulation of CCP gene expression could be mediated via it effects on EGFSP gene expression [31]. The present studies quantify the effects of luteolin and the EGFR antagonist, gefitinib, on the expression of EGFSP and CCP genes in PC-3 human prostate cancer cells. Luteolin [32] and gefitinib [33] are reported to inhibit EGFR-dependent protein kinases and autophosphorylation of EGFR. Therefore, if their effects on CCP genes are mediated via modulation of gene expression in the EGFSP, the response profiles of the genes in these two pathways to luteolin and gefitinib should be very similar.
Materials and Methods
Reagents and Materials
Luteolin was purchased from Indofine Chemicals (Hillsborough, NJ) and gefitinib was kindly provided by AstraZeneca, UK, Ltd. (Preclinical Evaluation Agreement with Baylor College of Medicine and Dr. Markaverich). PC-3 cells were obtained from the American Type Culture Collection (ATCC).
PC-3 Cell Growth and Experimental Conditions
Stock cultures of PC-3 human prostate cancer cells were grown and maintained in T-150 flasks [18]. The details of the microarray analyses for assessing luteolin effects on gene expression in PC-3 cells were described [30]. A brief description of these studies is provided here as background. For the microarray analyses, the cells were seeded into triplicate T-75 flasks for each treatment group. Each flask contained 10 mls of DMEM-F12 media supplemented with 10% fetal calf serum (FCS) and 1% penicillin-streptomycin. Twenty-four hours following plating, the attached cells were treated for 6 hours with 2 µL EtOH (controls) or luteolin (5 µg/mL) added to the DMEM-F12 media in 2 µL of EtOH [30]. RNA was prepared from these cells for the microarray studies as described briefly below and in detail our recent publication [30].
For dose response studies with luteolin and gefitinib, PC-3 cells were plated in 24-well plates (20,000 cells/well) and allowed to attach for 24 hours. At this time (time 0), the cells were treated with the indicated concentrations of luteolin or gefitinib (2–50 µM) added to the media in 2 µL of DMSO. DMSO was substituted for EtOH in these studies to solubilize gefitinib. Viable, attached cell numbers were monitored by hemocytometer counts, trypan blue dye exclusion [16] or by crystal violet dye uptake [34]. The latter assay consisted of staining the cells with 0.2% crystal violet dissolved in 20% EtOH, washing the fixed monolayers with water, and reading the absorbance at 560 nM in water: MeOH:EtOH (5:1:4).
To obtain sufficient quantities of RNA and protein for qPCR and western blots for validation of the microarray data on EGFSP genes, and for assessing the effects of luteolin and/or gefitinib on EDGSP and CCP gene expression at various times following luteolin and/or gefitinib treatment, PC-3 cells were grown in 100 mm Petri-dishes under the conditions described above and in the text and figure legends. Triplicate 100 mm plates were seeded with 8–10 × 106 cells for each control or treatment group. Twenty-four hours following seeding, the media was replaced with fresh media and attached cells were treated with either the low (17.5 µM luteolin and 22 µM gefitinib) or high (35 µM luteolin and 44 µM gefitinib) doses of luteolin or gefitinib as described in the text and figure legends. Cells for all studies were harvested by mild trypsinization and cell numbers were determined as described above [26]. RNA or protein analyses were as described below.
One study was performed to compare the response of EGFSP genes to luteolin in DU-145 prostate cancer cells to that obtained in PC-3 cells. DU-145 cells were grown in Minimal Essential Medium (MEM) supplemented with 10% Fetal Calf Serum. The cells were seeded into triplicate 100 mm plates (8 × 106 cells/well) and 24-hours following plating (time 0), the media was changed and attached DU-145 cells were treated for 6 hours with 2 µL EtOH (controls) or 5 µg/mL luteolin (added in 2 µL EtOH). The cells were harvested and the RNA prepared from the DU-145 cells was subjected to qPCR [30] to validate the EGFSP and CCP gene expression response to luteolin. PC-3 cells were predominantly used for the studies described here because the majority of the background work for these studies utilized the PC-3 model system and luteolin inhibits PC-3 cell xenografts in vivo [18, 26, 30].
RNA Preparation
The methods used for the preparation of RNA are validated techniques used in our lab [16, 30]. Cells from flasks or plates of luteolin and/or gefitinib treated cells or controls were washed with PBS and collected with 0.25% trypsin-0.02%EDTA (4 mls). Following 5-minute incubation, the trypsin was inactivated with 10 mLs of media containing 10% FCS. Approximately 5.0 × 106 cells from each flask or plate were centrifuged (2000 rpm × 5 minutes) in RNAse/DNAse free tubes, resuspended in 1 ml of PBS and 4 mls of RNAlater (Qiagen) and stored at −20°C. The frozen cells were thawed on ice, collected by centrifugation and lysed by resuspension in 0.6 mls of RTL (Qiagen) containing β-mercaptoethanol. The lysed cells from various treatment groups were homogenized by centrifugation through Qiashredders (18,000 × g × 2 minutes). Pass through from the Qiashredders was diluted with an equal volume of 70% EtOH and loaded onto RNeasy spin columns. The column was washed with RW1 followed by RNAse-free DNAse digestion to remove residual DNA and further washed with RPE buffers according to the manufacturer’s instructions. Purified total RNA was eluted with 50 µl of RNAse-free water following 5-minute incubation at 22°C. RNA integrity was routinely verified on an Agilent 2100 Bioanalyzer in the Baylor College of Medicine Microarray Core Facility headed by Dr. Lisa White.
Brief Description of Microarray Analyses [30]
RNA from controls or luteolin treated PC-3 cells was subjected to oligo(deoxythymidine)-Reverse Transcription, in vitro transcription and biotin-labeling of cRNA (Enzo Biochem, Farmingdale, NY) and cRNA hybridization to Human Genome U133 Plus 2.0 oligonucleotide arrays (Affymetrix, Santa Clara, CA). The array contained approximately 54,000 probe sets (38,500 genes). All experiments were performed in triplicate with independent pools of cRNA from EtOH controls and luteolin-treated PC-3 cells using six separate microarray chips. Following low-level quantification of the scanned data using GeneChip Operating System (GCOS, Affymetrix), data were analyzed with dChip 2006 (Harvard) to adjust the arrays to a common baseline, to estimate expression using the PM-only model [35, 36] and to normalize all 6 Gene Chips to the same baseline. Differentially expressed genes in the luteolin treated groups versus controls were selected using a two-sample comparison with a lower boundary 90% confidence interval of fold change greater than 1.2 and a value difference between group means of >50 [30]. GenMAPP (Gene Pap Annotator and Pathway Profiler, Version 2.1, Gladstone Institutes, University of CA at San Francisco) identified identified 23 genes in the Cell Cycle Pathway up-or down-regulated by luteolin [30]. Ingenuity Pathway Analysis (Redwood City, CA) of the dChip data from the above microarray studies was used to identify the EGFSP (Figure 1) genes as targets for luteolin regulation.
Figure 1. Effects Luteolin on Gene Expression in the EGF Signaling Pathway.
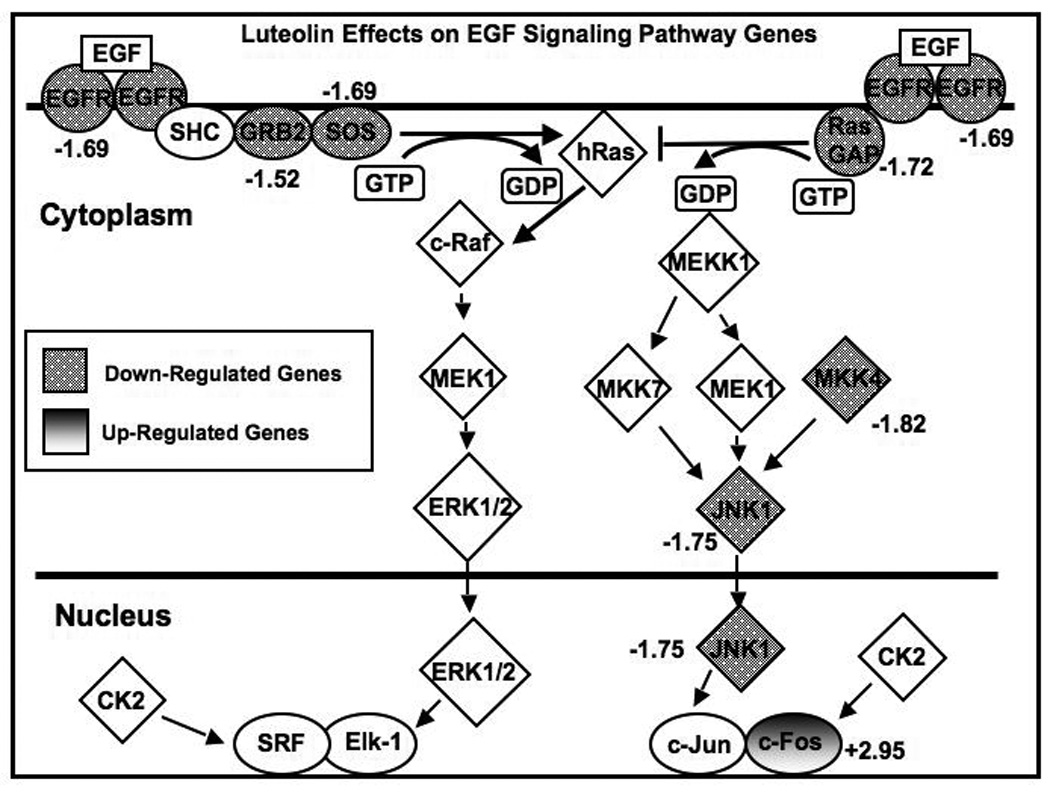
Microarray data from luteolin-treated PC-3 cells (17.5 µM for 6 hours) relative to controls described in detail in Shoulars et al, J Steroid Biochem Mol Biol,118: 41–50, 2010 were analyzed with Ingenuity Analysis Software. A number of genes in the pathway were significantly (p<0.01) upregulated (shaded) or downregulated (dotted) by luteolin treatment. The numbers adjacent to the gene symbols are the Luteolin-induced change in gene expression as a percent of EtOH controls (100%). Regulated genes were as follows: EGFR (Epidermal growth factor receptor (erythroblastic leukemia viral (v-erb-b) oncogene homolog, avian); AffyID 201983_ s at, Entrez Gene 1956, -1.69 fold change, adjusted P value = 0.001459), JNK1 (Mitogen-activated protein kinase 8; AffyID 229664_at, Entrez Gene 5599, -1.75 fold change, adjusted P value = 0.007131), MKK4 (MAP2K; Mitogen-activated protein kinase kinase 4, AffyID 203266_s at, Entrez Gene 6416, -1.82 fold change, adjusted P value = 0.000685), RasGAP (Ras-GTPase-activating protein SH3-domain-binding protein, AffyID 225007_at, Entrez Gene 10146, -1.72 fold change, adjusted P value = 0.013305), SOS (Son of sevenless homolog 1 (Drosophila), AffyID 227426_at, Entrez Gene 6654, -1.69 fold change, adjusted P value = 0.003924), c-FOS (v-fos FBJ murine osteosarcoma viral oncogene homolog, AffID 209189_at, Entrez Gene 2353, 2.95 fold change, adjusted P value = 0.036354).
Real-Time Quantitative Polymerase Chain Reaction (qPCR)
Pre-validated commercially available primers (Qiagen) for EGFSP and CCP genes were used. qPCR was performed using the MyiQ SYBR Green Supermix (Bio-Rad) and quantified on MyiQ Single Color Real-Time PCR Detection System using MyiQ Optical System Software, version 2.0 (Bio-Rad). Validation of each primer pair was accomplished by generating standard serial dilution and melt curves on cDNA from PC-3 cells to ensure that reaction efficiencies of 90–110% and correlation coefficients of > 0.995 are obtained. Melt curves demonstrating a single reaction product with an appropriate melting temperature confirmed that primer dimerization was contributing to the signal. Results from qPCR on triplicate pools of RNA from EtOH controls, luteolin, gefitinib or luteolin plus gefitinib treated cells were normalized to 18S. Products of the optimized reactions were analyzed by agarose gel electrophoresis to ensure that the size of the amplicon corresponds to the data provided by Qiagen for each primer pair (not shown).
