

Abstract
In a continuing investigation into the pharmacophores and structure-activity relationship (SAR) of (3′R,4′R)-3′,4′-di-O-(S)-camphanoyl-(+)-cis-khellactone (DCK) as a potent anti-HIV agent, 2′-monomethyl substituted 1′-oxa, 1′-thia, 1′-sulfoxide and 1′-sulfone analogs were synthesized and evaluated for inhibition of HIV-1 replication in H9 lymphocytes. Among them, 2′S-monomethyl-4-methyl DCK (5a) and 2′S-monomethyl-1′-thia-4-methyl DCK (7a) exhibited potent anti-HIV activity with EC50 values of 40.2 and 39.1 nM and remarkable therapeutic indexes of 705 and 1000, respectively, which were better than those of the lead compound DCK in the same assay. In contrast, the corresponding isomeric 2′R-monomethyl-4-methyl DCK (6) and 2′R-monomethyl-1′-thia-4-methyl DCK (8) showed much weaker inhibitory activity against HIV-1 replication. Therefore, the bioassay results suggest that the spatial orientation of the 2′-methyl group in DCK analogs can have important effects on anti-HIV activity of this compound class.
Keywords: 2′-Monomethyl-4-methyl-(3′R, 4′R)-3′; 4′-di-O-(S)-camphanoyl-(+)-cis-khellactone (DCK) analogs; 1′-Thia-4-methyl-(3′R,4′R)-3′; 4′-Di-O-(S)-camphanoyl-(+)-cis-khellactone (DCK) analogs; Anti-HIV activity; Structure-activity relationship (SAR)
1. Introduction
Since first reported in the 1980s, acquired immunodeficiency syndrome (AIDS) has spread rapidly through the human population and become one of the most devastating diseases facing mankind.1,2 This disease is caused by infection with the human immunodeficiency virus (HIV) and results in life-threatening opportunistic infections and malignancies. The increasing incidence of HIV drug resistance together with many serious side effects and long-term complications in patients diminish the action of current anti-HIV drugs. Consequently, the development of new anti-HIV agents continues to focus on novel structures or new action mechanisms.
In our previous research, 3′,4′-di-O-(−)-camphanoyl-(+)-cis-khellactone (DCK, 1, Figure 1) demonstrated extremely potent inhibitory activity against HIV-1 replication in H9 lymphocytic cells.3 In subsequent structural modification studies, numerous DCK derivatives were synthesized and more than 20 DCK analogs have shown promising inhibitory activity against HIV-1 replication in H9 lymphocytes, MT-2 cell lines and MT-4 cell lines, respectively.4 Among them, 3-methyl, 4-methyl, and 5-methyl substituted DCKs were much more potent than DCK and zidovudine (AZT) in the same assay.5 Initial structure-activity relationships (SAR) have been established for this compound type.4 In addition, a preliminary mechanistic study showed that DCK is a unique inhibitor of HIV-1 reverse transcriptase (RT). It did not significantly affect RNA-dependent DNA polymerase activity when poly-rA or poly-rC was used as template, while it did affect DNA-dependent DNA polymerase activity of HIV-1 RT when poly-dA or poly-dC was used as template.6 Moreover, a time-of-addition study suggested that DCK analogs are strongly synergistic with approved drugs, such as AZT, and act at a point in the virus life cycle immediately following the target for AZT and nevirapine.7
Figure 1.

Structures of previously synthesized DCK analogs (1–4)
Design
In our prior SAR study of the DCK C-ring, we found that the size of the 2′-alkyl substituent(s) had a significant effect on the antiviral activity. Gem-dimethyl substitution (2) was preferable, and in contrast, either no substitution (3) or larger alkyl substituents dramatically decreased activity. The rank order of activity was two hydrogen atoms (unsubstituted) ≈ methyl+propyl < gem-diethyl < gem-dimethyl.8 In a continuing effort to identify the pharmacophores of the 2′-position, 2′-monomethyl substituted 4-methyl DCK analogs were designed to further explore SAR at this position. We also extended our new design to 1′-thia derivatives, because bioisosteric replacement of sulfur for oxygen has also resulted in potent inhibitory effects on HIV-1 replication.9 The structures of the newly designed compounds (5–12) are shown in Figure 2. This paper reports their syntheses and bioassay data, as well as the X-ray crystallographic structure of 5a.
Figure 2.

Structures of 2′-monomethyl DCK and 1′-thia DCK analogues.
2. Results and discussion
2.1. Chemistry
Analogs 5a, 5b, and 6 were synthesized as illustrated in Scheme 1. 3-Chloro-1-butyne (14) was prepared by reacting 3-butyne-2-ol (13) with thionyl chloride in pyridine.10 4-Methyl-7-hydroxycoumarin (15) was treated with 14 in dimethyl formamide (DMF) or acetone in the presence of anhydrous potassium carbonate and potassium iodide at 75–80°C to produce the corresponding racemic mono-methyl propargyl ether 16, followed by thermal rearrangement in refluxing N,N-diethylaniline to form intermediate 17. Sharpless asymmetric dihydroxylation (AD) of 17 afforded two racemic mixtures 18 and 19, with yields of 64% and 24%, respectively.11 After acylation of 18 with optically pure (S)-(−)-camphanic chloride in anhydrous CH2Cl2 at room temperature with 4-(dimethylamino)pyridine (DMAP) as acid scavenger, diastereoisomers 5a and 5b were separated by column chromatography on silica gel [petroleum ether-EtOAc, 3:1(v/v)] in 68% and 18% yields, respectively. Compound 6 was obtained by acylation of 19 with (S)-(−)-camphanic chloride in anhydrous CH2Cl2 at room temperature in 84% yield. In Sharpless AD, osmylation preferentially takes place at the less hindered carbon of the double bond neighboring a methyl group, and the chiral auxiliary agent plays a decisive role on the stereoselectivity.12–16 In our synthetic scheme, hydroquinine 1,4-phthalazinediyl diether ((DHQ)2PHAL) was used as the chiral auxiliary; therefore, diol 18a was obtained as the main product. The absolute configuration of 5a (2′S,3′S,4′R) was confirmed by X-ray diffraction spectroscopy (Figure 3). Because of the directing effect of (DHQ)2PHAL and the steric effect of the 2′α-methyl (2′S), formation of diol 19b was unfavorable; therefore, the amount of corresponding camphanoyl ester was too small to quantify. The three chiral centers of the C ring in 6 were defined as 2′R, 3′S, and 4′R.
