

Abstract
Epidemiological studies have reported an increased risk of cancer in those with type 2 diabetes (T2DM) and obesity, related in part to hyperinsulinemia, secondary to insulin resistance. Hyperinsulinemia leads to increased insulin-like growth factor-1 (IGF-I) expression. In fact, increased insulin, IGF-I and IGF-II levels are associated with tumor growth in vitro, in animal models and in epidemiological studies in humans. Herein, we discuss insulin, IGF-I and IGF-II, their interaction with the insulin receptor (IR) and IGF-I receptor (IGF-IR), and their signaling pathways and regulation as it pertains to tumor growth. We explain how these pathways have been deciphered by in vitro and in vivo studies and how they are being exploited in the development of targeted cancer therapies.
The Association between Diabetes and Cancer
Cancer is currently a leading cause of death worldwide and in the United States, is second only to heart disease (www.who.int, www.cdc.gov). Epidemiological studies have shown an increased risk of cancer in those with diabetes, specifically liver, colorectal, pancreatic, breast, endometrial and kidney cancer, as well as multiple myeloma [1]. Obesity has also been linked to an increased risk of cancer. The results of the Cancer Prevention Study suggest that 14% of cancer deaths in men and 20% in women can be attributed to obesity [2]. In the United States, 33.8% of the population is obese and almost 8% have diabetes; furthermore, the World Health Organization predicts that over 300 million people worldwide will have diabetes by 2030, emphasizing that this increased risk of cancer is of great concern [3]. Moreover, recent epidemiological studies suggested a link between cancer and the use of exogenous insulin therapy, as well as insulin secretagogues, bringing this issue into the public arena [4, 5]. The link between obesity, type 2 diabetes (T2DM) and cancer may be related in part to hyperinsulinemia secondary to insulin resistance, as well as increased levels of insulin-like growth factor-1 (IGF-I). In vitro and in vivo studies have focused on understanding the mechanistic links between increased circulating levels of insulin, IGF-I and cancer, unraveling the insulin and IGF-I signaling pathways, and facilitating the development of novel cancer therapies that target specific points in these pathways. Many of these agents are undergoing preclinical testing and some have entered early phase clinical trials. In this review, we discuss insulin, IGF-I and IGF-II signaling as it relates to cancer development and describe the animal models that have deciphered this relationship and led to the development of new targeted anti-cancer therapies.
Insulin, Insulin-Like Growth Factors and Cancer
Insulin and Cancer
The administration of insulin was first reported in the 1970s to induce growth of mammary tumors in mice, and subsequent studies showed that exogenous insulin promoted colonic abnormalities in rats [6, 7]. As hyperinsulinemia is seen in obesity and early T2DM in the setting of insulin resistance, animal models of insulin resistance were studied to investigate whether endogenous insulin would also induce tumor growth. Insulin resistance, induced in rats fed a high fat diet, correlated with a greater number of colonic abnormalities, compared to those observed in more insulin sensitive rats [8]. Similarly, in mice, inducing obesity and insulin resistance increased the growth of transplanted lung and colon cancer cells, associated with higher levels of insulin and IGF-I [9]. Therefore, in rodents both exogenous and endogenous insulin induced tumor growth. These observations incited further questions. It was unclear if tumor growth was due to a direct effect on insulin receptor (IR) signaling or if it was that insulin induced IGF-1 receptor (IGF-IR) signaling which led to cancer growth. It also remained to be determined if these observations were relevant to humans and could explain the association between diabetes and cancer.