Cell Cycle Analysis by Flow Cytometry
Flow cytometry studies were performed as previously described by our lab with slight modifications [18, 26]. PC-3 cells (8.0 × 106) were plated in 6 well plates and grown in DMEM/F12 media for 24 hours. At time 0 (24 hours following plating), the cells were treated with either the low (17.5 µM luteolin and/or 22 µM gefitinib) or high (35 µM luteolin and/or 44 µM gefitinib) doses of luteolin or gefitinib, alone or in combination, as described above and in the text and Figure legends. After 24 hours of treatment, the cells were collected by trypsinization and washed twice in cold PBS. Cells were fixed in 70% cold ethanol on ice and stored at −20° C for analysis by flow cytometry. Ethanol suspended cells were centrifuged and the cell pellets were resuspended in 25ug/ml of propidium iodide and 50ug/ml of DNase free Rnase. Stained cells were kept for 20 min. at 22°C and the cell fluorescence determined by flow cytometry (BDFACS CANTOII; BD Biosciences, Franklin Lake, NJ). The experiments were performed in the Cytometry and Cell sorting Core Facility at Baylor College of Medicine. The flow cytometer was set for excitation with blue light at 488 nm and PI emission at red wavelength at 633 nm. Data were analyzed using FACS Diva software (version 6.1.3, BD Biosciences) that de-convolutes DNA content frequency histograms. Each experiment was replicated three times.
Western Blot Analyses
After treatment with the luteolin and/or gefitinib, PC-3 cell monolayers from triplicate control or treatment plates were washed twice with cold PBS. Cell lysates were prepared in lysis buffer (20mM Tris- HCl pH 7.4, 137 mM NaCl, 10% Glycerol, 5mM EDTA, 1% NP-40 with Complete Mini, EDTA-free protease inhibitor cocktail, Roche Diagnostics, Indianapolis, IN). Lysates were centrifuged at 13000 rpm for 10min at 4°C to remove insoluble material and protein concentration in the supernatants was determined using Bio-rad Protein Assay. Samples from each treatment group containing 25 µg of total protein were resolved by SDS-PAGE (12% acrylamide) and transferred onto PVDF or membrane as described by our laboratory [27, 30]. After blocking the membrane with 5.0% nonfat dry milk, the membranes were incubated with a 1:3000 dilution of primary antibodies against the selected EGFSP genes (EGFR, c-FOS, MKK4) and CCP genes (CDKN1B, CDC25A, CCNA2) obtained from Santa Cruz Biotechnologies. Membranes were washed and incubated with corresponding horseradish peroxide (HRP) conjugated secondary antibodies (Santa Cruz Biotechnology) as per the manufacturer’s instructions. As a loading control, membranes were stripped (62.5 mM Tris, 2%SDS, 100 mM β-mercaptoethanol) and reprobed with β-actin antibody (Santa Cruz Biotechnologies). Proteins were detected by enhanced chemiluminescence (ECL detection kit, GE Healthcare, UK) on Hyblot CL Autoradiography Film (Denville Scientific Inc, Metuchen, NJ). All blots were quantified with UNSCAN- IT Gel Software (Silk Scientific Software). Each experiment was replicated 3 times.
Statistical Analyses
The details of the statistical analysis of microarray studies to identify luteolin changed genes are published [30]. The Ingenuity analysis to identify luteolin-regulated EGFSP genes is described in detail in the Materials and Methods section. For all other studies in this paper, each experiment was repeated a minimum of 3 times. Thus, the qPCR analyses were performed on at least 3 pools of RNA from replicate controls or replicate luteolin and/or gefitinib treated PC-3 or DU-145 prostate cancer cells. Quadriplicate reactions from each pool were analyzed by qPCR for each gene and normalized to 18S RNA. The qPCR data (mean ± SEM) were analyzed by Analysis of Variance (ANOVA) and Tukey’s test on the treatment means or by a two-tailed T-test utilizing Instat (GraphPad Software). Cell proliferation assays were replicated a minimum of three times and cell numbers (mean ± SEM) were determined from triplicate (wells or plates) for each of the replicate studies. Cell proliferation data (mean ± SEM) were analyzed by ANOVA and Tukey’s Test on the treatment means. Western blot analyses were also performed on protein extracts from three replicate experiments. For brevity, only a single representative replicate of the immunoblot data (Panels A and B) for low or high dose studies was presented in Figures 8 and 9. However, the gel scans (Panels C and D in Figures 8 and 9) were obtained for each of the three replicate blots and therefore, represent the mean ± the SEM for three separate determinations. The data for controls and each treatment group for each gene were analyzed by ANOVA and Tukey’s Test on the treatment means (Instat). Each flow cytometry study was repeated at least 3 times the data represent the mean ± SEM for three separate determinations analyzed by ANOVA and Tukey’s Test on the treatment means (Instat).
Figure 8. Western Blot Analysis of Selected EGFSP and CCP Genes Treated with the Low Doses of Luteolin and/or Gefitinib.
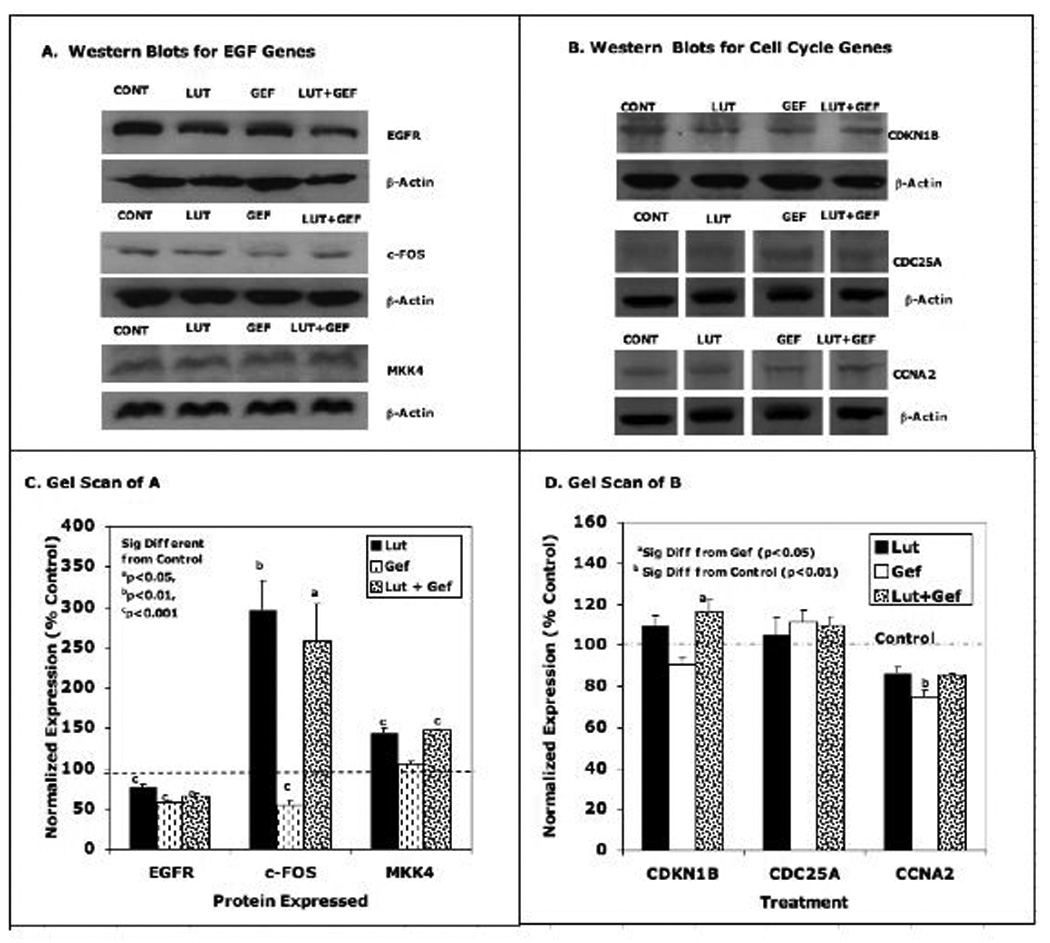
PC-3 cells were treated with the low doses of luteolin (17.5 µM) or gefitinib (22 µM) alone or in combination as described in Figure 5. Twenty-four hours following treatment, the protein extracted from luteolin and/or gefitinib treated cells or EtOH controls was subjected to western blot analysis with antibodies for EGFSP Genes (Panel A) or CCP Genes (Panel B). Bands were also analyzed by UN-SCAN-IT gel to determine band intensity, normalized to β-actin and graphed according to the normalized expression as a percent of the EtOH control that represented 100%. Data for Panel C are the scans for the EGF Genes (Panel A) and Panel C contains the scans for the CCP genes (Panel B
Figure 9. Western Blot Analysis of Selected EGFSP and CCP Genes Treated with the High Doses of Luteolin and/or Gefitinib.
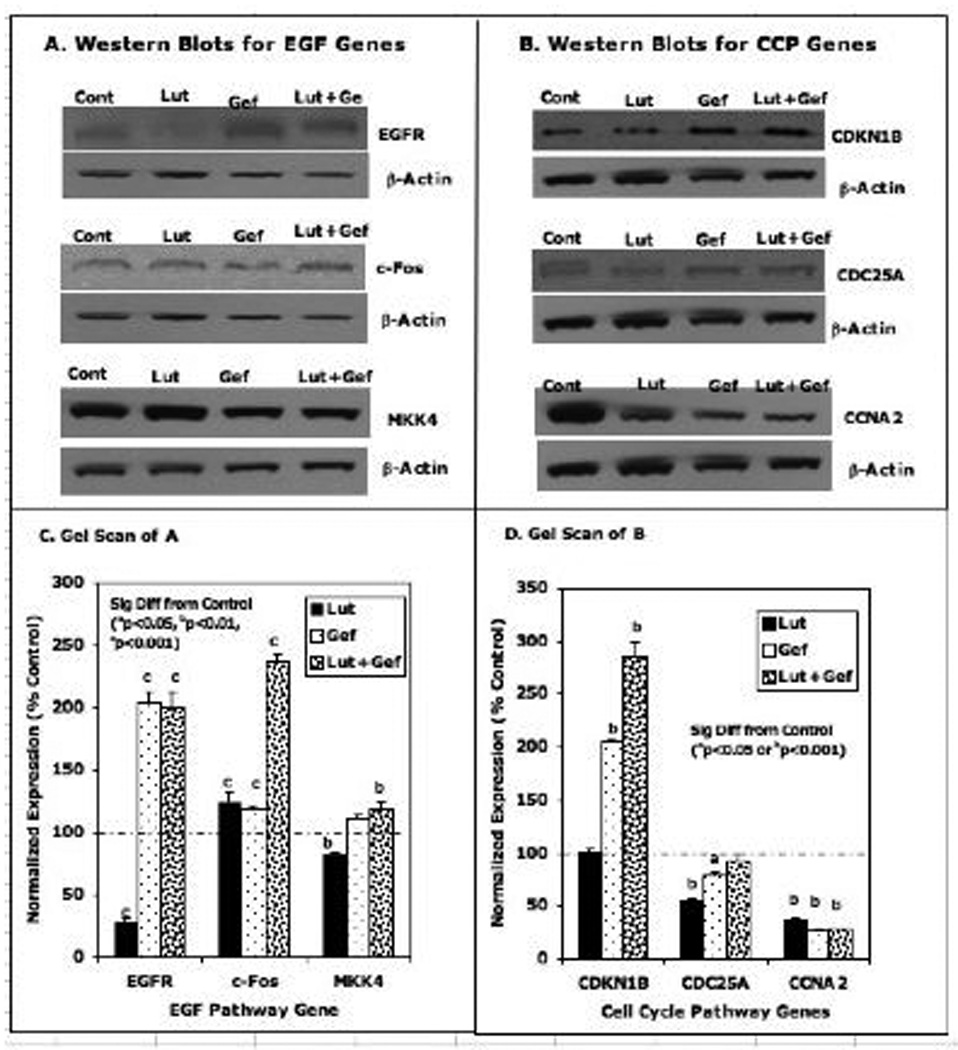
PC-3 cells were treated with the high doses of luteolin (35 µM) or gefitinib (44 µM) alone or in combination as described in Figure 5. Twenty-four hours following treatment, the protein extracted from luteolin and/or gefitinib treated cells and EtOH controls was subjected to western blot analysis with antibodies for EGFSP Genes (Panel A) or CCP Genes (Panel B). Bands were also analyzed by UN-SCAN-IT gel to determine band intensity, normalized to β-actin and graphed according to the change in fold expression as a percent of the EtOH control that represented 100%. Data for Panel C are the scans for the EGF Genes (Panel A) and Panel C contains the scans for the CCP genes (Panel B).