Scheme 1.

Reagents and conditions: (i) SOCl2, pyridine; (ii) K2CO3, KI in DMF or acetone, 75–80°C; (iii) N,N-diethylaniline, reflux; (iv) K2OsO2(OH)4, (DHQ)2-PHAL, K3Fe(CN)6, K2CO3 in t-butanol/H2O (v/v=1:1), ice bath; (v) (S)-camphanic chloride, DMAP in CH2Cl2, rt.
Figure 3.
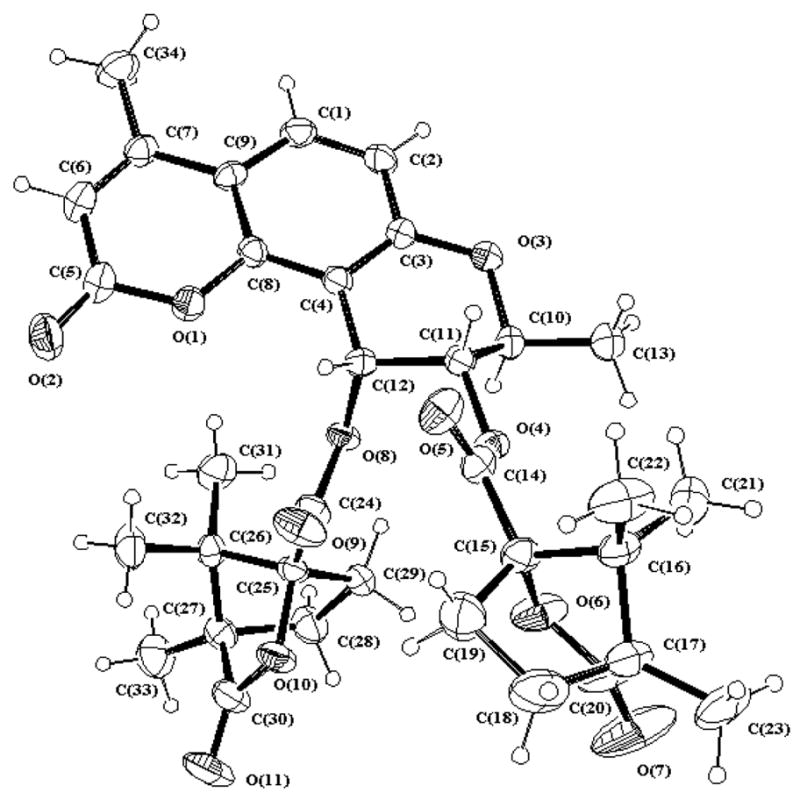
X-ray structure of 2′S-monomethyl-4-methyl DCK (5a).
The preparation of 7a, 7b and 8 is illustrated in Scheme 2. In a modified procedure,17,18 intermediate 4-methyl-7-mercaptocoumarin (22) was obtained by reacting 4-methyl-7-hydroxy coumarin (15) with dimethylthiocarbamoyl chloride in DMF in the presence of anhydrous potassium carbonate, followed by a rearrangement at 220–230°C, then hydrolysis with methanolic KOH and acidification with HCl. The remaining synthetic steps were similar to those in Scheme 1 detailed above. However, diastereoisomers 7a and 7b could not be separated by flash chromatography on silica gel or crystallization. 3H NMR spectroscopy showed that the ratio of diastereoisomers 7a and 7b was about 1:1. Finally, after exploring many different HPLC columns and conditions, diastereoisomers 7a and 7b were successfully separated on an Alltima HP column (Grace, 22mm×150mm, 10μm) with acetonitrile-water 70:30. The chemical shifts of protons at 2′-, 3′-, and 4′-positions in 3H NMR spectra of 5a, 5b, and 6 showed the same patterns as those of the corresponding 7a, 7b, and 8.
Scheme 2.

Reagents and conditions: (i) dimethylthiocarbamoyl chloride, K2CO3 in DMF, 55–60°C; (ii) heating at 220–230°C; (iii) KOH in MeOH/N2, reflux; concentrated HCl; (iv) 3-chloro-1-butyne, K2CO3, KI in DMF or acetone/N2, 75–80°C; (v) N,N-diethylaniline, reflux; (vi) K2OsO2(OH)4, (DHQ)2-PHAL, K3Fe(CN)6, K2CO3 in t-butanol/H2O (v/v=1:1), ice bath; (vii) (S)-camphanic chloride, DMAP in CH2Cl2, rt.
Scheme 3 illustrates the synthesis of 9a, 9b, 10, 11a, 11b, and 12. The diasteromeric mixture of 7a and 7b was reacted with one equivalent of 3-chloroperoxybenzoic acid (MCPBA) in CH2Cl2 to afford a diastereomeric mixture of sulfoxides 9a and 9b, which was again treated with one equivalent MCPBA in CH2Cl2 to give a diasteromeric mixture of sulfones 11a and 11b.19 Compounds 10 and 12 were prepared by similar oxidation of 8. The pure diastereomers of mixtures 9 and 11 were not isolated, because the mixtures did not show antiviral activity (Table 1).
Scheme 3.

Reagents and conditions: (i) one equivalent 3-chloroperoxybenzoic acid in CH2Cl2.
Table 1.
Anti-HIV activity of analogs 5–12a
| Compound | IC50 (μM) | EC50 (μM) | TI |
|---|---|---|---|
| 5a | 28.4 | <0.040 | >705.6 |
| 5b | 40.2 | 2.260 | 17.78 |
| 6 | 40.2 | no suppression | no suppression |
| 7a | 39.1 | <0.039 | >1000 |
| 7b | 39.1 | no suppression | no suppression |
| 8 | 39.1 | 1.260 | 31.06 |
| 9a+9b (2.4:1) | 38.2 | no suppression | no suppression |
| 10 | 38.2 | 24.400 | 1.57 |
| 11a+11b (3.2:1) | 37.3 | 3.370 | 11.07 |
| 12 | 37.3 | no suppression | no suppression |
| 2 (4-Me-DCK) | >39.2 | 0.126 | 301.2 |
| 3 | 41.1 | 16.200 | 2.54 |
| 4 | 40.1 | 0.990 | 40.58 |
| AZT | 500 | 0.010 | 43,848 |
All data presented are averages of at least three separate experiments performed by Panacos Pharmaceuticals Inc., Gaithersburg, MD. EC50: concentration that inhibits HIV-1IIIB replication by 50%. IC50: concentration that inhibits uninfected H9 cell growth by 50%. TI = IC50/EC50. No suppression: there was no inhibition at concentrations below the IC50.