Epidemiological studies investigated the association between insulin and the risk of certain cancers in humans without diabetes. The Women’s Health Initiative (WHI) study demonstrated that individuals with higher insulin levels were more likely to develop colorectal and endometrial cancer [10, 11]. C-peptide levels were also correlated with greater risk of developing colorectal and endometrial cancer, in addition to a greater risk of mortality from prostate cancer, in the Physicians’ Health Study and European Prospective Investigation into Cancer and Nutrition (EPIC) [12–15]. The results of breast cancer studies have varied, with the EPIC and WHI studies reporting a greater risk of breast cancer amongst postmenopausal women with higher C-peptide and insulin levels, respectively [16, 17]. The Nurses’ Health Study however, reported no association with either insulin or C-peptide levels in pre-menopausal women [18]. These divergent results may be partly explained by the differences in breast cancer hormone receptor status, the effects of hyperinsulinemia on bioavailable estrogens and androgens, and variations in screening and self reporting of breast cancer diagnosis in epidemiological studies. In pre-menopausal women, hyperinsulinemia can stimulate ovarian androgen synthesis and decrease hepatic production of sex hormone binding globulin (SHBG), leading to increased levels of bioavailable androgens and lower progesterone levels [19,20]. In contrast, in post-menopausal women, adipose tissue is the main source of estrogens through aromatization of adrenal androgens. With obesity and hyperinsulinemia, there is increased aromatization of androgens and decreased production of SHBG, leading to increased levels of bioavailable estrogen [19]. Breast cancer cells may overexpress the IR, and animal studies have demonstrated that estrogen enhances IGF-I signaling in breast cancer cells [21,22]. Postmenopausal women are more likely to have estrogen receptor and progesterone receptor positive breast cancer and so the effect of hyperinsulinemia on hormone levels may partly explain the epidemiological study results showing increased postmenopausal breast cancer risk correlating with insulin and C-peptide levels [23].
Observational studies recently reported an increased risk of cancer in patients with T2DM taking insulin or sulfonylureas, compared to those taking metformin. However, whether these results represent an increased risk with insulin and secretagogues, or a protective effect from metformin remains a matter of debate [5, 24]. A combination of both factors is likely. The chronic administration of large doses of insulin to insulin resistant individuals may potentially stimulate cell proliferation by signaling through the IR and IGF-I receptor (IGF-IR), similar to exogenous insulin administration in animals. However, the studies that described the association between insulin, oral hypoglycemic agents and cancer were cohort and case control studies with significant risk of selection bias, so prospective studies controlling for confounding factors are needed to investigate this association further in humans. Evidence from in vitro studies suggest that metformin attenuates tumor growth through its activation of AMP kinase (AMPK) and by decreasing insulin levels [25,26]. Metformin is currently being studied in non diabetic women as adjuvant therapy in breast cancer (NCT01101438). Studying animal models of diabetes and cancer may reveal whether metformin protects against breast cancer in diabetic patients.
IGF-I and Cancer
In hyperinsulinemic states, higher insulin levels in the portal circulation upregulate the growth hormone receptor (GHR) and augment GHR signaling, increasing hepatic IGF-I production [27]. Higher IGF-I levels have been correlated with an increased risk of colorectal, pre-menopausal breast and prostate cancer, in prospective epidemiological studies and meta-analyses [28, 29]. In the Rancho Bernardo Study, after 18 years of follow up, men with baseline IGF-I levels of >100ng/ml had a risk of cancer mortality of 1.82 compared to those with lower levels; those with IGF-I levels of ≥200ng/ml had a risk of 2.61 [30]. Therefore, higher IGF-I levels in humans are associated with increased risk of certain cancers. This implies that individuals with hyperinsulinemia may develop tumors due to increased hepatic production of IGF-I. Animal models have corroborated the link between IGF-I and cancer growth. IGF-I deficiency, induced by caloric restriction in p53 deficient mice, decreased bladder tumor growth. Similarly, transgenic mice with liver specific IGF-I deficiency (LID) had decreased growth and metastasis of transplanted colonic adenocarcinomas, and mammary tumors, induced either by the carcinogen 7, 12-dimethybenz(a)anthracene or by crossing the LID mice with C3(1)/SV40 large T-antigen transgenic mice [31–33]. In both p53 deficient mice with bladder tumors and LID mice with colonic adenocarcinomas, administration of IGF-I reversed the protective effect of IGF-I deficiency on tumor growth [31,33]. IGF-I not only increased tumor growth, but also increased expression of vascular endothelial growth factor (VEGF) and led to the development of neovascularization and metastases in the LID model of colon cancer [31,33]. IGF-I prevented apoptosis by inhibiting p53 and led to angiogenesis by inducing hypoxia-inducible factor 1α (HIF1α), which is involved in the expression of VEGF [34,35]. In addition, metastatic tumor spread, induced by IGF-I, may be related to the relocation of integrins to the edge of migrating cells and the extension of lamellipodia, as demonstrated in colon cancer and neuroblastoma cells [36, 37]. Hence, hyperinsulinemia can increase circulating levels of IGF-I, which in turn can induce tumor growth and metastasis. Additionally, prostate cancer cells in Noble rats with hormone sensitive prostate cancer, have been shown to upregulate their intrinsic production of IGF-I, suggesting that malignant cells may not only rely on circulating IGF-I levels, but may also be able to regulate their own growth through IGF-I production [38].