Results
Ingenuity Analysis of Microarray data
Our recently published microarray studies identified 3331 genes in PC-3 cells whose expression was changed by luteolin treatment [30]. Of these, GenMAPP analysis identified 23 genes in the CCP as luteolin targets. qPCR and western blotting studies confirmed that a selected number of these CCP genes including cyclin A2 (CCNA2), cyclin E2 (CCNE2), cell division cycle 25A (CDC25A), cyclin-dependent kinase inhibitor 1B (CDKN1B), and polo-like kinase I (PLK1) were regulated by luteolin in PC-3 cells [30]. In the present study, we analyzed the dChip microarray data with Ingenuity Pathway Analysis Software (Ingenuity Systems, Inc, Redwood City, CA). Ingenuity Analysis identified 7 genes in the EGF Signaling Pathway (EGFSP) subject to luteolin regulation. These included epidermal growth factor receptor (EGFR), mitogen-activated protein kinase 8 (JNK1), mitogen-activated protein kinase 4 (MKK4), Ras-GTP-ase activating protein SH3-Domain Binding Protein (RasGAP), Son of sevenless homolog 1 (SOS), v-fos FBJ murine osteosarcoma viral oncogene homolog (c-FOS) and growth factor receptor bound protein 2 (GRB2) as shown in Figure 1. Treatment with luteolin for 6 hours resulted in a 2.95-fold stimulation of c-Fos (Figure 1), and inhibited the expression of EGFR, JNK1, MKK4, RasGAP, SOS and GRB2 relative to controls. The down-regulation of 6 of these genes by luteolin is consistent with the inhibitory effects of this compound on CCP gene expression and PC-3 cell proliferation [30], as EGFSP genes are closely coupled to the regulation of CCP genes in a variety of cell systems [31, 37–39].
Validation of Microarray Studies by Assessment of Luteolin Effects on EGFSP Gene Expression in PC-3 and DU-145 Human Prostatic Cancer Cells by qPCR
To further validate the Ingenuity Analysis of the microarray data, we assessed the effects of luteolin on the EGFSP genes identified as targets for luteolin regulation by qPCR in PC-3 cells. This analysis was to DU-145 cells for further validation of luteolin regulation of these genes prostate cancer cells. RNA prepared from PC-3 or DU-145 cells treated for 6 hours with luteolin was analyzed by qPCR. The data in Figure 2A demonstrate that luteolin modulation of EGFSP gene expression essentially mirrors that derived from the Ingenuity Analysis (Figure 1). Luteolin treatment inhibited (p<0.001) the expression of 6 EGFSP genes (EGFR, SOS, GRB2, JNK1, MKK4, RasGAP) and stimulated c-Fos gene expression. This response pattern was replicated in the DU-145 cells and the induction of c-Fos was approximately 4-fold greater than that observed in PC-cells. The induction of c-Fos appears to be a key component in the response to luteolin in both cell lines.
Figure 2. Comparison of Luteolin Effects on Gene Expression in PC-3 and DU-145 Human Prostate Cancer Cells.
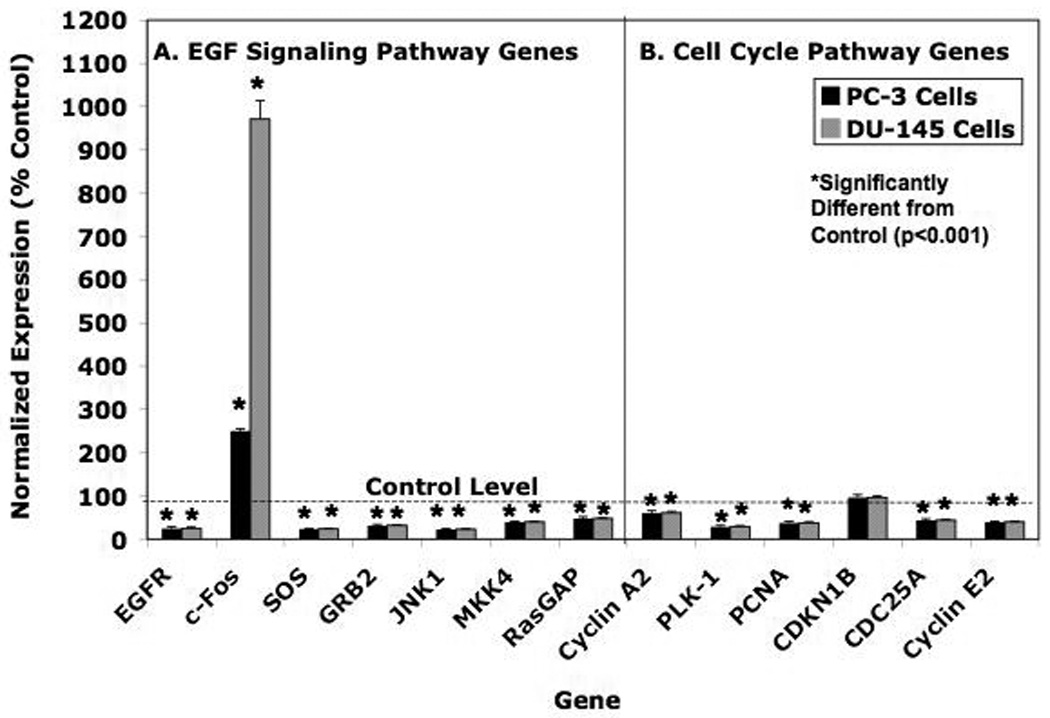
PC-3 and DU-145 cells were grown as described in Material and Methods in either DMEM-F12 Media (PC-3 cells) or Minimal Essential Medium (MEM) supplemented with 10% Fetal Calf Serum (FCS). Twenty-four hours following plating, the cells were treated for 6 hours with with 17.5 µM luteolin (added in EtOH). RNA prepared from these cells was analyzed by real-time PCR (qPCR). Relative expression values are the mean ± SEM for three independent RNA sets normalized to 18S RNA as described in detail [30] presented as % control where the EtOH controls represent 100%. Panel A = EGF Signaling Pathway Genes. Panel B = Cell Cycle Pathway Genes. Data from three replicate experiments were analyzed statistically by ANOVA with Tukey’s Test on the means with Instat Software as described in Methods.
As noted in the Introduction, we previously identified 4 genes in the CCP as targets for luteolin [30]. Therefore, the expression of these CCP genes in PC-3 and DU-145 cells was compared for further validation. The data in Figure 2B show that luteolin inhibited (p<0.001) the expression of CCNA2, PLK-1, CDC25A and CCNE2 in PC-3 and DU-145 cells. Luteolin failed to affect CDKN1B gene expression in PC-3 or DU-145 cells, even though we previously reported a small but significant increase in the expression of this gene in PC-3 cells under similar conditions [30]. That CDKNIB expression is modulated by growth factors during G1 would likely explain this minor discrepancy and slight variability in response of this gene to luteolin in the present studies [40]. Otherwise, the response profile of EGFSP and CCP genes to luteolin at 6 hours was very similar in the two different prostate cancer cell lines [31, 37–39].
Temporal Response of EGFSP and CCP Genes in PC-3 Cells to Luteolin
The data in Figures 1 and 2 identified EGFSP and CCP genes in PC-3 cells whose expression was blocked by luteolin 6 hours following treatment. Time studies were also performed to define a window where the genes in both pathways are most responsive to luteolin. PC-3 cells were treated with 17.5 µM luteolin at time 0 (24 hours after plating) and collected at various times (1, 8, 16, 24, 48, 72 hours) after treatment. RNA was prepared from these cells and the effects of luteolin on EGFSP and CCP gene expression was evaluated by qPCR (Figure 3). At early times following treatment (1–8 hours), the response to luteolin essentially mirrored that observed for CCP genes at 6 hours in our previous study [30] and the EGFSP genes identified by Ingenuity Analysis in the present study (Figures 1 and 2). Luteolin inhibited gene expression in both pathways at these early times. However, between 24 and 72 hours following treatment, marked changes in the expression of EGFSP and CCP genes were observed. In both pathways, this response peaked at 24–48 hours and declined at longer times. At this dose level (17.5 µM) luteolin treatment caused enhanced expression of a number of the EGFSP genes (c-FOS, RasGAP, MKK4, GRB2, JNK1) and CCP (CDKN1B, CCNA2, PCNA, CCNE2, CDC25A) and only EGFR and SOS expression was repressed by this low dose of luteolin. The peak response time for most of these genes to luteolin was approximately 24 hours. This time was used for the experiments described below with various doses of luteolin and the EGFR antagonist, gefitinib.
Figure 3. Temporal Effects of Luteolin on EGFSP and CCP Gene Expression in PC-3 Cells.
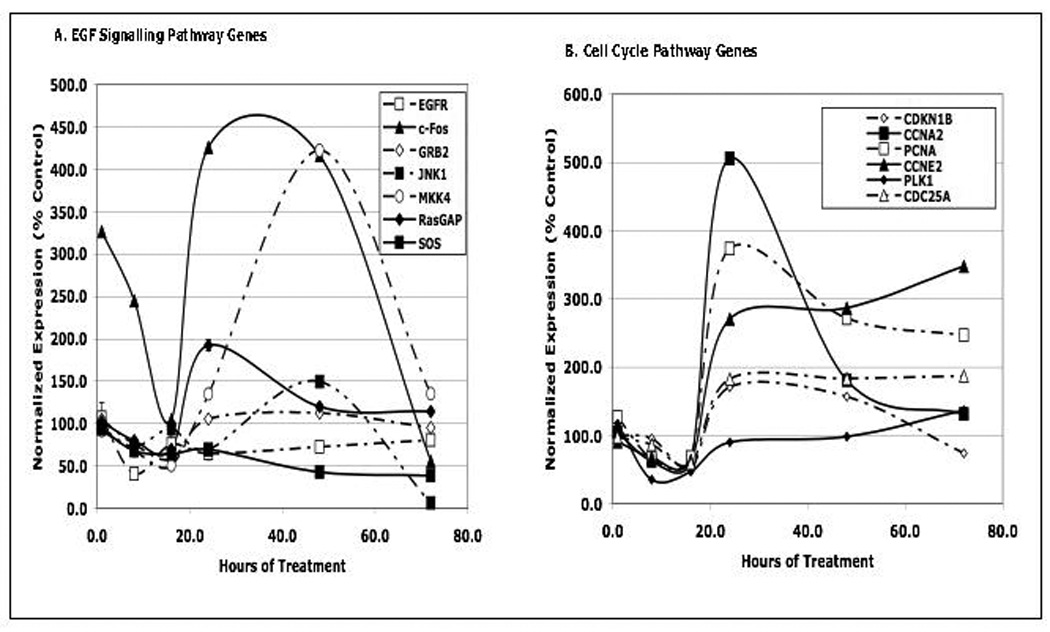
PC-3 cells were plated and grown and described in methods. Twenty four hours following plating, the cells were treated with 17.5 µM luteolin in 2 µL of EtOH. At various times following treatment (1, 8, 16, 24, 48, 72 hours) the cells from time matched controls or luteolin treated cells were harvested and RNA was prepared and analyzed by qPCR. Relative Expression values represent the mean ± SEM for a minimum of three independent RNA sets from time matched EtOH controls and luteolin treated PC-3 cells, each normalized to 18S RNA as described in detail [30]. To simplify the figure, the control for all time points was represented at 100%. Statistical analyses were performed on quadruplicate aliquots from each of three replicated experiments for each luteolin sample time point and its corresponding control.