2.2. Biological activity
Compounds 5–12 were evaluated in HIV-1 infected H9 lymphocytes in parallel with AZT and prior DCK analogs 2–4. The bioassay data are summarized in Table 1.
2′S-Methyl-4-methyl DCK (5a) showed significant anti-HIV activity with an EC50 value of 0.0402 μM and remarkable TI value of 705, which were slightly better than those of the 2′-dimethyl lead compound (2, 4-Me-DCK, EC50: 0.126μM, TI 301) in the same assay system. The 2′R-methyl isomer (6) was inactive at the test concentrations, and compound 3, with no substitution at the 2′-position, exhibited dramatically reduced activity (EC50: 16.2 μM). These results demonstrated that the spatial location of the 2′-methyl has a strong impact on the anti-HIV activity of DCK analogs. The same trend was also observed in the 1′-thia DCK series, where the EC50 values of 7a (2′S-Me), 8 (2′R-Me), and 4 (2′-H2) were 0.0391, 1.26, and 0.99 μM, respectively. Because the same rank order of potency was found in both DCK and 1′-thia DCK series [2′S-methyl (5a and 7a) > 2′-H2 (3 and 4) > 2′R-methyl (6 and 8)], we concluded that 2′S-methyl (α-methyl) substitution was essential for enhanced antiviral potency among Type I compounds (see Figure 2). In comparison, 2′R-methyl (β-methyl) was unfavorable and led to decreased antiviral activity. Moreover, we observed that 1′-thia DCK analogs generally had a slightly better antiviral profile than the DCK series analogs. With oxygen at the 1′-position, 6 and 3 showed significantly reduced activity compared with 5a, whereas the corresponding 1′-thia analogs, 8 and 4, retained more activity (lower fold reduction) compared with 7a. This result may possibly be due to a better interaction of S rather than O with the binding pocket, considering the extra size and electrons carried by the S atom, which may fit into the binding pocket better. Based on these results, we postulated that the 1′ and 2′ positions are important pharmacophores for the anti-HIV activity of DCK analogs.
The low potency and inactivity of analogs 5b (1′-oxa; 2′R,3′R,4′S) and 7b (1′-thia; 2′R,3′S,4′S), respectively, in which the 2′-methyl is β and 3′,4′-dicamphanoyl groups are α (Type II in Figure 2), were in accord with the preferred configuration (β) and spatial demand for the two camphanoyls found previously with DCK compounds.3, 4 In addition, all 1′-sulfoxide and 1′-sulfone analogs (9–12) showed much weaker activity than the corresponding 1′-thia-4-methyl DCK analogs (7a+7b or 8) or were inactive. The reason for the poor activities of these analogs might be that sulfone and sulfoxide have extra oxygen atoms that may cause some steric repulsion or loss of hydrogen bonding accepter function.
To confirm RT is the biological target of the newly synthesized 2′-monomethyl DCK analogs, compound 5a was evaluated against viral enzyme RT and compared with 2. From the results, we can see 5a potently inhibited RT activity with an IC50 value of 6.7 μM compared to the previous best hit 2 (IC50: 10.4 μM), suggesting that the monomethyl analogs may function similarly to DCKs as RT inhibitors.
3. Conclusion
In conclusion, 2′-monomethyl substituted DCKs, as well as 1′-thia, 1′-sulfoxide and 1′-sulfone DCK analogs, were designed and synthesized in this study. We discovered the following SAR conclusions. (1) In both the DCK and 1′-thia DCK Type I series, 2′S configuration (α-methyl substitution) was preferable to 2′R (β-methyl substitution) for anti-HIV activity. 2′S-Methyl analogs also showed more potent activity than corresponding 2′-H analogs, suggesting that the stereochemistry at the 2′-position has an important influence on the DCK series. (2) 1′-Thia DCK analogs generally showed a better antiviral profile than the DCK analogs. (3) Converting 1′-thia-4-methyl DCK analogs to the corresponding 1′-sulfoxide- and 1′-sulfone-4-methyl DCK analogs resulted in decreased or completely abolished activity. (4) Consistent with prior SAR, β orientations of the two camphanoyls (Type I) at the 3′ and 4′ positions were favorable for anti-HIV activity.
4. Experimental Section
4.1. Chemistry
Melting points were measured with the capillary tube method without correction. The proton nuclear magnetic resonance (3H NMR) spectra were measured on a Bruker-DPX 400 MHz spectrometer. The solvent used was CDCl3 unless indicated. Elemental analyses were measured with Elementar Vario EL. Mass spectra were measured with HP5973N analytical mass spectrometers. High resolution mass spectra (HRMS) were measured on a Shimadzu LCMS-IT-TOF with ESI interface. All target compounds were analyzed for C, H and gave values within ±0.4% of the theoretical values. Optical rotations were measured with a Jasco P-1020 digital polarimeter at 17.5°C at the sodium D line. The diastereoisomeric excess percentages were determined from intensity of protons at the 3′-position in the 3H NMR spectra. Thin-layer chromatography (TLC) was performed on PLC silica gel 60 F254 plates.
4.2. 3-Chloro-1-butyne (14)
Under stirring, 3-butyne-2-ol (13) (7.8 mL, 0.1 mol) was added dropwise to a mixture of thionyl chloride (7.3 mL, 0.1 mol) and anhydrous pyridine (0.8 g) at 0 °C over 0.5 h. The mixture was stirred for additional 2 h at 0 °C, allowed to warm to rt within 0.5 h, heated at 50°C for 1 h. Then pyridine was added to adjust the pH of the mixture to 7–8. After filtration, the filtrate was directly used in the next reaction.