IGF-II and Cancer
In addition to the growth promoting effects of insulin and IGF-I, IGF-II overexpression is also associated with tumor development. IGF-II is produced in the liver and many other tissues in adult humans, while in rats, IGF-II expression decreases in postnatal life and is only expressed to a significant degree in the adult rat brain [39]. IGF-II expression is controlled by genetic imprinting, i.e, it is normally only expressed on the paternal chromosome under the control of the differentially methylated region (DMR) associated with the H19 gene, located upstream on chromosome 11. When DMR is methylated, IGF-II is expressed. Loss of imprinting (LOI) results from methylation of the DMR on the maternal allele, leading to overexpression of IGF-II, as seen in many tumors [40]. In mouse models of colon cancer, overexpression of IGF-II led to increased tumor development in the presence of the adenomatous polyposis coli (APC) gene mutation [41]. This effect was shown to be due, not only to the increased IGF-II expression, but also to enhanced sensitivity to IGF-II signaling with increased expression of proliferation-related genes [42]. IGF-II signaling occurs through the IR (in particular the IR-A subtype that is more mitogenic than metabolic) and the IGF-IR, as discussed next.
IGF Binding Proteins
IGF-I and IGF-II (but not insulin) circulate bound to insulin like growth factor binding proteins (IGFBPs), of which there are 6, named IGFBP-1 to IGFBP-6, with IGFBP-3 the most predominant. These binding proteins increase the circulating half-lives of IGF-I and IGF-II by protecting them from degradation; while IGFBPs may be important for delivery of IGFs to the target tissue, they may also reduce their availability for receptor binding [43]. IGFBP-2 overexpression has been observed in breast cancer cell lines [44]. SNPs in the 3’ region of the IGFBP-2 gene have been found in genome wide association studies in women with estrogen receptor (ER) positive breast cancer and in women carrying the breast cancer associated mutation, BRCA2 [45–47]. Studies in MCF-7 breast cancer cells show that IGFBP-2 suppressed the tumor suppressor phosphatase and tensin homolog deleted on chromosome 10 (PTEN) [44]. PTEN regulates phosphatidyinositol 3-kinase (PI3K) signaling and IGF-II signaling and inhibits protein synthesis and cell cycle progression [48, 49]. IGFBP-2 overexpression may therefore promote tumor growth by enhancing IGF-II signaling and inhibiting the tumor suppressing effects of PTEN.
IGFBP-3 is believed to be protective from cancer development, as demonstrated in vitro and in mouse models of prostate cancer (LBP-Tag mice) overexpressing IGFBP-3 [50]. This protective effect may be due to its binding of IGF-I and IGF-II, preventing their interaction with the IGF-IR and decreasing their signaling, resulting in cell cycle arrest and upregulation of apoptotic signals [43]. IGFBP-3 is upregulated by p53 and may induce apoptosis through p53 dependent mechanisms. Additionally, IGFBP-3 may have p53 independent interactions with pro and anti-apoptotic members of the Bcl-2 family of proteins [51, 52]. IGFBP-3 interacts with the nuclear retinoid X receptor (RXR)-α, altering its binding to the transcription factor Nur77, activating the apoptosis cascade [44)]. A cell surface IGFBP-3 receptor has recently been identified that also appears to mediate the apoptotic effects of IGFBP-3 through caspase-8 [53]. Despite the results of in vitro and animal studies demonstrating a possible protective effect of IGFBP-3, the association of lower serum IGFBP-3 levels with increased cancer risk in humans has had inconsistent results [28,29,54]. One of the factors thought to contribute to differing results in these epidemiological studies is the variability of the IGFBP-3 assay [55]. IGFBP-3 levels are increased in the presence of GH excess and decreased in GH deficiency [56]. Hyperinsulinemia and increased IGF-I levels can reduce GH levels and therefore lead to decreased IGFBP-3. As IGFBP-3 is the most abundant of the IGFBPs, decreased IGFBP-3 may lead to increased levels of free IGF-I in addition to decreased direct activity of IGFBP-3 on cells.