Dose Response Studies with Luteolin and Gefitinib
Gefitinib is a well-known EGFR antagonist, and both gefitnib and luteolin inhibit the EGFR-associated tyrosine protein kinase responsible for autophosphorylation of EGFR [32, 33]. Thus, the two compounds share this common effect on EGFR. In the present studies, Ingenuity Analysis identified the EGFSP as a target for luteolin regulation and qPCR studies confirmed that EGFR gene expression in PC-3 cells was blocked by luteolin (Figures 2 and 3). Therefore, we suspected that the response of EGFSP and CCP genes to luteolin would mirror that obtained with the EGFR antagonist, gefitinib, if luteolin modulation of CCP genes was mediated via its effects on EGFSP gene expression. To evaluate these possibilities, dose response studies were performed to define concentrations of gefitinib and luteolin required for combination studies to assess their singular and combined effects on gene expression and cell proliferation. PC-3 cells were plated, allowed to attach for 24 hours (time 0) and then treated with a range of gefitinib or luteolin concentrations (2–50 µM). Cell number was determined for EtOH control or treated cells at either 24 hours (not shown) or 48 hours (Figure 4) following treatment. Equivalent dose response curves for the two compounds were obtained at either time, but more complete inhibition (90–100%) was observed at 48 hours. The inhibition curves for the two compounds were remarkably similar and approximately 80–90% inhibition was achieved with doses of either compound ranging from 20–50 µM and these doses will be used for the studies below.
Figure 4. Effects of Luteolin and Gefitinib on PC-3 Human Prostate Cancer Cell Proliferation.
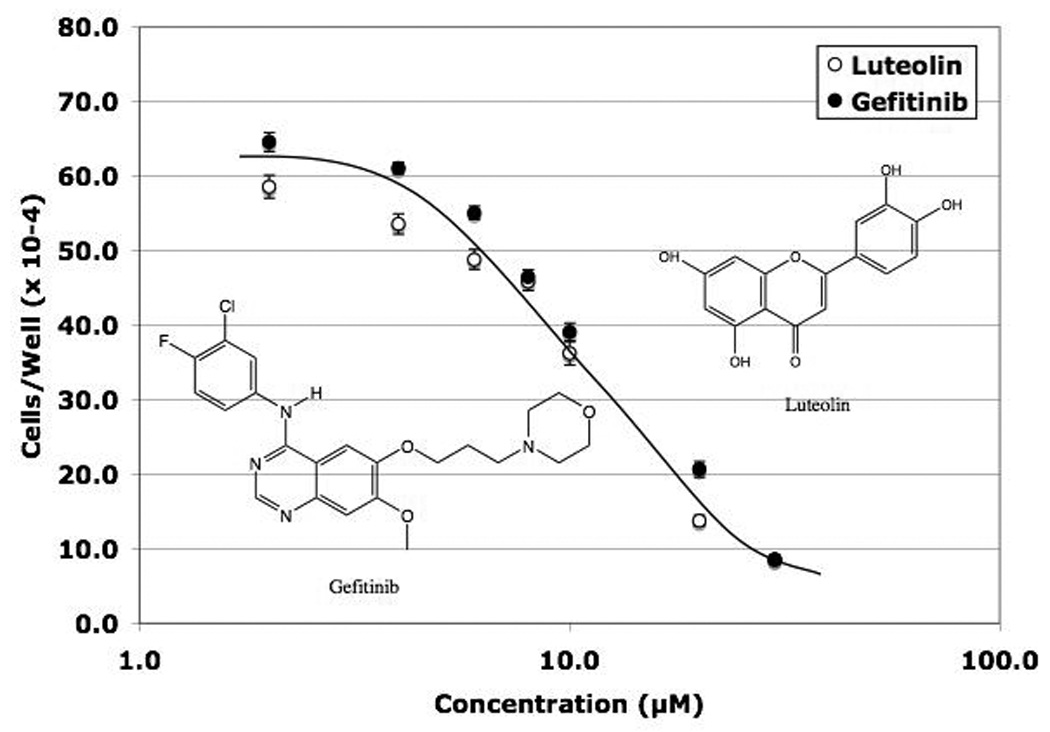
PC-3 cells were grown as described in Figure 1 and the Methods section Twenty-four hours following plating, the attached cells were treated with the indicated concentrations of luteolin or gefitinib added to the media in 2 µL of DMSO and 48 hours following treatment the cells were collected and counted as described in detail [30] and in Methods. There were three wells per concentration of luteolin or gefitinib and each experiment was replicated three times. The results thus represent the mean ± the SEM for 9 separate determinations for each concentration.
Luteolin and Gefitinib Effects on PC-3 Cell Cycle
Based upon the dose response study, we initially evaluated the effects of a low dose (17.5 µM luteolin; 22 µM gefitinib) or high dose (35 µM luteolin; 44 µM gefitinib) of luteolin and/or gefitinib on the cell cycle progression in PC-3 cells. Two doses were chosen so that response profiles for antagonistic and additive interactions with luteolin and/or gefitinib could be evaluated when examining treatment effects on RNA and protein by qPCR and western blotting. The data in Figure 5 and Table 1 summarize the flow cytometry findings. The response of the PC-3 cells to the lower doses of the compounds was not as substantial as that observed with the higher dose. The 17.5 µM luteolin concentration decreased (p<0.01) the numbers of cells in G0/G1 and this was reflected by a slight G2/M arrest and a non-significant increase in the numbers of apoptotic cells relative to controls. The low dose of gefitinib caused G0/G1 arrest (p<0.001) and significant decreases in the numbers of cells in G2/M and undergoing apoptosis. When the PC-3 cells were treated with a combination of these compounds, the low dose of luteolin blocked (p<0.01) the G0/G1 arrest caused by gefitinib, suggesting that response pathways regulated by luteolin were required for the response to gefitinib.
Figure 5. Assessment of Luteolin Effects on PC-3 Cell Proliferation by Flow Cytometry.
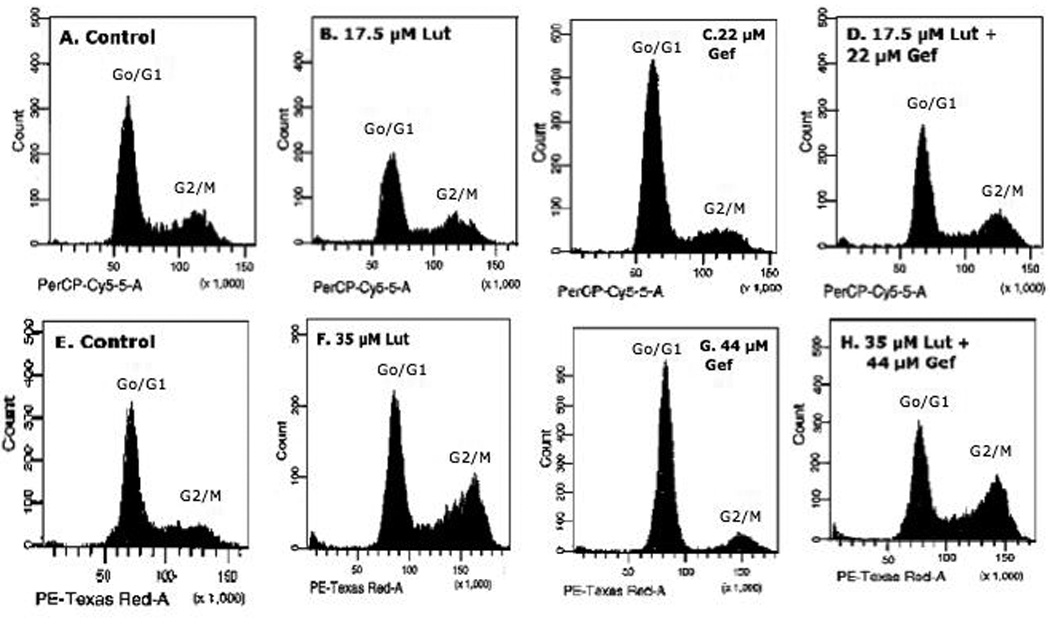
PC-3 cells were grown and plated as described in Methods. Twenty-four hours following plating, the cells were treated with two doses of luteolin (17.5 µM; 35 µM) or Gefitinib (22 µM; 44 µM) alone or in combination (as indicated in each panel) and 24 hours following treatment, the cells were collected, stained with propidium iodide and analyzed by flow cytometry (see Methods for details) as previously described by our laboratory [26]
Table 1.
Effects of Luteolin and Gefitinib on Cell Cycle of PC-3 Cells
| TREATMENT GROUP |
G0/G1 | S-PHASE | G2/M | APOPTOSIS |
|---|---|---|---|---|
| Control | 60.1 ± 0.78 | 13.7 ± 1.01 | 23.2 ± 0.18 | 1.3 ± 0.15 |
| Low Dose Lut | 55.2 ± 0.24a | 14.1 ± 0.83 | 26.3 ± 0.60 | 3.3 ± .012 |
| Low Dose Gef | 73.2 ± 0.29b | 9.5 ± 1.00 | 14.4 ± 0.96b | 1.27 ± 0.09 |
| Low Dose Lut+Gef | 56.0 ± 0.92a | 16.4 ± 1.50 | 22.2 ± 1.17 | 4.1 ± 0.42 |
| Control | 61.8 ± 0.60 | 16.8 ± 0.80 | 19.1 ± 1.13 | 1.6 ± 0.03 |
| High Dose Lut | 45.2 ± 1.04b | 15.3 ± 1.09 | 35.4 ± 1.10b | 2.6 ± 0.31b |
| High Dose Gef | 82.0 ± 0.13b | 2.4 ± 0.07b | 13.5 ± 0.03a | 1.2 ± 0.15 |
| High Dose Lut+Gef | 42.1 ± 0.50b | 18.2 ± 1.28 | 35.8 ± 1.88b | 2.7 ± 0.15b |
Low Dose Luteolin =17.5 µM; High Dose Luteolin =35 µM; Low Dose Gefitinib =22 µM; High Dose Gefitinib =44 µM. Significantly Different from Control (ap<0.01 or bp<0.001)
Significantly Different from Control (p<0.01 or
p<0.001)
The responses to the higher doses of luteolin and gefitinib were more profound. Treatment of PC-3 cells with 35 µM luteolin caused significant (p<0.001) G2/M arrest and this was reflected by a reduction (p<0.001) in the number of cells in G0/G1. Luteolin also increased (p<0.001) the number of apoptotic cells relative to controls. The high dose of gefitinib (44 µM) caused G0/G1 arrest (p<0.001) and decreased (p<0.001) the numbers of cells in G2/M. The response of PC-3 cells to the higher dose level of luteolin was not altered by gefitinib treatment. In the presence of 44 µM gefitinib, the response to 35 µM luteolin (Figure 5 and Table 1) was not different from that obtained with 35 µM luteolin alone. Thus, although luteolin and gefitinib regulate EGFR expression through a common mechanism (EGFR-dependent tyrosine kinase) as previously described [32, 33], luteolin modulation of PC-3 cell proliferation likely involves an additional mechanism(s) not regulated by gefitinib.
Effects of Low and High Dose Luteolin and/or Gefitinib Treatment on EGFSP and CCP Gene Expression in PC-3 Cells at 24 Hours Following Treatment
In keeping with the flow cytometry studies, we evaluated the effects of low and high doses of luteolin and/or gefitinib on gene expression in the EGFSP (Figure 6) and CCP (Figure 7) in PC-3 cells 24 hour following treatment. Although these are a complex series of experiments, they were performed for comparison of the EGFSP and CCP gene response profiles to luteolin and/or gefitinib at two different doses. Our hypothesis was that if luteolin regulation of the CCP genes was mediated via EGFR (noted in Figure 1), then the response profiles of genes in the EGFSP and CCP to luteolin and gefitinib should be very similar. The data in Figure 6 demonstrate that this is not the case for the EGFSP genes. With the exception of c-Fos (which was stimulated maximally with either luteolin dose), EGFR, SOS, GRB2, JNK-1, MKK4 and RasGAP expression was more substantially inhibited with 35 µM luteolin. This was not necessarily the case for gefitinib, which inhibited c-Fos expression and failed to significantly affect the expression of EGFR, GRB2, JNK-1, MKK4 and RasGAP relative to controls at either dose level. Surprisingly, the low dose of gefitinib reduced SOS expression relative to control and this response was not observed with the higher dose.