4.3. 7-(But-3-yn-2-yloxy)-4-methyl-2H-chromen-2-one (16)
A mixture of 15 (1.41 g, 8 mmol), K2CO3 (4.41 g, 32 mmol), KI (266 mg, 1.6 mmol), and 3-chloro-1-butyne in DMF (15 mL) was stirred for 2 h at 75–80°C. Ice water (120 mL) was added. The precipitate was filtered and purified by column chromatography on silica gel (petroleum ether-EtOAc, 5:1) to provide 16 as a white solid (1.42 g, 78%), mp 134–135 °C. 3H NMR δ 1.71 (3H, d, J = 6.65 Hz, 8-CH3), 2.40 (3H, d, J = 1.17 Hz, 4-CH3), 2.52 (1H, d, J = 1.96 Hz, -CCH), 4.92 (1H, m, 8-CH), 6.16 (1H, d, J = 1.17 Hz, 3-H), 6.93 (1H, d, J = 2.74 Hz, 8-H), 6.97 (1H, dd, J = 8.00, 2.35 Hz, 6-H), 7.52 (1H, d, J = 8.60 Hz, 5-H). EI-MS m/z (%): 228 (M +, 45).
4.4. 2′,4-Dimethylseselin (17)
A mixture of 16 (1.3 g, 5.7 mmol) and N,N-diethylaniline (12 mL) was refluxed for 4 h. The solvent was removed in vacuo and the residue was purified by column chromatography on silica gel (petroleum ether-EtOAc, 7:1) to provide 17 as a white solid (1.0 g, 77%), mp 115–116 °C. 3H NMR δ 1.48 (3H, d, J = 6.65 Hz, 2′-CH3), 2.37 (3H, d, J = 1.17 Hz, 4-CH3), 5.10 (1H, m, 2′-CH), 5.76 (1H, dd, J1 = 9.98 Hz, J2 = 3.33 Hz, 3′-H), 6.12 (1H, d, J = 1.17 Hz, 3-H), 6.74 (1H, d, J = 8.61 Hz, 6-H), 6.95 (1H, dd, J1 = 9.78 Hz, J2 = 1.18 Hz, 4′-H), 7.34 (1H, d, J = 8.61 Hz, 5-H). EI-MS m/z (%): 228 (M +, 21).
4.5. 2′,4-Dimethyl-(+)-cis-khellactone (18) and (19)
Compound 17 (684 mg, 3.0 mmol), (DHQ)2-PHAL (82 mg, 0.105 mmol), K3Fe(CN)6 (4.94 g, 15 mmol), K2CO3 (2.08 g, 15 mmol) were dissolved in t-BuOH/H2O (120 mL, v/v, 1:1) at rt. Then the solution was cooled to 0 °C and K2OsO2(OH)4 (38 mg, 0.105 mmol) was added under stirring. The mixture was stirred at 0 °C for 70 h. Then sodium sulfite (4 g) was added and stirred at rt for 1 h. The mixture was extracted with EtOAc three times. The combined organic layer was dried over Na2SO4, and then solvent was removed. The residue was purified by column chromatography on silica gel (CHCl3-MeOH, 140:1) to provide 19 (188 mg, 24%, CHCl3-MeOH, 60:1, Rf 0.07) and 18 (503 mg, 64%, CHCl3-MeOH, 60:1, Rf 0.05) as white solids. Compound 18, mp 231–234 °C. 3H NMR (DMSO) δ 1.38 (3H, d, J = 6.26 Hz, 2′-CH3), 2.36 (3H, s, 4-CH3), 3.42 (1H, m, 3′-CH), 4.25 (1H, m, 2′-CH), 4.85 (1H, m, 4′-CH), 5.12 (1H, d, J = 6.65 Hz, 3′-OH), 5.36 (1H, d, J = 4.69 Hz, 4′-OH), 6.19 (1H, s, 3-H), 6.79 (1H, d, J = 8.60 Hz, 6-H), 7.58 (1H, d, J = 9.0 Hz, 5-H). EI-MS m/z (%): 262 (M +, 28). Compound 19, mp 252–255 °C. 3H NMR (DMSO) δ 1.36 (3H, d, J = 6.65 Hz, 2′-CH3), 2.36 (3H, s, 4-CH3), 3.80 (1H, m, 3′-CH), 4.27 (1H, m, 2′-CH), 4.96 (1H, m, 4′-CH), 5.09 (1H, d, J = 5.09 Hz, 3′-OH), 5.13 (1H, d, J = 6.26 Hz, 4′-OH), 6.18 (1H, s, 3-H), 6.78 (1H, d, J = 9.0 Hz, 6-H), 7.56 (1H, d, J = 8.6 Hz, 5-H). EI-MS m/z (%): 262 (M +, 37).
4.6. (2′S,3′S,4′R)-2′,4-Dimethyl-3′,4′-di-O-(−)-camphanoyl-(+)-cis-khellactone (5a) and (2′R,3′R,4′S)-2′,4-Dimethyl-3′,4′-di-O-(−)-camphanoyl-(+)-cis-khellactone (5b)
Compound 18 (152 mg, 0.58 mmol) was acylated with (S)-(−)-camphanic chloride (504 mg, 2.32 mmol) in anhydrous CH2Cl2 (15 mL) in the presence of DMAP (426 mg, 3.48 mmol) for 0.5 h at rt. After the removal of the solvent under reduced pressure, the residue was purified by using silica gel chromatography (petroleum ether-EtOAc, 3:1) to give product 5b (65 mg, 18%, petroleum ether-EtOAc, 5:2, Rf 0.76) and product 5a (245 mg, 68%, petroleum ether-EtOAc, 5:2, Rf 0.75) as white solids. Compound 5a, mp 263–266 °C. Crystals suitable for single crystal X-ray diffraction were grown from EtOH. 3H NMR δ 0.96–1.11 (18H, m, 6×-CH3 in camphanoyl), 1.61–2.54 (8H, m, 4×-CH2 in camphanoyl), 1.46 (3H, d, J = 6.26 Hz, 2′-CH3), 2.39 (3H, d, J = 1.18 Hz, 4-CH3), 4.51 (1H, m, 2′-CH), 5.15 (1H, m, 3′-H), 6.13 (1H, d, J = 1.57 Hz, 3-H), 6.71 (1H, d, J = 3.52 Hz, 4′-H), 6.86 (1H, d, J = 8.99 Hz, 6-H), 7.54 (1H, d, J = 8.99 Hz, 5-H). [α]D +28.9 (c 0.9, CH2Cl2); EI-MS m/z (%): 622 (M +, 8). Anal. calcd for C34H38O11: C 65.58, H 6.15; found C 65.44, H 6.24.