Decreased IGFBP-1 levels were found in female MKR mice (a non obese hyperinsulinemic mouse model) with polyoma virus middle T antigen (PyVmT) induced breast cancer, compared to wild-type mice [57]. In mice with MCF-7 breast cancer xenografts, IGFBP-1 inhibits breast cancer cell growth; IGFBP-1 also inhibited hepatocellular cancer growth in mice overexpressing IGF-I and IGF-II [58, 59]. Again, epidemiological studies have been inconsistent regarding IGFBP-1 levels and cancer risk [28]. Hepatic IGFBP-1 expression is decreased by insulin and therefore, hyperinsulinemia may lead to decreased IGFBP-1 and contribute to increased tumor growth [60]. Ongoing studies are investigating the degree to which different IGFBPs affect tumor growth, as well as their IGF dependent and independent mechanisms, and how their activity is altered in hyperinsulinemia and T2DM.
Insulin Receptor, IGF-I Receptor and Signaling
The mechanisms by which increased levels of insulin, IGF-I and IGF-II promote cancer growth has been revealed by examining their interaction with the IR and IGF-IR and subsequent signaling pathways.
The Insulin and IGF-I Receptors
Insulin signals through the IR, of which there are 2 isoforms, IR-A and IR-B, formed by the absence or presence of exon 11, respectively. IGF-II can interact with IR-A, and primarily mediates proliferative effects through this receptor and IGF-IR. Compared with insulin binding to IR-A, binding of IGF-II to IR-A results in the expression of different genes, as well as a temporal separation in gene expression from that occurring after insulin binding [61]. IR-A expression has been demonstrated in fetal cells and many tumor cells, and signaling through IR-A results in more mitogenic effects than IR-B signaling, which activates the metabolic signaling pathway [62]. Hyperinsulinemia therefore can activate signaling pathways that lead to both metabolic and mitogenic effects. Overexpression of IGF-II in tumor cells can also lead to increased mitogenic signaling through IR-A [63]. IGF-I primarily signals through the IGF-IR, and mediates mitogenic effects. Additionally, IGF-II can bind to the IGF-IR. Cells that express the IR and IGF-IR can also express hybrid receptors, made of the α and β subunit of an IR (IR-A or IR-B) bound to the α and β subunit of the IGF-IR (Figure 1). IGF-I can bind to either of these hybrid receptors, while IGF-II binds only to the IGF-IR/IR-A hybrid, and insulin has negligible affinity for either [64]. Increased expression of IR-A in tumors therefore allows for the increased formation of IGF-IR/IR-A hybrid receptors in tumors, permitting mitogenic signaling by IGF-I and IGF-II through the hybrid receptor or insulin and IGF-II through IR-A. So, hyperinsulinemia, increased IGF-I or IGF-II can augment tumor growth by signaling through these receptors.
Figure 1.