Figure 6. Effects of Luteolin and Gefitinib on EGFSP Gene Expression.
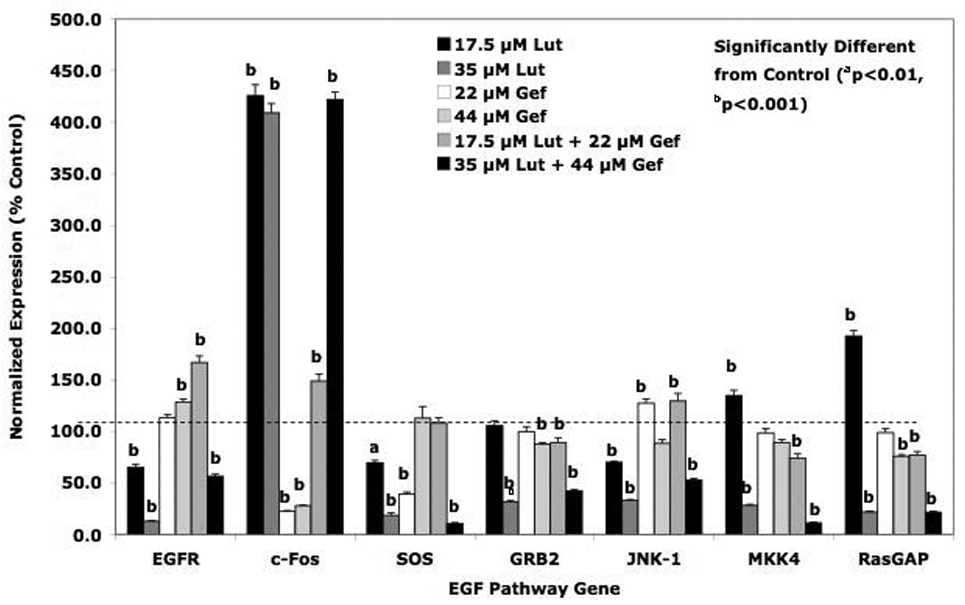
PC-3 cells were treated with low or high doses of luteolin or gefitinib as described in Figure 5. Twenty-four hours following treatment, RNA was prepared from controls, luteolin, gefitinib or luteolin plus gefitinib treated cells and analyzed by qPCR. Relative expression values are the mean ± SEM for three independent RNA sets normalized to 18S RNA and presented as % Control where the control is 100 %.
Figure 7. Effects of Luteolin and Gefitinib on CCP Gene Expression.
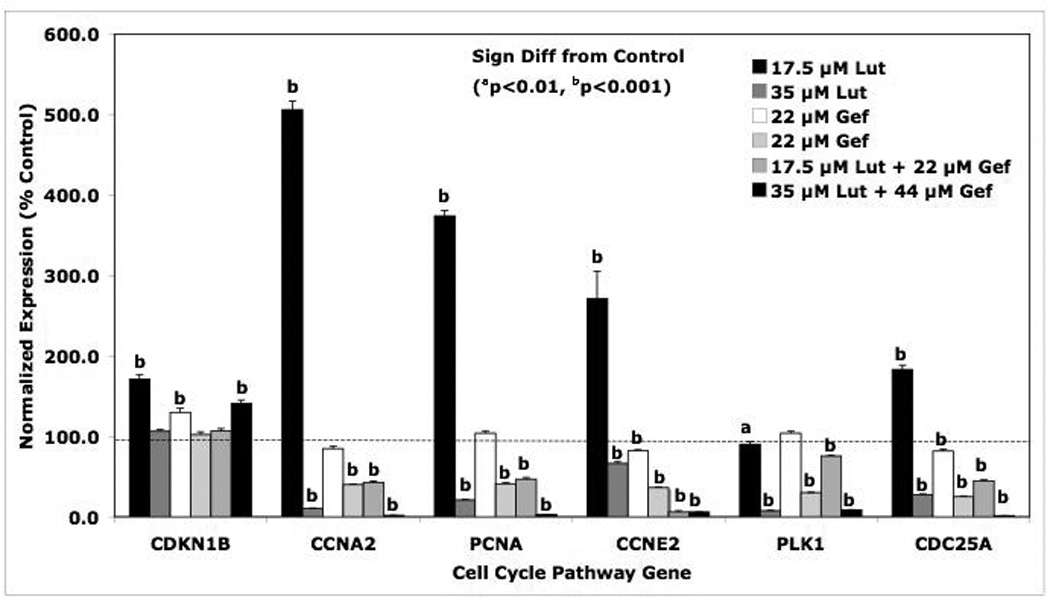
PC-3 cells were treated with low or high doses of luteolin or gefitinib as described in Figure 5. Twenty-four hours following treatment, RNA was prepared from controls, luteolin, gefitinib or luteolin plus gefitinib treated cells and analyzed by qPCR. Relative expression values are the mean ± SEM for three independent RNA sets normalized to 18S RNA and presented as % Control where the control is 100 %.
Central to this study is the comparison of the response profiles to the low and high doses of luteolin alone and in combination with the low and high doses of gefitinib to determine whether gefitinib was capable of blocking the response to luteolin. It is clear from the data presented in Figure 6 for the EGFSP genes, that gefitinib was unable to block the response of the EGFR, c-Fos, SOS, GRB2, JNK-1, MKK4 or RasGAP to luteolin. These findings strongly suggest that luteolin is regulating the expression of all of these EGFSP genes at a level not directly involving EGFR. Thus, even though luteolin [32] and gefitinib [33] inhibit EGFR-dependent tyrosine kinase cells in human cancer cells, the present data demonstrate that luteolin likely regulates genes in the EGFSP through an EGFR independent mechanism not antagonized by gefitinib.
Examination of the response profiles of the CCP genes to the low and high doses of luteolin and/or geftinib was also very interesting (Figure 7). The overall pattern of the response of these genes to the compounds is probably what is most important. The low dose of luteolin resulted in marked stimulation of the expression of CCNA2, PCNA, CCNE2 and CDC25A. This was a somewhat surprising in view of the fact that the expression of these CCP genes are typically increased in proliferating cells. However, the cell cycle analysis data (Figure 5 and Table 1) indicated that the effects of the low dose of luteolin on cell cycle were not significant (with the exception of a slight decrease in the numbers of cells in G0/G1). We suspect the expression of CCNA2, PCNA, CCNE2 and CDC25A was enhanced by the slight, but non-significant, G2/M arrest in response to luteolin. This would likely cause and extension of the transcription and biosynthetic activity of the CCNA2, PCNA, CCNE2 and CDC25A during G1/S-phase of the cell cycle [38, 41, 42]. More importantly, the high dose of luteolin blocked the expression of CCNA2, PCNA, CCNE2, PLK1 and CDC25A, a finding consistent with the inhibitory effects of the compound on cell proliferation (Figures 4 and 5 and Table 1). Although the modulation of CDKN1B by either luteolin or gefitinib (alone or in combination) would be consistent with an inhibitory effect of these compounds on PC-3 cell proliferation [37], the results for this gene were inconsistent and therefore, inconclusive.
The data in Table 2 summarize the results of the high dose combination studies with luteolin and/or gefitinib on the expression of EGFSP and CCP genes (Figures 6 and 7). A column was created to characterize the nature of effect of gefitinib on the response to luteolin. Gefitinib failed to substantially modify (antagonize or enhance) the effects of luteolin on the expression of all of the EGFSP genes (EGFR, c-FOS, SOS, GRB2, JNK-1, MKK4 and RasGAP) and two CCP genes (CDKN1B, PLK1). Failure of gefitinib to block the response to luteolin suggests that luteolin is not regulating the expression of these genes at the level of EGFR, even though the bioflavonoid is capable of blocking EGFR expression. Gefitinib, however, enhanced luteolin effects on the expression of 4 CCP genes including CCNA2, CCNE2, PCNA and CDC25A. The additive nature of this interaction supports a common mechanism of action, perhaps involving the control of CCP genes via genes in the EGFSP regulated by EGFR [31, 38, 43].
Table 2.
Summary of Gefitinib Effects on Response of EGF Signalling and Cell Cycle Pathway Genes to Luteolin
| Gene | 35 µM Luteolin |
44 µM Gefitinib |
35 µM Luteolin + 44 µM Gefitinib |
Gefitinib Effect on Luteolin Response |
|---|---|---|---|---|
| EGFR | 12.7 ± 0.5 | 128.3 ± 3.2 | 56.3 ± 1.8 | M to N |
| c-Fos | 409.0 ± 9.1 | 27.7 ± 0.7 | 422.0 ± 7.4 | N |
| SOS | 18.3 ± 2.3 | 112.7 ± 11.3 | 10.3 ± 1.0 | N |
| GRB2 | 31.3 ± 1.2 | 87.3 ± 1.4 | 42.0 ± 1.1 | N |
| JNK-1 | 32.7 ± 1.0 | 88.3 ± 3.5 | 52.3 ± 1.6 | M to N |
| MKK4 | 28.0 ± 0.9 | 89.0 ± 2.8 | 11.0 ± 0.5 | N |
| RasGAP | 21.7 ± 0.8 | 75.7 ± 1.8 | 21.3 ± 0.8 | N |
| CDKN1B | 106.3 ± 1.9 | 102.0 ± 3.5 | 141.0 ± 4.0 | N |
| CCNA2 | 10.3 ± 1.9 | 40.0 ± 0.7 | 1.7 ± 0.2 | E |
| PCNA | 21.0 ± 0.6 | 41.0 ± 1.3 | 2.7 ± 0.1 | E |
| CCNE2 | 66.3 ± 2.0 | 36.3 ± 0.9 | 5.7 ± 0.2 | E |
| PLK1 | 7.7 ± 0.2 | 30.0 ± 0.5 | 8.7 ± 0.3 | N |
| CDC25A | 27.3 ± 1.0 | 25.0 ± 0.9 | 1.3 ± 0.1 | E |
N=None, M to N= minimal to None, E=Enhance
Values represent the numerical data used to generate Figure 8.
Assessment of Luteolin and Gefitinib Effects on EGFSP and CCP Proteins in PC-3 Cells
Western blotting studies were performed on a few select proteins from each pathway following luteolin and/or gefitinib treatment (Figures 8 and 9). For most of the EGFSP or CCP genes, very small protein responses were observed following treatment with the low doses of luteolin and/or gefitinib and protein expression did not always correlate with RNA expression as expected [44]. However, luteolin substantially increased (250% of control) the level of the c-Fos protein and gefitinib failed to block this response. This increase in the c-Fos protein was consistent with the 400–500% increase in the RNA for the c-Fos gene determined by qPCR (Figure 6) and likely represents a key component in the inhibitory response to luteolin. More dramatic effects on the protein response were obtained with the high doses of luteolin and/or gefitinib (Figure 9). Luteolin (35 µM) significantly decreased (20% of control) the level of the EGFR protein (a finding consistent with the inhibitory effects of luteolin on EGFR gene expression; Figs 6). Conversely, gefitinib (44 µM) increased the level of the EGFR protein (200% relative to control) and this response was not blocked by luteolin. Thus, gefitinib effects on the EGFR protein are likely independent of luteolin regulation. Surprisingly, gefitinib significantly increased the CDKN1B protein and luteolin enhanced this response, even though luteolin alone failed to increase the CNKN1B protein relative to control. We have no good explanation for this observation at the present time.
In order to directly compare the RNA to protein data, Figure 10 was prepared as a summary from Figures 6, 7, 8 and 9). In the low dose studies (Figure 10A), only small changes in the RNA and protein expression patterns relative to controls for MKK4, CDKN1B and CDC25A were observed. Luteolin treatment caused marked stimulation in c-Fos RNA and protein expression, and this response was not blocked by gefitinib. Luteolin treatment also resulted in a marked stimulation in CCNA2 RNA, but failed to increase the CCNA2 protein relative to control at 24 hours and neither gefitinib nor luteolin plus gefitinib substantially changed the expression of this gene (RNA or protein).
Figure 10. Summary of the Response Profiles for RNA and Protein for EGFSP and CCP Genes to the Low (Panel A) and High (Panel B) Doses of Luteolin and/or Gefitinib.