Compound 5b, mp 237–239 °C. 3H NMR δ 0.82 (3H, s, -CH3 in Camphanoyl), 1.04–1.12 (15H, m, -CH3×5 in camphanoyl), 1.64–2.57 (8H, m, 4×-CH2 in camphanoyl), 1.45 (3H, d, J = 6.26 Hz, 2′-CH3), 2.39 (3H, d, J = 1.17 Hz, 4-CH3), 4.62 (1H, m, 2′-CH), 5.27 (1H, m, 3′-H), 6.11 (1H, d, J = 1.17 Hz, 3-H), 6.81 (1H, d, J = 3.52 Hz, 4′-H), 6.86 (1H, d, J = 8.61 Hz, 6-H), 7.54 (1H, d, J = 8.61 Hz, 5-H). [α]D -24.0 (c 1.0, CHCl3); EI-MS m/z (%): 622 (M +, 6). Anal. calcd for C34H38O11: C 65.58, H 6.15; found C 65.37, H 6.28.
4.7. (2′R,3′S,4′R)-2′,4-Dimethyl-3′,4′-di-O-(−)-camphanoyl-(+)-cis-khellactone (6)
Compound 19 (63 mg, 0.245 mmol) was acylated with (S)-(−)-camphanic chloride (171 mg, 0.78 mmol) in anhydrous CH2Cl2 (10 mL) in the presence of DMAP (150 mg, 1.23 mmol) for 0.5 h at rt. After the removal of the solvent under reduced pressure, the residue was submitted to silica gel chromatography separation (CHCl3-CH3OH, 120:1) to afford product 6 as a white solid (128 mg, 84%), mp >280 °C. 3H NMR δ 0.98–1.11 (18H, m, -CH3×6 in camphanoyl), 1.64–2.57 (8H, m, 4×-CH2 in camphanoyl), 1.46 (3H, d, J = 6.60 Hz, 2′-CH3), 2.39 (3H, d, J = 1.22 Hz, 4-CH3), 4.48 (1H, m, 2′-CH), 5.63 (1H, m, 3′-H), 6.12 (1H, d, J = 0.98 Hz, 3-H), 6.73 (1H, d, J = 4.88 Hz, 4′-H), 6.88 (1H, d, J = 8.79 Hz, 6-H), 7.51 (1H, d, J = 9.03 Hz, 5-H). [α]D +111 (c 1.0, CH2Cl2); ESI-MS m/z (%): 645 (M+Na+). Anal. calcd for C34H38O11: C 65.58, H 6.15; found C 65.56, H 6.36.
4.8. O-4-Methyl-2-oxo-2H-chromen-7-yl dimethylcarbamothioate (20)
Compound 15 (4.4 g, 25 mmol), K2CO3 (7.6 g, 55 mmol), and dimethylthiocarbamoyl chloride (6.18g, 50 mmol) were dissolved in DMF (45mL). After stirring at 65–70 °C for 1 h, ice water (300 mL) was added, and the precipitate was filtered and recrystallized from EtOAc to afford 20 as a yellow solid (5.8 g, 88%), mp 214–216°C (lit.17 mp 208–209 °C). 3H NMR 2.44 (3H, s, 4-CH3), 3.38 (3H, s, N-CH3), 3.47 (3H, s, N-CH3), 6.28(1H, s, 3-H), 7.07(1H, s, 8-H), 7.26(1H, 6-H), 7.60 (1H, d, J =8.18Hz, 5-H).
4.9. S-4-Methyl-2oxo-2H-chromen-7-yl dimethylcarbamothioate (21)
Compound 20 (5.0 g, 19.0 mmol) was heated to 220–230°C for 1 h. The crude product was recrystallized from EtOH to give 21 as a yellow solid (4.2 g, 84%), mp 160–162°C (lit.17 mp 159–160°C).
4.10. 4-Methyl-7-mercapto-2H-chromen-2-one (22)
Under nitrogen, compound 21 (3.6 g, 13.7 mmol) and potassium hydroxide (2.34 g, 41.6 mmol) were dissolved in MeOH (800 mL). The mixture was refluxed for 1.5 h with stirring. After cooling to rt, the mixture was acidified with concentrated HCl, and MeOH was removed under vacuum. Then ice water (400 mL) was added, and the reaction mixture was filtered. The precipitate was recrystallized from MeOH to afford 22 as a yellow solid (2.1 g, 81%), mp 137–138°C (lit.17 mp 138–139 °C).
4.11. 7-(But-3-yn-2-ylthio)-4-methyl-2-methylene-2H-chromene (23)
Following the same procedure for the preparation of 16, except under nitrogen, 23 was obtained from 22 as a white solid (yield, 75%): mp 124–126 °C. 3H NMRδ 1.62 (3H, d, J = 6.65 Hz, 8-CH3), 2.42 (3H, d, J = 1.56 Hz, 4-CH3), 2.37 (1H, d, J = 1.35 Hz, -CCH), 4.04 (1H, m, 8-CH), 6.26 (1H, d, J = 1.17 Hz, 3-H), 7.43 (1H, d, J = 2.35 Hz, 8-H), 7.32 (1H, dd, J1 = 8.60 Hz, J2 = 1.95 Hz, 6-H), 7.52 (1H, d, J = 8.60 Hz, 5-H). EI-MS m/z (%): 244 (M +, 50), 229 (base).
4.12. 1′-Thia-2′,4-dimethylseselin (24)
Following the same procedure for the preparation of 17, 24 was obtained from 23 as a yellow solid (yield, 56%, estimated by H NMR). The crude 24 was used directly in the preparation of 25 and 26.