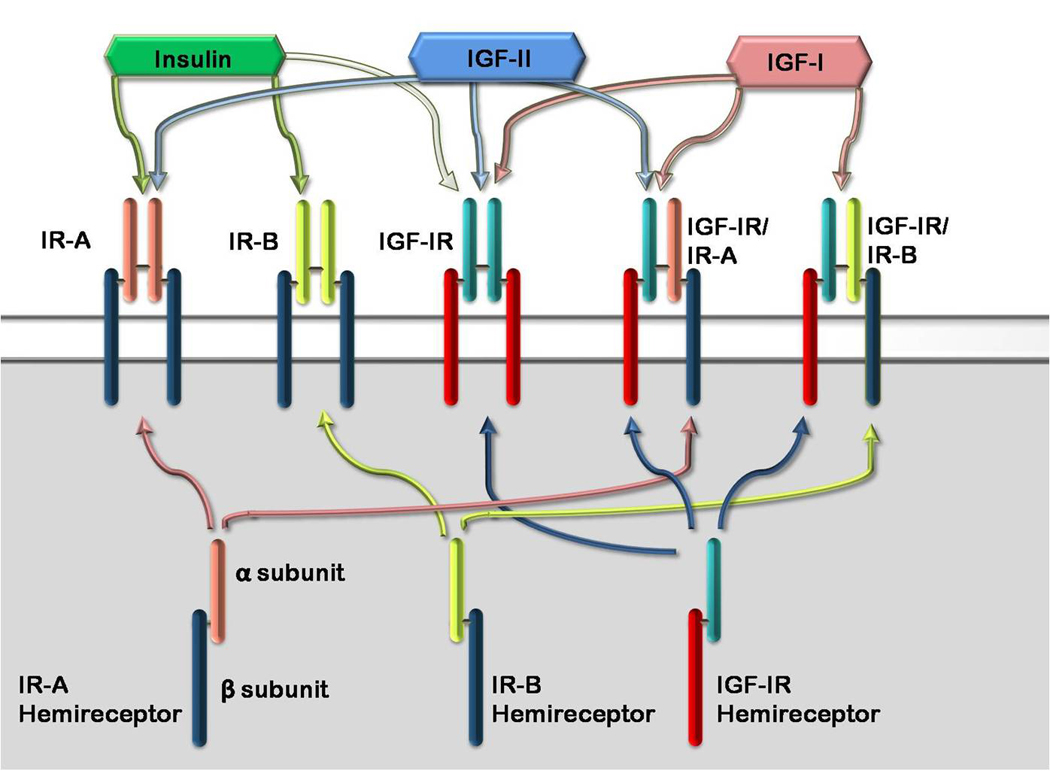
Insulin Receptor, IGF-IR and Hybrid Receptors. Insulin binds with greatest affinity to the insulin receptors (IR-A and IR-B). IR-A and IR-B are splice variants of the IR, with IR-A lacking exon 11. Insulin has low affinity for the IGF-IR. Each ‘hemireceptor’ made up of one α and one β subunit of the IR-A, IR-B or IGF-IR can form a hybrid with another hemireceptor, forming a hybrid receptor. IGF-I binds to the IGF-IR, and the IGF-IR/IR-A and IGF-IR/IR-B hybrid receptors. IGF-II can bind with the IGF-IR, IR-A and IGF-IR/IR-A hybrid receptor. Insulin does not bind with the hybrid receptors.
The IR and IGF-IR Signaling Pathways and Tumor Development
Binding of insulin, IGF-I or IGF-II to the extracellular portion of the IR, IGF-IR or hybrid receptor leads to autophosphorylation of the β subunit tyrosine kinase, followed by the phosphorylation of additional tyrosine residues. This leads to recruitment of insulin receptor substrates (IRS) 1–4 and other proteins allowing for activation of the PI3K and mitogen activated protein kinase (MAPK) signaling pathways. These pathways are outlined in Figure 2.
Figure 2.