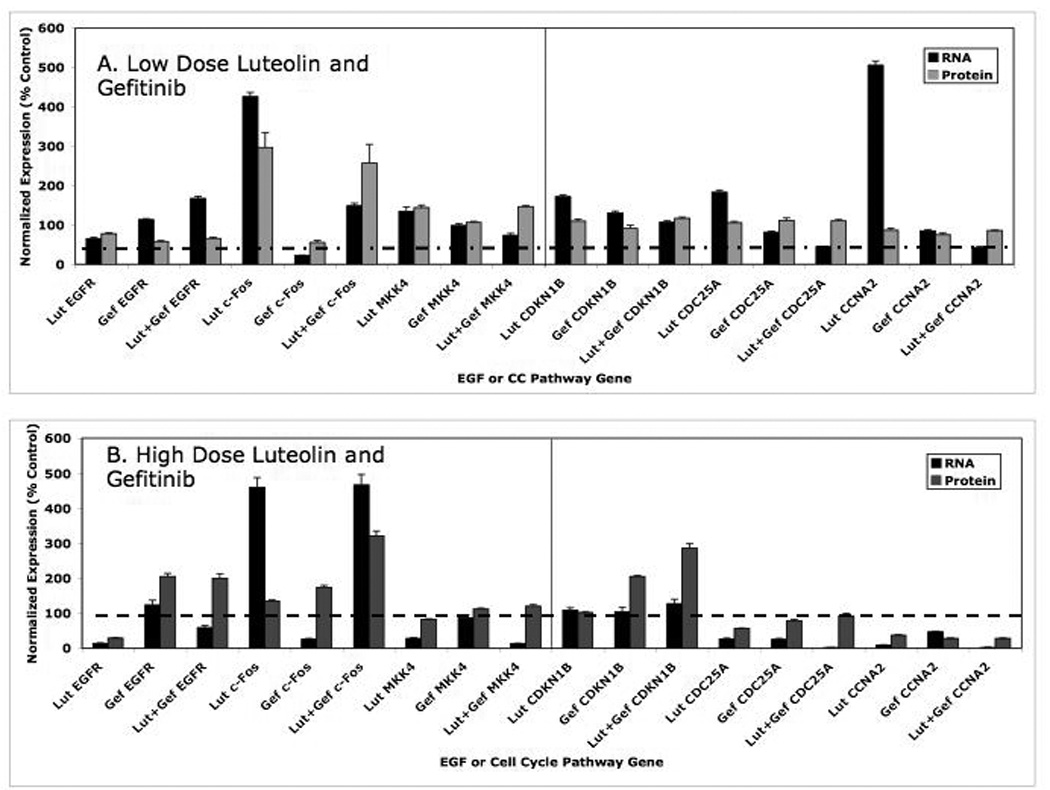
These data are the summary of the quantitative data presented in Figures 6–9 for direct comparison.
The high dose data in Figure 10B were more enlightening. Luteolin significantly reduced the level of EGFR RNA and protein. Gefitinib stimulated EGFR RNA and EGFR protein and luteolin failed to block this response. Thus, gefitinib effects on the EGFR RNA or protein were not blocked by luteolin even though both compounds decrease EFGR-dependent tyrosine kinases in malignant cells [33, 38, 43]. This further suggests that luteolin inhibition of EGFR gene expression (RNA and protein; Figure 10) is independent of its effects on EGFR-associated tyrosine protein kinase. The high dose luteolin also increased c-Fos RNA and protein and the response was not blocked by gefitinib, again confirming a separate mechanism(s) for the two compounds with respect to the regulation of c-Fos. Luteolin failed to significantly change the CDKN1B RNA or protein levels in PC-3 cells, in the absence or presence of gefitinib. In general fairly good correlations between RNA and protein in response to the various high dose treatment groups were observed for MKK4, CDC25A and CCNA2. The RNA and protein data in Figure 10 B suggest that the key components of the response to the high dose of luteolin in the EGFSP are a marked stimulation of c-Fos RNA and reduced expression MKK4 RNA. These responses were reflected by the inhibition RNA and protein for key genes involved in the regulation of the cell cycle including CDC25A and CCNA2. Although we did not run western blots on all of the EGFSP or CCP genes, it is clear from the data in Figures 2 and 7 that luteolin inhibition of PC-3 cell proliferation also involves the down regulation of PCNA and PLK1.
Luteolin Effects on CDKN1A (p21) gene expression in PC-3 cells
The observation that luteolin inibition of cell proliferation (G2/M arrest) was associated with a marked stimulation of c-FOS expression (Figures 1–3, 6, 8–10) without corresponding changes in CDKN1B (p27; Figures 7–10), suggested that luteolin may cause G2/M arrest by modulating p21 gene expression. In normal cells, the latter protein is under p53 regulation. However, PC-3 cells are p53 null due to a single copy of the gene containing codon 138 deletion which prevents them from expressing the p53 protein [45]. Nevertheless, treatment of PC-3 cells [46] or gastric cancer cells [47] with luteolin stimulates c-FOS gene expression causing G2/M arrest and this is attributed to the stimulation of p21. c-FOS was upregulated by luteolin in our studies and therefore, we also assessed p21 (CDKN1A) gene expression in PC-3 cells by qPCR. The data in Figure 11 demonstrate that luteolin significantly increased the expression of p21 approximately 4-fold relative to controls 24 hours following treatment. Thus, luteolin-induced G2/M arrest in PC-3 cells likely resulted from the induction of c-FOS and p21.
Figure 11. Luteolin Effects on CDKN1A (p21) Gene Expression in PC-3 Cells.
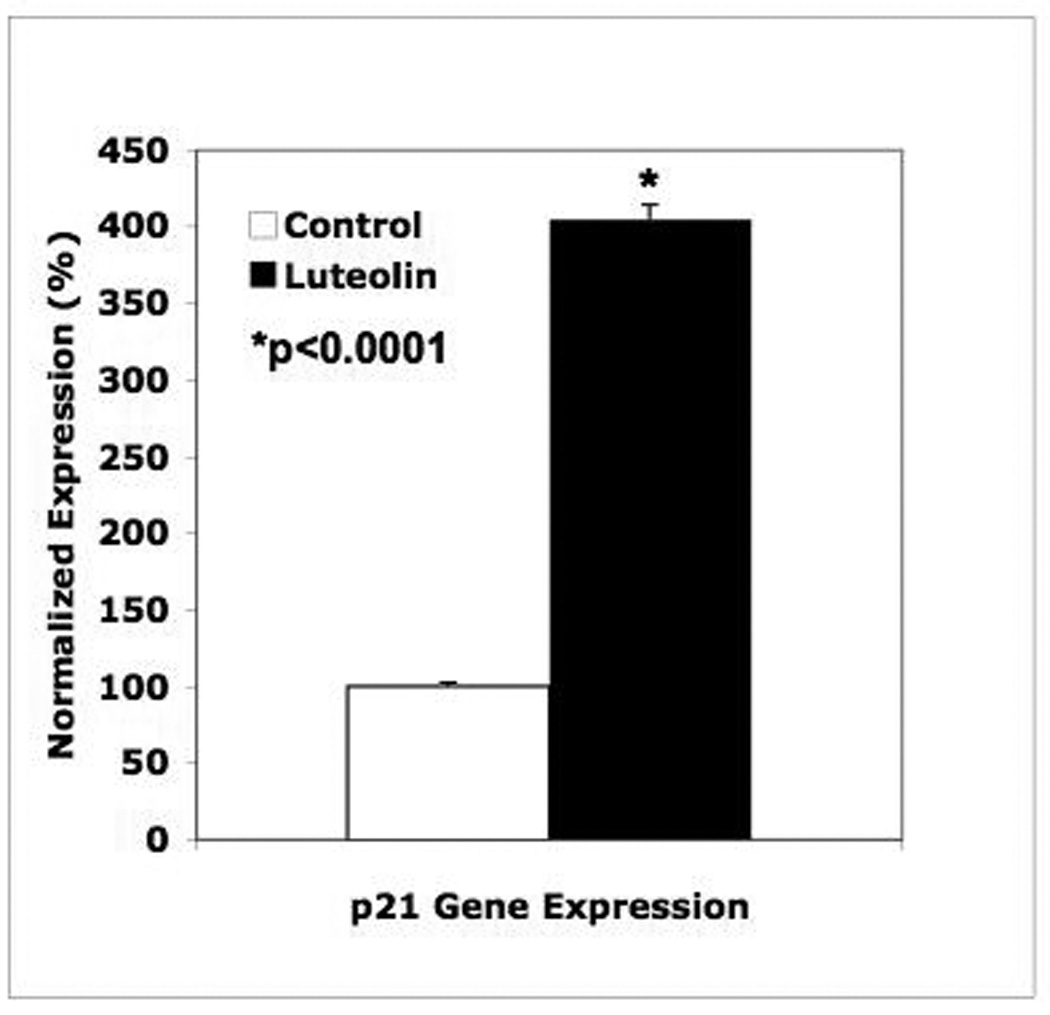
RNA from the PC-3 cells treated with 35 µM luteolin in Figure 5 was analyzed for CDK1NA (p27) gene expression by qPCR as described in the methods and figure legends above. Data represent the mean ± SEM for triplicate samples.
Discussion
A major focus of our laboratory has been determining the mechanism of action of bioflavonoids such as luteolin and quercetin in the regulation of normal and malignant cell growth and proliferation. We discovered that compounds such as luteolin and quercetin block estrogenic response (uterine growth) in the rat even though they failed to interfere with the function of the estrogen receptor [48]. Compounds such as luteolin and quercetin were found to inhibit the proliferation of estrogen-dependent and estrogen-independent breast [16] and prostate cancer [26] cells, further establishing these bioflavonoids as cell growth regulating agents. In addition, we also reported that luteolin, quercetin and a number of other bioflavonoids bind to type II sites in a variety of normal and abnormal tissues and cells, and nuclear type II site occupancy and binding affinity was directly related to the abilities of these compounds to inhibit cell proliferation [3, 25, 26, 49]. The importance of these findings was enhanced by the identification of a bioflavonoid metabolite, MeHPLA (methyl-p-hydroxyphenyllactate), as the endogenous ligand for type II sites [10] and the demonstration that esterase inactivation of this compound [10, 13, 14] was responsible for the deficiency of MeHPLA in malignant cells [11, 12], the reported high levels of nuclear type II sites and the loss of regulatory control. Thus, luteolin and other naturally occurring bioflavonoids are likely mimicking MeHPLA as cell growth regulating agents by binding to nuclear type II sites.
The identification of nuclear type II sites as histone H4 [27–29, 50] suggests that the ligand-binding domain for MeHPLA and related ligands (including luteolin) may be involved in the regulation of genes that control normal and abnormal cell proliferation. This hypothesis was confirmed by microarray studies in our lab suggesting that luteolin and related ligands regulate RNA transcription genes and CCP genes in PC-3 cells through an epigenetic mechanism involving the binding to type II sites and the acetylation of histone H4 associated with their promoters [30]. In the present studies, more extensive analysis of this microarray data with Ingenuity software (Figure 1) identified a number of genes in the EGFSP as targets for luteolin regulation. These are very exciting findings because of the well-known association of the EGFSP and CCP genes to the control of cell proliferation [38, 51, 52]. The studies in this manuscript validate the Ingenuity Pathway Analysis identifying EGFSP genes as luteolin targets and also explore and define the nature of the mechanisms involved in the regulation of EGFSP and CCP genes by type II site ligands that control of prostate cancer cell proliferation.
The Ingenuity Pathway Analysis identified EGFR, GRB2, SOS, RasGAP, MKK4, JNK1 and c-FOS as targets for luteolin regulation in PC-3 cells. The qPCR data in Figure 2 confirmed the Ingenuity analysis. In both PC-3 and DU-145 human prostatic cancer cell lines, the pattern of the response profile of these EGFSP genes was virtually indistinguishable, even though c-Fos induction in DU-145 cells (> 900% above control) was more pronounced than that observed (>250% of control) in PC-3 cells. In addition, the level of luteolin inhibition of CCNA2, PLK-1, PCNA, CKDN1B, CDC25A and CCNE2 was indistinguishable in both cell types. Thus, on the basis of data in two extensively studied human prostatic cancer cell lines, it appears that the EGFSP and CCP genes are targets for luteolin regulation.