4.13. 1′-Thia-2′,4-dimethyl-(+)-cis-khellactone (25) and (26)
Following the same procedure for the preparation of 18 and 19, 25 (yield 52%, mp 226–227 °C, CHCl3-MeOH, 80:1, Rf 0.14) and 26 (yield 16%, mp 242–243 °C, CHCl3-MeOH, 80:1, Rf 0.15) were obtained from 24 as white solids. Compound 25. 3H NMR (DMSO) δ 1.31 (3H, d, J = 6.25 Hz, 2′-CH3), 2.38 (3H, s, 4-CH3), 3.47 (1H, m, 3′-CH), 3.63 (1H, m, 2′-CH), 5.07 (1H, m, 4′-CH), 5.19 (1H, d, J = 6.65 Hz, 3′-OH), 5.44 (1H, d, J = 4.7 Hz, 4′-OH), 6.30 (1H, d, J = 1.18 Hz, 3-H), 7.06 (1H, d, J = 8.60 Hz, 6-H), 7.55 (1H, d, J = 8.22 Hz, 5-H). EI-MS m/z (%): 278 (M +, 26). Compound 26. 3H NMR (DMSO) δ 1.50 (3H, d, J = 7.04 Hz, 2′-CH3), 2.38 (3H, d, J = 1.17 Hz, 4-CH3), 3.31 (1H, m, 3′-CH), 3.97 (1H, m, 2′-CH), 5.07 (1H, m, 4′-CH), 5.14 (1H, d, J = 4.30 Hz, 3′-OH), 5.32 (1H, d, J = 5.48 Hz, 4′-OH), 6.30 (1H, d, J = 1.18 Hz, 3-H), 7.08 (1H, d, J = 8.22 Hz, 6-H), 7.55 (1H, d, J = 8.21 Hz, 5-H). EI-MS m/z (%): 278 (M +, 22).
4.14. (2′S,3′R,4′R)-1′-Thia-2′,4-dimethyl-3′,4′-di-O-(−)-camphanoyl-(+)-cis-khellactone (7a) and (2′R,3′S,4′S) -1′-Thia-2′,4-dimethyl-3′,4′-di-O-(−)-camphanoyl-(+)-cis-khellactone (7b)
Following the same procedure for the preparation of 5a and 5b, 7a (yield 42%, mp 175–177 °C) and 7b (yield 42%, mp 153–155 °C) were obtained from 25 as yellow solids. The diastereoisomers (7a) and (7b) were separated by semi-preparative isolation on Alltima HP column with CH3CN-water 70:30. Compound 7a (HPLC, CH3CN-water 70:30, Rf 0.49). 3H NMRδ 0.95 (3H, s, -CH3 in camphanoyl), 1.05–1.12 (15H, m, -CH3×5 in camphanoyl), 1.58–2.60 (8H, m, 4×-CH2 in camphanoyl), 1.33 (3H, d, J = 6.6 Hz, 2′-CH3), 2.38 (3H, 4-CH3), 3.85 (1H, m, 2′-CH), 5.33 (1H, dd, J1 = 11.7 Hz, J2 = 2.7 Hz, 3′-H), 6.17 (1H, 3-H), 6.85 (1H, d, J = 3.0 Hz, 4′-H), 7.04 (1H, d, J = 8.4 Hz, 6-H), 7.46 (1H, d, J = 8.4 Hz, 5-H). [α]D -171.9 (c 0.70, CH2Cl2); HRMS (MALDI-DHB) calcd mass for C34H38O10S [M+ + Na] 661.2083, found 661.2085. Compound 7b (HPLC, CH3CN-water 70:30, Rf 0.39). 3H NMR δ 0.75 (3H, s, -CH3 in camphanoyl), 1.05–1.12 (15H, m, -CH3×5 in camphanoyl), 1.58–2.60 (8H, m, 4×-CH2 in camphanoyl), 1.34 (3H, d, J = 6.65 Hz, 2′-CH3), 2.38 (3H, d, J = 1.0 Hz, 4-CH3), 3.90 (1H, m, 2′-CH), 5.42 (1H, dd, J1 = 11.4 Hz, J2 = 3.0 Hz, 3′-H), 6.17 (1H, d, J=1.0 Hz, 3-H), 6.95 (1H, d, J = 3.0 Hz, 4′-H), 7.04 (1H, d, J = 8.7 Hz, 6-H), 7.46 (1H, d, J = 8.7 Hz, 5-H). [α]D +135.6 (c 0.83, CH2Cl2); HRMS (MALDI-DHB) calcd mass for C34H38O10S [M+ + Na] 661.2083, found 661.2086.
4.15. (2′R,3′R,4′R)-1′-Thia-2′,4-Dimethyl-3′,4′-di-O-(−)-camphanoyl-(+)-cis-khellactone (8)
Following the same procedure for the preparation of 6, 8 was obtained from 26 as a yellow solid (yield, 78%), 260–263 °C. 3H NMR δ: 0.82–1.11(18H, m, -CH3×6 in camphanoyl), 1.64–2.58 (8H, m, 4×-CH2 in camphanoyl), 1.10 (3H, d, J = 5.86 Hz, 2′-CH3), 2.39 (3H, d, J = 1.10 Hz, 4-CH3), 3.32 (1H, m, 2′-CH), 5.80 (1H, m, 3′-H), 6.18 (1H, d, J = 1.10 Hz, 3-H), 6.76 (1H, d, J = 4.40 Hz, 4′-H), 7.08 (1H, d, J = 8.44 Hz, 6-H), 7.49 (1H, d, J = 8.43 Hz, 5-H). [α]D +102.5 (c 0.8, CH2Cl2); ESI-MS m/z (%): 661 (M+Na+). Anal. calcd for C34H38O10S: C 63.93, H 6.00, S 5.02; found C 63.72, H 6.12, S 5.15.
4.16. 1′-Sulfoxide-2′,4-dimethyl-3′,4′-di-O-(−)-camphanoyl-(+)-cis-khellactone (9a) and (9b)
A mixture of 7a and 7b (150 mg, 0.235 mmol) was dissolved in anhydrous CH2Cl2 (10 mL). Then the solution was cooled to 0 °C and 90% MCPBA (44 mg, 0.235 mmol) was added portionwise and stirred for 15 min. The solution was stirred for an additional 10 min at rt. After dilution with water (60 mL), the mixture was extracted with CH2Cl2. The combined organic layer was dried over Na2SO4, and then solvent was removed in vacuo. The residue was recrystallized from MeOH to afford a mixture of 9a and 9b as a white solid (138 mg, 90%), mp 273–276°C. 3H NMR spectra showed the ratio of diastereoisomers (9a) and (9b) was about 2.4:1. 3H NMR δ 0.76–1.25 (18H, s, -CH3×6 in camphanoyl), 1.55–2.59 (8H, m, 4×-CH2 in camphanoyl), 1.55 (3H, d, J = 7.15 Hz, 2′-CH3), 2.49 (3H, s, 4-CH3), 3.59 (0.69H, m, 2′-CH), 3.72 (0.31H, m, 2′-CH), 6.04 (1H, m, 3′-H), 6.40 (1H, 3-H), 6.95 (0.71H, d, J = 3.30 Hz, 4′-H), 7.08 (0.29H, d, J = 3.12 Hz, 4′-H), 7.75 (1H, d, J = 8.24 Hz, 6-H), 7.86 (1H, d, J = 8.24 Hz, 5-H). [α]D +22.2 (c 0.9, CH2Cl2); ESI-MS m/z (%): 677.2 (M+Na+). Anal. calcd for C34H38O11S: C 62.37, H 5.85, S 4.90; found C 62.54, H 5.79, S 4.93.