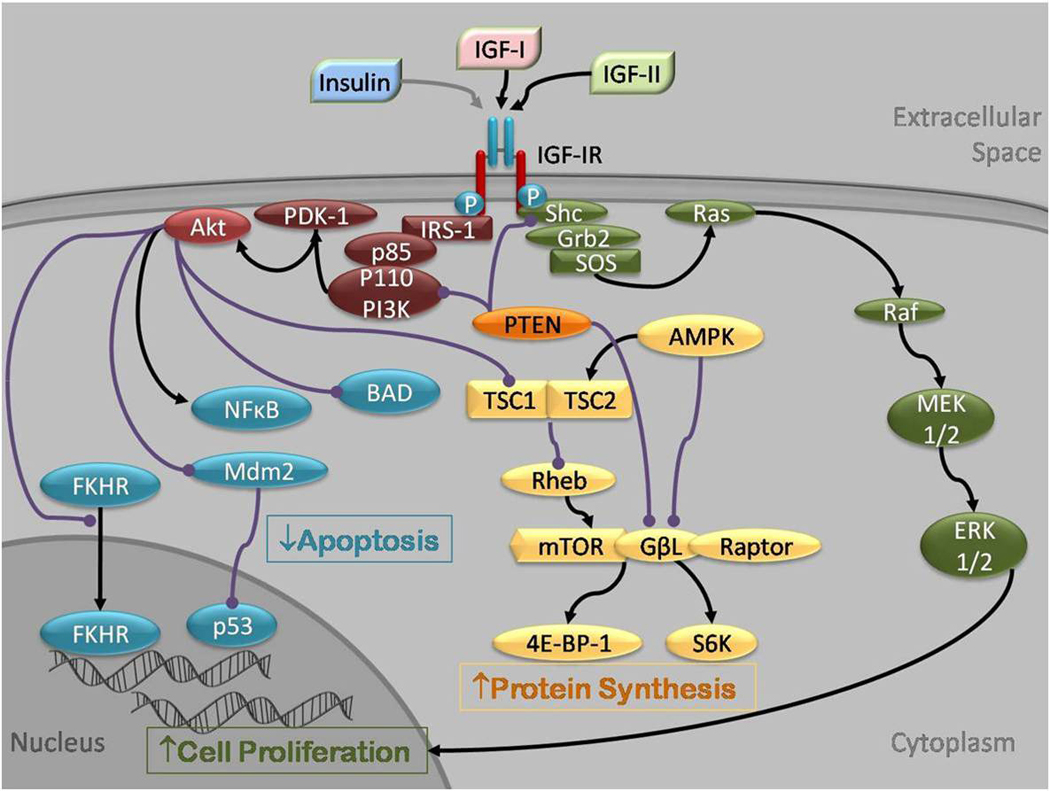
Insulin, IGF-I and IGF-II signaling through the IGF-IR. IGF-I, IGF-II and, to a lesser extent insulin, bind IGF-IR at the α subunit, leading to autophosphorylation of β subunit residues, which then act as docking sites for the insulin receptor substrates (IRS)1–4 and other signaling proteins, such as Shc, phospholipase C γ1 (PLCγ1) and Gab1. IRS-1 recruits the p85 regulatory subunit of phosphatidylinositol 3-kinase (PI3K) which through the catalytic effects of its p110 subunit, phosphorylates to activate protein kinase B (Akt), S6 kinase (S6K) and the mitogen activated protein kinase/ extracellular signal regulated kinase (MAP/ERK) pathway. Akt is recruited to the cell membrane and is phosphorylated by phosphoinositide-dependent protein kinase (PDK)-1. Activated Akt has many substrates, including the forkhead transcription factors (FKHR), Mouse double minute 2 (Mdm2), Bcl2-antagonist of cell death (BAD), NFκB and p53. These factors are chiefly involved in the regulation of apoptosis. Akt also regulates protein synthesis by phosphorylating tuberous sclerosis protein (TSC2), releasing its inhibition of Rheb to activate the mammalian target of rapamycin (mTOR). mTOR targets S6 kinase (S6K) and eukaryotic initiation factor 4E binding protein-1 (4E-BP-1). The phosphorylation of IRS-1 and Shc can also lead to recruitment of growth factor receptor bound protein 2 (Grb2), Son of Sevenless (SOS) and Ras, with subsequent activation of Raf-1, MEK1/2 (MAP/ERK kinase 1/2) and ERK 1/2 by phosphorylation. Activation of this pathway leads to cell proliferation. The tumor suppressor gene PTEN inhibits mTOR, PI3K and Shc. AMPK also inhibits mTOR, by activating the TSC1/2 complex in addition to directly inhibiting mTOR.
The activity of PI3K is regulated by the tumor suppressor gene PTEN. PTEN dephosphorylates PI3K, inhibits MAPK signaling, cell growth and cell cycle progression through its interaction with cyclin D [65]. PTEN is induced by IGF-II, as well as p53 and in turn, feeds back to inhibit IGF-II signaling, as demonstrated in the MMTV mouse overexpressing IGF-II (MMTV-IGF-II) [49]. PTEN is the second most common tumor suppressor gene to be mutated in human cancer, after p53. At least 50% of women with breast cancer have a mutation or inactivation of at least one copy of PTEN [66]. Loss of PTEN in MCF-7 breast cancer cells has been shown to lead to increased IGF-II signaling through the IGF-IR or IR-A [44]. So, in the presence of PTEN mutations, increased IR and IGF-IR signaling could stimulate tumor growth.