In order to explore the nature of the interaction of genes in the EGFSP and CCP controlled by luteolin, we assessed the effects of luteolin and/or gefitinib on the expression of these genes and effects on cell proliferation. It is clear from the flow cytometry data that luteolin and gefitinib control cell cycle transition through different mechanisms. The high dose luteolin studies indicate that treatment of PC-3 cells with this bioflavonoid causes G2/M arrest (p<0.001), decreases the numbers of cells in G0/G1 (p<0.001) and causes small but significant increase in apoptotic cells (p<0.001), even though the apoptotic cells represent only 2.6% of the population (Figure 5 and Table 1). Alternatively, gefitinib caused G0/G1 arrest (p<0.001) and decreased the number of cells in S-phase (p<0.001) and G2/M (p<0.01) relative to controls. These results were consistent with the published effects of this compound on the cell cycle [33]. Gefitinib inhibition of cell proliferation is attributed to reduced c-Fos and increased p27 (CDKN1B) expression [53]. Our data are in agreement with these findings. Although the RNA and protein data did not always directly correlate, gefitinib significantly reduced c-Fos RNA levels (Figure 7 and 10) in PC-3 cells (even though the protein was not changed; Figure 9) and increased the level of CDKN1B protein (even though the RNA was not changed; Figures 7 and 10). CKN1B (p27) is a cyclin-dependent kinase inhibitor that blocks the G0/G1 cell cycle transition by binding to Cdk’s [54]. Thus, our results with gefitinib are consistent with those recently reported by others on the mechanism of action for this compound.
More important for our studies was the observation that gefitinib failed to block luteolin effects on the cell cycle. This observation strongly suggests that these effects are not mediated through the modulation of EGFR-associated tyrosine kinase activity. This was somewhat surprising given the fact that this enzyme is a target for both luteolin and gefitinib [32, 55]. However, further analysis of our data indicated luteolin induction of c-Fos (RNA and protein) expression (Figures 6, 8–10) is likely to be a key component in the anti-proliferative response to the bioflavonoid. It is clear that gefitinib was incapable of blocking this response (Figs 6, 8–10) and that luteolin is regulating c-Fos through a novel mechanism not mediated by EGFR and/or its associated tyrosine kinase [33, 53]. That gefitinib effects on cell proliferation are mediated through the down-regulation of c-Fos and up regulation of CDKN1B (as noted above) is clearly different from the response to luteolin where 130–400% increases in RNA and protein for c-Fos were observed. In addition, luteolin treatment did not consistently affect CDKN1B (Figures 6–10). Thus, luteolin regulation of gene expression involves “gefitinib-independent” mechanisms not directly involving the phosphorylation of EGFR by its associated kinase.
It is also important to point out the a recent report demonstrated G2 cell cycle arrest resulting from the down-regulation of cyclin-B1, CDC25A, and CDC2 involved the stimulation of cjun/c-fos-dependent upregulation of p21 [46]. As noted in the results section, treatment of PC-3 cells [46] or gastric cancer cells [47] with luteolin resulted in G2/M arrest resulting from luteolin stimulation of p21. These two studies are in good agreement with the results presented here. The marked stimulation of c-Fos and the inhibition of the CCP genes (RNA and protein) including CDC25A, CCNA2, PLK-1, PCNA, CCNA2 and the G2/M arrest were associated with a 4-fold stimulation in p21 gene expression by luteolin (Figure 11). It is well established that each of these cell cycle proteins (CDC25A, CCNA2, PLK-1, PCNA, CCNA2) are inhibited by p21 [46, 47, 56]. These data suggest that luteolin stimulation of c-Fos upregulated p21 gene expression (Figure 11) resulting in the inhibition of the CCP genes controlling cell proliferation. We are currently evaluating these possibilities via detailed SiRNA-knockdown studies for EGFR, c-Fos and p21.
A key finding from the present studies is the observation that the mechanism for luteolin regulation of the EGFSP genes is likely to be different from the gefitinib sensitive-mechanism involved in luteolin regulation of cell cycle genes. Luteolin inhibition of CCNA2, PCNA, CCNE2 and CDC25A gene expression was enhanced by gefitinib treatment (Fig 10), suggesting the two compounds were additive. This additive response is certainly consistent with the fact that both compounds block EGFR-associated tyrosine kinase [32, 33]. Thus, their combined effects would be expected to be additive. Conversely, luteolin regulation of EGFSP genes (c-Fos, EGFR, MKK4, SOS, GRB2, JNK1, RasGAP; Figures 2, 6, 10 and Table 2) was not modified by gefitinib treatment (Fig 10). This result suggests that EGFR, per se, may not be directly involved in this response to luteolin. These findings are also consistent with a previous report from our laboratory demonstrating that luteolin regulates target gene transcription via and epigenetic mechanism involving its association with type II binding sites on histone H4. This was recently shown to be the case for the PLK-1 gene promoter in PC-3 cells [30] where luteolin treatment decreased the acetylation state of histone H4 associated with the PLK-1 gene promoter. We suspect this could be the case for the genes in the EGFSP as well since luteolin regulation of the expression of these genes obviously involved a mechanism insensitive to gefitinib. This being the case, one has to wonder if luteolin is affecting the acetylation of promoter-associated histones with all of the genes in the EGFSP (Figure 1) through a similar mechanism of action. Whether the luteolin-induced modification (acetylation, methylation, phosphorylation, ubiquination) of these histones results from changes in the recruitment of specific co-regulators with histone acetyltransferase (HAT) or deacetylases (HDAC) to these promoters remains to be resolved. That luteolin and related type II site ligands are capable of causing G2/M arrest via a c-FOS-p21 regulatory pathway in p53 null malignant cells may have significant clinical implications.
Acknowledgements
These studies were supported NIH grants CA-128932 and CA-35480 from the National Cancer Institute awarded to Baylor College of Medicine and Dr. Markaverich.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Eriksson H, Upchurch S, Hardin JW, Peck EJ, Jr, Clark JH. Heterogeneity of Estrogen Receptors in the Cytosol and Nuclear Fractions of the Rat Uterus. Biochem. Biophys. Res. Commun. 1978;81(1):1–7. doi: 10.1016/0006-291x(78)91622-4. [DOI] [PubMed] [Google Scholar]
- 2.Clark JH, Markaverich B, Upchurch S, Eriksson H, Hardin JW. Nuclear binding of the estrogen receptor: heterogeneity of sites and uterotropic response. Adv Exp Med Biol. 1979;117:17–46. doi: 10.1007/978-1-4757-6589-2_2. [DOI] [PubMed] [Google Scholar]
- 3.Markaverich BM, Schauweker TH, Gregory RR, Varma M, Kittrell FS, Medina D, Varma RS. Nuclear type II sites and malignant cell proliferation: inhibition by 2,6-bisbenzylidenecyclohexanones. Cancer Res. 1992;52(9):2482–2488. [PubMed] [Google Scholar]
- 4.Syne JS, Markaverich BM, Clark JH, Panko WB. Estrogen binding sites in the nucleus of normal and malignant human tissue: characteristics of the multiple nuclear binding sites. Cancer Res. 1982;42(11):4449–4454. [PubMed] [Google Scholar]
- 5.McKenna N, Lanz RB, O'Malley BW. Nuclear Receptor Coregulators: Cellular and Molecular Biology. Endocrine Reviews. 1999;20(3):321–344. doi: 10.1210/edrv.20.3.0366. [DOI] [PubMed] [Google Scholar]
- 6.Markaverich BM, Clark JH. Two binding sites for estradiol in rat uterine nuclei: relationship to uterotropic response. Endocrinology. 1979;105(6):1458–1462. doi: 10.1210/endo-105-6-1458. [DOI] [PubMed] [Google Scholar]
- 7.Clark JH, Markaverich BM. Relationships between type I and II estradiol binding sites and estrogen induced responses. J Steroid Biochem. 1981;15:49–54. doi: 10.1016/0022-4731(81)90257-0. [DOI] [PubMed] [Google Scholar]
- 8.Markaverich B, Upchurch S, Clark J. Progesterone and dexamethasone antagonism of uterine growth: role for a second nuclear estrogen binding site for estradiol in estrogen action. J Steroid Biochem. 1981;14:125–132. doi: 10.1016/0022-4731(81)90164-3. [DOI] [PubMed] [Google Scholar]
- 9.Watson CS, Medina D, Clark JH. Characterization of progesterone receptors, estrogen receptors and estrogen (type ii)-binding sites in the hormone-independent variant of the mxt-3590 mouse mammary tumor. Endocrinology. 1982;107(1):1432–1437. doi: 10.1210/endo-107-5-1432. [DOI] [PubMed] [Google Scholar]
- 10.Markaverich BM, Gregory RR, Alejandro MA, Clark JH, Johnson GA, Middleditch BS. Methyl p-hydroxyphenyllactate. An inhibitor of cell growth and proliferation and an endogenous ligand for nuclear type-II binding sites. J Biol Chem. 1988;263(15):7203–7210. [PubMed] [Google Scholar]
- 11.Markaverich BM, Roberts RR, Finney RW, Clark JH. Preliminary characterization of an endogenous inhibitor of [3H]estradiol binding in rat uterine nuclei. J Biol Chem. 1983;258(19):11663–11671. [PubMed] [Google Scholar]
- 12.Markaverich BM, Roberts RR, Alejandro MA, Clark JH. An endogenous inhibitor of [3H]estradiol binding to nuclear type II estrogen binding sites in normal and malignant tissues. Cancer Res. 1984;44(4):1515–1519. [PubMed] [Google Scholar]
- 13.Markaverich BM, Gregory RR, Alejandro MA, Varma RS, Johnson GA, Middleditch BS. Estrogen regulation of methyl p-hydroxyphenyllactate hydrolysis: correlation with estrogen stimulation of rat uterine growth. J Steroid Biochem. 1989;33(5):867–876. doi: 10.1016/0022-4731(89)90234-3. [DOI] [PubMed] [Google Scholar]
- 14.Markaverich BM, Gregory RR, Alejandro M, Kittrell FS, Medina D, Clark JH, Varma M, Varma RS. Methyl p-hydroxyphenyllactate and nuclear type II binding sites in malignant cells: metabolic fate and mammary tumor growth. Cancer Res. 1990;50(5):1470–1478. [PubMed] [Google Scholar]
- 15.Maybruck WM, Markaverich BM. Partial purification and characterization of methyl-p-hydroxyphenyllactate esterase in rat uterine cytosol. Steroids. 1997;62(3):321–330. doi: 10.1016/s0039-128x(96)00227-9. [DOI] [PubMed] [Google Scholar]
- 16.Markaverich BM, Shoulars K, Alejandro MA. Nuclear type II [3H]estradiol binding site ligands: inhibition of ER-positive and ER-negative cell proliferation and c-Myc and cyclin D1 gene expression. Steroids. 2006;71(10):865–874. doi: 10.1016/j.steroids.2006.05.017. [DOI] [PubMed] [Google Scholar]
- 17.Carbone A, Ranelletti F, Rinelli A, Vecchio F, Lauriola L, Piantelli M, Capelli A. Type II Estrogen Receptors in the Papillary Cystic Tumor of the Pancreas. Am. J. Cancer Research. 1989;92:572–576. doi: 10.1093/ajcp/92.5.572. [DOI] [PubMed] [Google Scholar]