4.17. 1′-Sulfone-2′,4-dimethyl-3′,4′-di-O-(−)-camphanoyl-(+)-cis-khellactone (11a) and (11b)
A mixture of compound 9a and 9b (105 mg, 0.16 mmol), 90% MCPBA (44 mg, 0.21 mmol) in anhydrous CH2Cl2 (8 mL) was stirred for 30 min at rt. Then diluted with water (50 mL), the mixture was extracted with CH2Cl2. The combined organic layer was dried over Na2SO4, and then solvent was removed. The residue was recrystallized from MeOH to afford a mixture of 11a and 11b as a white solid (92 mg, 86%), mp >270°C. 3H NMR spectra showed the ratio of diastereoisomers (11a) and (11b) was about 3.2:1. 3H NMRδ 0.77–1.24 (18H, s, -CH3×6 in camphanoyl), 1.55–2.58 (8H, m, 4×-CH2 in camphanoyl), 1.55 (3H, d, J = 7.04 Hz, 2′-CH3), 2.49 (3H, d, J =1.17 Hz, 4-CH3), 3.96 (0.77H, m, 2′-CH), 4.10 (0.23H, m, 2′-CH), 5.75 (1H, m, 3′-H), 6.40 (1H, d, J =1.18 Hz, 3-H), 6.97 (0.76H, d, J = 3.25 Hz, 4′-H), 7.11 (0.24H, d, J = 3.13 Hz, 4′-H), 7.91 (2H, 6-H and 5-H). [α]D -5.0 (c 1.0, CH2Cl2); ESI-MS m/z (%): 669 (M+-1). Anal. calcd for C34H38O12S: C 60.88, H 5.71, S 4.78; found C 60.96, H 5.84, S 4.80.
4.18. (8R,9R,10R)-1′-Sulfoxide-2′,4-dimethyl-3′,4′-di-O-(−)-camphanoyl-(+)-cis-khellactone (10)
Following the same procedure for the preparation of 9a and 9b, 10 was obtained from 8 as a white solid (yield, 82%), mp 246–248°C. 3H NMR δ 0.85–1.25 (18H, m, -CH3×6 in camphanoyl), 1.60–2.58 (8H, m, 4×-CH2 in camphanoyl), 1.46 (3H, d, J = 7.43 Hz, 2′-CH3), 2.49 (3H, s, 4-CH3), 3.48 (1H, m, 2′-CH), 6.38 (1H, m, 3′-H), 6.39 (1H, s, 3-H), 6.89 (1H, d, J = 4.30 Hz, 4′-H), 7.78 (1H, d, J = 8.21 Hz, 6-H), 7.89 (1H, d, J = 8.21 Hz, 5-H). [α]D +21.8 (c 1.1, CH2Cl2); ESI-MS m/z (%): 677 (M+Na+).
4.19. (8R,9R,10R)-1′-Sulfone-2′,4-dimethyl-3′,4′-di-O-(−)-camphanoyl-(+)-cis-khellactone (12)
A mixture of compound 8 (64 mg, 0.10 mmol), 90% MCPBA (60 mg, 0.30 mmol) in anhydrous CH2Cl2 (6 mL) was stirred for 45 min at rt. The reaction mixture was diluted with CH2Cl2 (60 mL), and washed with 10% Na2CO3 and brine. The organic layer was dried over Na2SO4. After removal of the solvent, the residue was separated by column chromatography on silica gel (petroleum ether-acetone, 3:1) to provide product 12 as a white solid (57 mg, 82%), mp 203–205°C. 3H NMRδ 0.79–1.12 (18H, m, -CH3×6 in camphanoyl), 1.68–2.58 (8H, m, 4×-CH2 in camphanoyl), 1.65 (3H, d, J = 6.65 Hz, 2′-CH3), 2.49 (3H, s, 4-CH3), 3.59 (1H, m, 2′-CH), 6.29 (1H, m, 3′-H), 6.41 (1H, s, 3-H), 6.91 (1H, d, J = 4.30 Hz, 4′-H), 7.92 (2H, 6-H and 5-H). [α]D −40.0 (c 1.0, CH2Cl2); ESI-MS m/z (%): 693 (M+Na+).
4.20. Biological assays
4.20.1. HIV-1 infectivity assay against non-drug-resistant strain in H9 lymphocytes
This assay was performed by Panacos Pharmaceuticals, Inc as follows. The human T-cell line, H9, was maintained in continuous culture with L-glutamine at 5% CO2 and 37°C. Test samples were first dissolved in dimethyl sulfoxide. The following were the final drug concentrations routinely used for screening 100, 20, 4 and 0.8 μg/mL. For agents found to be active, additional dilutions were prepared for subsequent testing so that an accurate EC50 value could be determined. Test samples were prepared, and to each sample well was added 90 μL of media containing H9 cells at 3 × 105 cells/mL and 45 μL of virus inoculum (HIV-1 IIIB isolate) containing 125 TCID50. Control wells containing virus and cells only (no drug) and cells only (no virus or drug) were also prepared. A second set of samples was prepared identical to the first and added to cells under identical conditions without virus (mock infection) for toxicity determinations (IC50 defined below). In addition, AZT and 4-methyl DCK was also assayed during each experiment as positive drug controls. On days 1 and 4 post-infection (PI), spent media was removed from each well and replaced with fresh media. On day 6 PI, the assay was terminated and cultured supernatants were harvested for analysis for virus replication by p24 antigen capture. The compound toxicity was determined by XTT using the mock-infected sample wells. If a test sample inhibited virus replication and was not toxic, its effects were reported in the following terms: IC50, the concentration of test sample that was toxic to 50% of the mock-infected cells; EC50, the concentration of test sample that was able to suppress HIV replication by 50%; and the therapeutic index (TI), the ratio of the IC50 to EC50.