Akt activation of the mammalian target of rapamycin (mTOR) leads to protein synthesis, allowing protein accumulation, cell growth and the preparation of cells for mitosis, all events that favor tumor growth. The targets of mTOR include S6K1, S6K2 and the eukaryotic initiation factor 4E (eIF4E)-binding protein 1 (4E-BP1), resulting in increased translation, cell growth and proliferation. In addition, mTOR phosphorylates protein phosphorylase 2A (PP2A), which prevents the dephosphorylation of S6K1 and 4E-BP1[48]. mTOR forms two complexes, TORC1 and TORC2. TORC1 is formed by mTOR-GβL-Raptor, is rapamycin-sensitive and is involved in protein translation in response to growth factor and nutrient signals. mTOR is regulated by the tumor suppressor genes tuberous sclerosis (TSC)1 and TSC2. The complex formed by the TSC1 and TSC2 proteins has GTPase activity toward the small GTPase Rheb and inhibits its activity. Rheb activates mTOR, while the TSC1/TSC2 complex, by inhibiting Rheb, avidly inhibits mTOR and therefore prevents protein synthesis. Insulin and IGF-I signaling activate Akt, leading to phosphorylation of TSC-2, releasing its inhibitory influence on mTOR [48]. mTOR is regulated by PTEN, and loss of PTEN leads to constitutively active mTOR [67]. Activation of mTOR in breast cancer cell lines is associated with resistance to the chemotherapeutic agents trastuzumab and tamoxifen [68].
In states of glucose deprivation, ATP levels decrease and AMP levels rise, leading to phosphorylation and activation of AMPK. LKB1 is a tumor suppressor gene that phosphorylates and activates AMPK. The activation of AMPK prevents the cellular production of proteins for cell growth and proliferation. AMPK phosphorylates TSC2, increasing GTPase activity on Rheb and inhibiting mTOR. AMPK also directly phosphorylates and inhibits mTOR [67]. In mice with colon cancer induced by a high fat diet, metformin decreased insulin levels, decreased Akt, activated AMPK and attenuated the effects of the high fat diet on tumor growth [69]. In human epidemiological studies, metformin has been associated with a decreased risk of cancer development and a better response to chemotherapy in patients with breast cancer [70, 71].
Linking Insulin Signaling and Diabetes to Cancer in Animal Models
Increased levels of insulin, IGF-I and IGF-II and dysregulation of their signaling pathways by mutations in tumor suppressor genes lead to tumor development. However, clear evidence to tie these signaling pathways to cancer development, particularly in obesity and T2DM is required. In addition, it remains to be seen if inhibiting insulin, IGF-1 and IGF-II signaling prevents tumor growth.
Studies using animal models have investigated the role of IGF-IR in tumor development. For example, studies in the transgenic adenocarcinoma of mouse prostate (TRAMP) model demonstrated that selective overexpression of IGF-I in the basal epithelial cells of the prostate led to increased expression of IGF-IR and development of early prostate cancer [72]. A transgenic mouse model that formed a constitutively active IGF-IR homodimer, by the formation of a CD8-IGF-IR fusion protein under the control of the mouse mammary tumor virus (MMTV) promoter, was also shown to have rapid development of aggressive salivary and mammary adenocarcinomas [73]. Dominant-negative mutations of IGF-IR inhibited the growth of lung cancer cell lines, and dysfunctional IGF-IR was shown to decrease proliferation in mammary glands in mice and Ewing sarcoma cells [74–76].