- 18.Markaverich BM, Alejandro MA. Type II [3H]estradiol binding site antagonists: inhibition of normal and malignant prostate cell growth and proliferation. Int J Oncol. 1998;12(5):1127–1135. doi: 10.3892/ijo.12.5.1127. [DOI] [PubMed] [Google Scholar]
- 19.Piantelli M, Ricci R, Larocca L, Capelli A, Rizzo S, Scambia G, Ranelletti F. Type II Estrogen Binding Sites in Human Colorectal Carcinoma. J. Clin Path. 1990;43:1004–1006. doi: 10.1136/jcp.43.12.1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Scambia G, Ranelletti FO, Benedetti Panici P, Piantelli M, Rumi C, Battaglia F, LaRocca LM, Capelli A, Manusco S. Type II Estrogen Binding Sites in a Lymphoblastoid Cell Line and Growth Inhibitory Effects of Estrogen, Anti-Estrogen and Bioflavonoids. Int. J. Cancer. 1990;46:1112–1116. doi: 10.1002/ijc.2910460627. [DOI] [PubMed] [Google Scholar]
- 21.Leone LLTLMNPMRF, G Quercetin and the growth of leukemic progenitors. Leukemia & Lymphoma. 1996;23:49–53. doi: 10.3109/10428199609054801. [DOI] [PubMed] [Google Scholar]
- 22.Adlercreutz H, Mousavi Y, Hockerstedt K. Diet and Breast Cancer. Acta Oncologica. 1992;31:175–181. doi: 10.3109/02841869209088899. [DOI] [PubMed] [Google Scholar]
- 23.Severson RK, Nomura AMY, Grove JS, Stemmermann GN. A Prostpective Study of Demographics, Diet, and Prostate Cancer Among Men of Japanese Ancestory in Hawaii. Cancer Res. 1989;49:1857–1860. [PubMed] [Google Scholar]
- 24.Steinmets KA, Potter JD. Food-Group Consumption and Colon Cancer in Adelaide Case-Control Study I. Vegetables and Fruit. Int. J. Cancer. 1993;53:711–719. doi: 10.1002/ijc.2910530502. [DOI] [PubMed] [Google Scholar]
- 25.Markaverich BM, Roberts RR, Alejandro MA, Johnson GA, Middleditch BS, Clark JH. Bioflavonoid interaction with rat uterine type II binding sites and cell growth inhibition. J Steroid Biochem. 1988;30(1–6):71–78. doi: 10.1016/0022-4731(88)90078-7. [DOI] [PubMed] [Google Scholar]
- 26.Markaverich BM, Alejandro MA. Bioflavonoids, Type II [3H]Estradiol Binding Sites and Prostatic Cancer Cell Proliferation. International J. of Oncology. 1997;11:1311–1319. doi: 10.3892/ijo.11.6.1311. [DOI] [PubMed] [Google Scholar]
- 27.Shoulars K, Alejandro M, Thomson T, Markaverich B. Preliminary Identification of Rat Uterine Nuclear Type II [3H]Estradiol Binding Sites as Histone H4, Colorodo State University. Denver. 2001 [Google Scholar]
- 28.Shoulars K, Rodrigues MA, Crowley JR, Turk J, Thompson T, Markaverich BM. Nuclear type II [3H]estradiol binding sites: a histone H3–H4 complex. J Steroid Biochem Mol Biol. 2005;96(1):19–30. doi: 10.1016/j.jsbmb.2004.12.047. [DOI] [PubMed] [Google Scholar]
- 29.Shoulars K, Rodriguez MA, Crowley J, Turk J, Thompson T, Markaverich BM. Reconstitution of the type II [3H]estradiol binding site with recombinant histone H4. J Steroid Biochem Mol Biol. 2006;99(1):1–8. doi: 10.1016/j.jsbmb.2005.11.009. [DOI] [PubMed] [Google Scholar]
- 30.Shoulars K, Rodriguez MA, Thompson T, Markaverich BM. Regulation of Cell Cycle and RNA Transcription Genes Identified by Microarray Analysis of PC-3 Human Prostate Cancer Cells Treated with Luteolin. J Steroid Biochem Mol Biol. 2010;118(1–2):41–50. doi: 10.1016/j.jsbmb.2009.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nass SJ, Dickson RB. Epidermal growth factor-dependent cell cycle progression is altered in mammary epithelial cells that overexpress c-myc. Clin Cancer Res. 1998;4(7):1813–1822. [PubMed] [Google Scholar]
- 32.Huang YT, Hwang JJ, Lee PP, Ke FC, Huang JH, Huang CJ, Kandaswami C, Middleton E, Jr, Lee MT. Effects of luteolin and quercetin, inhibitors of tyrosine kinase, on cell growth and metastasis-associated properties in A431 cells overexpressing epidermal growth factor receptor. Br J Pharmacol. 1999;128(5):999–1010. doi: 10.1038/sj.bjp.0702879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Teraishi F, Kagawa S, Watanabe T, Tango Y, Kawashima T, Umeoka T, Nisizaki M, Tanaka N, Fujiwara T. ZD1839 (Gefitinib, 'Iressa'), an epidermal growth factor receptor-tyrosine kinase inhibitor, enhances the anti-cancer effects of TRAIL in human esophageal squamous cell carcinoma. FEBS Lett. 2005;579(19):4069–4075. doi: 10.1016/j.febslet.2005.06.031. [DOI] [PubMed] [Google Scholar]
- 34.Sharma RK, Chalam KV. In vitro evaluation of bevacizumab toxicity on a retinal ganglion cell line. Acta Ophthalmol. 2009;87(6):618–622. doi: 10.1111/j.1755-3768.2008.01410.x. [DOI] [PubMed] [Google Scholar]
- 35.Li C, Wong W. Model-based analysis of oligonucleotide arrays: expression index computation and outlier detection. Proc Natl Acad Sci USA. 2001;98:31–36. doi: 10.1073/pnas.011404098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li C, Wong W. Model-based analysis of oligonucleotide arrays: model validation, design issues and standard error application. Genome Biol 2:RESEARCH0032. 2001 doi: 10.1186/gb-2001-2-8-research0032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Agarwal R. Cell signaling and regulators of cell cycle as molecular targets for prostate cancer prevention by dietary agents. Biochem Pharmacol. 2000;60(8):1051–1059. doi: 10.1016/s0006-2952(00)00385-3. [DOI] [PubMed] [Google Scholar]
- 38.Santiskulvong C, Sinnett-Smith J, Rozengurt E. EGF receptor function is required in late G(1) for cell cycle progression induced by bombesin and bradykinin. Am J Physiol Cell Physiol. 2001;281(3):C886–C898. doi: 10.1152/ajpcell.2001.281.3.C886. [DOI] [PubMed] [Google Scholar]
- 39.Way TD, Lee JC, Kuo DH, Fan LL, Huang CH, Lin HY, Shieh PC, Kuo PT, Liao CF, Liu H, Kao JY. Inhibition of epidermal growth factor receptor signaling by Saussurea involucrata, a rare traditional Chinese medicinal herb, in human hormone-resistant prostate cancer PC-3 cells. J Agric Food Chem. 2010;58(6):3356–3365. doi: 10.1021/jf903793p. [DOI] [PubMed] [Google Scholar]
- 40.Toyoshima H, Hunter T. p27, a novel inhibitor of G1 cyclin-Cdk protein kinase activity, is related to p21. Cell. 1994;78(1):67–74. doi: 10.1016/0092-8674(94)90573-8. [DOI] [PubMed] [Google Scholar]
- 41.Paunesku T, Mittal S, Protic M, Oryhon J, Korolev S, Joachimiak A, Woloschak G. Proliferating cell nuclear antigen (PCNA): ringmaster of the genome. International J Radiation Biology. 2001;77(10):1007–1021. doi: 10.1080/09553000110069335. [DOI] [PubMed] [Google Scholar]
- 42.Johnson DG, Walker CL. Cyclins and cell cycle checkpoints. Annu Rev Pharmacol Toxicol. 1999;39:295–312. doi: 10.1146/annurev.pharmtox.39.1.295. [DOI] [PubMed] [Google Scholar]
- 43.Sgambato A, Camerini A, Faraglia B, Ardito R, Bianchino G, Spada D, Boninsegna A, Valentini V, Cittadini A. Targeted inhibition of the epidermal growth factor receptor-tyrosine kinase by ZD1839 ('Iressa') induces cell-cycle arrest and inhibits proliferation in prostate cancer cells. J Cell Physiol. 2004;201(1):97–105. doi: 10.1002/jcp.20045. [DOI] [PubMed] [Google Scholar]
- 44.Gry M, Rimini R, Stromberg S, Asplund A, Ponten F, Uhlen M, Nilsson P. Correlations between RNA and protein expression profiles in 23 human cell lines. BMC Genomics. 2009;10:365. doi: 10.1186/1471-2164-10-365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Scott SL, Earle JD, Gumerlock PH. Functional p53 increases prostate cancer cell survival after exposure to fractionated doses of ionizing radiation. Cancer Res. 2003;63(21):7190–7196. [PubMed] [Google Scholar]
- 46.Chan QK, Lam HM, Ng CF, Lee AY, Chan ES, Ng HK, Ho SM, Lau KM. Activation of GPR30 inhibits the growth of prostate cancer cells through sustained activation of Erk1/2, c-jun/c-fos-dependent upregulation of p21, and induction of G(2) cell-cycle arrest. Cell Death Differ. 2010 doi: 10.1038/cdd.2010.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wu B, Zhang Q, Shen W, Zhu J. Anti-proliferative and chemosensitizing effects of luteolin on human gastric cancer AGS cell line. Mol Cell Biochem. 2008;313(1–2):125–132. doi: 10.1007/s11010-008-9749-x. [DOI] [PubMed] [Google Scholar]
- 48.Markaverich BM, Roberts RR, Alejandro MA, Johnson GA, Middleditch BS, Clark JH. Bioflavonoid Interactions with Rat Uterine Type II Binding Sites and Cell Growth Inhibition. J. Steroid Biochem. 1988;30:71–78. doi: 10.1016/0022-4731(88)90078-7. [DOI] [PubMed] [Google Scholar]
- 49.Markaverich BM, Varma M, Densmore CL, Schauweker TH, Gregory RR. Nuclear Type II [3H]Estradiol Binding Sites in MCF-7 Human Breast Cancer Cells: Binding Interactions with 2,6-Bis([3,4-dihydroxyphenyl]-methylene)-cyclohexanone Esters and Inhibition of Cell Proliferation. Internat. J. Oncol. 1994;4:1291–1300. doi: 10.3892/ijo.4.6.1291. [DOI] [PubMed] [Google Scholar]
- 50.Shoulars K, Brown T, Alejandro MA, Crowley J, Markaverich BM. Identification of nuclear type II [(3)H]estradiol binding sites as histone H4. Biochem Biophys Res Commun. 2002;296(5):1083–1090. doi: 10.1016/s0006-291x(02)02042-9. [DOI] [PubMed] [Google Scholar]
- 51.Ang K, Berkey B, Tu X, Zhang H-Z, Katz R, Hammond E, Fu K, Milas L. Impact of epidermal growth factor receptor expression on survival and pattern of relapse in patients with advanced head and neck carcinoma. Cancer Research. 2002;62:7350–7356. [PubMed] [Google Scholar]
- 52.Tari AM, Hung MC, Li K, Lopez-Berestein G. Growth inhibition of breast cancer cells by Grb2 downregulation is correlated with inactivation of mitogen-activated protein kinase in EGFR, but not in ErbB2, cells. Oncogene. 1999;18(6):1325–1332. doi: 10.1038/sj.onc.1202422. [DOI] [PubMed] [Google Scholar]
- 53.Suzuki T, Yonemura K, Maruyama Y, Takahashi T, Takita T, Furuhashi M, Hishida A. Impact of serum parathyroid hormone concentration and its regulatory factors on arterial stiffness in patients undergoing maintenance hemodialysis. Blood Purif. 2004;22(3):293–297. doi: 10.1159/000078700. [DOI] [PubMed] [Google Scholar]
- 54.Glover CE, Gurley KE, Kim KH, Storer B, Fero ML, Kemp CJ. Endocrine dysfunction in p27Kip1 deficient mice and susceptibility to Wnt-1 driven breast cancer. Carcinogenesis. 2009;30(6):1058–1063. doi: 10.1093/carcin/bgp089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bianco R, Caputo R, Caputo R, Damiano V, De Placido S, Ficorella C, Agrawal S, Bianco AR, Ciardiello F, Tortora G. Combined targeting of epidermal growth factor receptor and MDM2 by gefitinib and antisense MDM2 cooperatively inhibit hormone-independent prostate cancer. Clin Cancer Res. 2004;10(14):4858–4864. doi: 10.1158/1078-0432.CCR-03-0497. [DOI] [PubMed] [Google Scholar]
- 56.Fang J, Zhou Q, Shi XL, Jiang BH. Luteolin inhibits insulin-like growth factor 1 receptor signaling in prostate cancer cells. Carcinogenesis. 2007;28(3):713–723. doi: 10.1093/carcin/bgl189. [DOI] [PubMed] [Google Scholar]