4.20.2. Assay for RT enzymatic inhibition
The RT activity of HIV-1 DH012 was determined in the presence of various concentrations of the tested compounds using a Roche colorimetric HIV-1 RT assay kit following the protocol provided by the manufacturer.
Supplementary Material
Acknowledgments
This research was supported by the grants from the National Natural Science Foundation of China awarded to P. Xia (No. 20272010) and Y. Chen (No. 30200348 and 30873164, respectively, and the research grant (2002046069) for PhD program from the National Education Administration of China awarded to P. Xia, and Grant AI-33066 from the National Institute of Allergies and Infectious Diseases awarded to K. H. Lee. Thanks are also due to the National Drug Innovative Program(Grant No. 2009ZX09301-011) for partial support.
Abbreviations
- DCK
(3′R, 4′R)-3′,4′-di-O-(S)-camphanoyl-(+)-cis-khellactone
- SAR
structure activity relationship
- AIDS
acquired immunodeficiency syndrome
- HIV-1
human immunodeficiency virus type 1
- AZT
zidovudine
- SAR
structure-activity relationships
- RT
reverse transcriptase
- DMF
dimethyl formamide
- AD
asymmetric dihydroxylation
- DMAP
4-(dimethylamino)pyridine
- (DHQ)2PHAL
hydroquinine 1,4-phthalazinediyl diether
- MCPBA
3-chloroperoxybenzoic acid
Footnotes
Anti-AIDS Agents 84. For part 83, see T. Zhou, Q. Shi, and K.H. Lee. Anti-AIDS agents 83. Efficient microwave-assisted one-pot preparation of angular 2,2-dimethyl-2H-chromone containing compounds. Tetrahedron Lett., 2010, 4382-4386.
X-ray data of 5a: DEPOSIT NUMBER CCDC 678968
Supporting Information Available
Additional information on compound purity including elemental analysis, high-resolution mass spectral data, and HPLC analysis results of the target compounds.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Gottlieb MS, Schroff R, Schanker HM, Weisman JD, Fan PT, Wolf RA, Saxon A. N Engl J Med. 1981;305:1425. doi: 10.1056/NEJM198112103052401. [DOI] [PubMed] [Google Scholar]
- 2.Barre-Sinoussi F, Chermann JC, Rey F, Nugeyre MT, Chamaret S, Gruest J, Dauguet C, Axler-Blin C, Vezinet-Brun F, Rouzioux C, Rozenbaum W, Montagnier L. Science. 1983;220:868. doi: 10.1126/science.6189183. [DOI] [PubMed] [Google Scholar]
- 3.Huang L, Kashiwada Y, Cosentino LM, Fan S, Chen CH, McPhail AT, Fujioka T, Mihashi K, Lee KH. J Med Chem. 1994;37:3947. doi: 10.1021/jm00049a014. [DOI] [PubMed] [Google Scholar]
- 4.a) Yu D, Suzuki M, Xie L, Morris-Natschke SL, Lee KH. Med Res Rev. 2003;23:322. doi: 10.1002/med.10034. [DOI] [PubMed] [Google Scholar]; b) Yu D, Morris-Natschke SL, Lee KH. Med Res Rev. 2007;27:108. doi: 10.1002/med.20075. [DOI] [PubMed] [Google Scholar]; c) Xie L, Guo HF, Lu H, Zhuang XM, Zhang AM, Wu G, Ruan JX, Zhou T, Yu D, Qian X, Lee KH, Jiang S. J Med Chem. 2008;25:7689. doi: 10.1021/jm8003009. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Tang J, Qian K, Zhang BN, Chen Y, Xia P, Yu D, Xia Y, Yang ZY, Chen CH, Morris-Natschke SL, Lee KH. Bioorg Med Chem. 2010;18:4363. doi: 10.1016/j.bmc.2010.04.089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Xie L, Takeuchi Y, Cosentino LM, Lee KH. J Med Chem. 1999;42:2662. doi: 10.1021/jm9900624. [DOI] [PubMed] [Google Scholar]
- 6.Huang L, Yuan X, Yu D, Lee KH, Chen CH. Virology. 2005;332:623. doi: 10.1016/j.virol.2004.11.033. [DOI] [PubMed] [Google Scholar]
- 7.Xie L, Yu D, Wild C, Allaway G, Turpin J, Smith PC, Lee KH. J Med Chem. 2004;47:756. doi: 10.1021/jm030416y. [DOI] [PubMed] [Google Scholar]
- 8.Zhang Q, Chen Y, Xia P, Xia Y, Yang ZY, Yu DL, Morris-Natschke SL, Lee KH. Bioorg Med Chem Lett. 2004;14:5855. doi: 10.1016/j.bmcl.2004.09.032. [DOI] [PubMed] [Google Scholar]
- 9.Chen Y, Zhang Q, Zhang BN, Xia P, Xia Y, Yang ZY, Kilgore N, Wild C, Morris-Natschke SL, Lee KH. Bioorg Med Chem. 2004;12:6383. doi: 10.1016/j.bmc.2004.09.038. [DOI] [PubMed] [Google Scholar]
- 10.Jacobs TL, Petty WL, Teach EG. J Am Chem Soc. 1960;82:4094. [Google Scholar]
- 11.Sharpless KB, Amberg W, Bennani YL. J Org Chem. 1992;57:2768. [Google Scholar]
- 12.Zhou WS, Huang LF, Sun LQ, Pan XF. Tetrahedron Lett. 1991;32:6745. [Google Scholar]
- 13.Takatsuto S, Ikekawa N. Chem Pharm Bull. 1984;32:2001. doi: 10.1248/cpb.32.3866. [DOI] [PubMed] [Google Scholar]
- 14.McMorris TC, Patil PA. J Org Chem. 1993;58:2338. [Google Scholar]
- 15.Crispino GA, Sharpless KB. Synthesis. 1993;8:777. [Google Scholar]
- 16.Kolb HC, VanNieuwenhze MS, Sharpless KB. Chem Rev. 1994;94:2483. [Google Scholar]
- 17.Sebok P, Timar T, Eszenyi T, Patonay T. J Org Chem. 1995;59:6318. [Google Scholar]
- 18.Tanaka M, Kai T, Sun XL, Takayanagi H, Uda T, Furuhata K. Chem Pharm Bull. 1995;43:1844. [Google Scholar]
- 19.Gabbutt CD, Hepworth John D, Heron BM. Tetrahedron. 1995;51:13277. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.