In addition to IGF-IR upregulation in cancers, increased IR expression has been reported in breast and prostate cancer cell lines [62, 77]. This has led to studies examining the role of the IR in cancer. Examination of Met-1 breast cancer cells, overexpressing PyVmT, demonstrated that insulin and IGF-I augment the interaction of IR and IGF-IR with PyVmT, by tyrosine phosphorylation of PyVmT. When stimulated by insulin or IGF-I, the interaction of PyVmT with Src and phospholipase Cγ1 (PLCγ1) was increased. Src is involved in activating the MAPK pathway, and both Src and PLCγ1 are involved in tumor formation. Upon implantation of Met-1 cells into mice, disruption of IR or IGF-IR signaling by short hairpin RNA (shRNA) prevented tumor formation, showing that IR and IGF-IR function are required for tumor development in the presence of PyVmT [78]. Similar findings were reported from the highly metastatic LCC6 derivative of the MDA-MB-435 cell line and insulin responsive breast cancer cell line, T47D. shRNA was used to downregulate the IR in both cell lines, and cells with reduced IR activity had decreased insulin-stimulated Akt activation, with normal IGF-IR function. When transplanted into athymic mice, the LCC6 IR shRNA tumors had decreased growth, angiogenesis, lymphangiogenesis and metastases compared to wild-type cells, and were found to produce less HIF1α and VEGF [79]. Increased IR expression was detected on the endothelial cells of human breast, colon, pancreas, lung and kidney cancer specimens, suggesting that insulin may play an important role in angiogenesis. In vitro studies of human microvascular endothelial cells (hMVEC) have shown that insulin stimulated capillary-like tube formation [80]. From these studies it is evident that hyperinsulinemia, IGF-I and IGF-II may induce tumor growth, by signaling through the IR and IGF-IR.
To decipher the specific contribution of insulin resistance, independent of the inflammation associated with obesity, a non obese insulin resistant mouse was developed. The female MKR mouse is a non-obese mouse model of insulin resistance and hyperinsulinemia, with mild dysglycemia [81]. Mammary tumors were induced in this mouse model by crossing it with a mouse expressing PyVmT. The MKR mice were found to have accelerated mammary gland development and breast cancer progression. The IR, IGF-IR, PI3K/Akt signaling pathways were demonstrated to be the main mechanisms accounting for tumor development [57]. Decreasing the circulating levels of insulin, by using the β3 adrenergic receptor agonist Cl-316243 prevented the accelerated tumor growth, by reducing IR and IGF-IR signaling [82]. In addition, treating these mice with the IGF-IR/IR tyrosine kinase inhibitor BMS-536924 to block IR signaling, inhibited tumor growth [57]. In vitro, other IGF-IR/IR tyrosine kinase inhibitors inhibit tumor growth; for example, BMS-754807 inhibits the growth of osteosarcoma, rhabdomyosarcoma, neuroblastoma, liposarcoma, breast, lung, pancreatic, colon and gastric tumors and multiple myeloma and leukemia cell lines in addition to xenograft tumor models with tumor growth inhibition [83].
Conclusions
Concerns have been raised over the increasing prevalence of obesity and T2DM and their association with increased cancer risk. We have described how dysregulation of insulin and IGF-I signaling, associated with increased activation of IR and IGF-IR, may occur in tumors, with subsequent increases in signaling through the PI3K and MAPK pathways, leading to unregulated protein synthesis, cell cycle progression, cell growth and prevention of apoptosis. Animal models have helped us understand these pathways and how T2DM and obesity are linked to cancer. From these animal models and epidemiological studies, it appears that treating T2DM with the AMPK activator metformin may reduce the risk of developing cancer. In light of these findings, studies are ongoing to examine the effect of metformin on tumor proliferation and survival in patients with breast cancer (NCT00897884, NCT01101438), and other AMPK activators are being developed as potential cancer treatments.
The understanding gained from animal and cell studies has allowed for the development of novel anti-cancer therapies targeting specific points in the insulin, IGF-I and IGF-II signaling pathways. For example, monoclonal antibodies against IGF-I, IGF-II and the IGF-IR, in addition to inhibitors targeting IGFBP-2, IGF-IR, PI3K, Akt and mTOR and activators of AMPK are being investigated (Figure 3). The monoclonal antibodies directed against the IGF-IR bind to the α-subunit, blocking IGF-I and IGF-II binding and also appear to increase internalization of the IGF-IR, decreasing the number available for binding. Robatumumab is one such agent that inhibits growth of osteosarcoma, neuroblastoma and rhabdomyosarcoma in animal studies [84)]. Early clinical trials are ongoing to investigate these monoclonal antibodies in humans [85] (NCT00551213). Inhibition of signal transduction can be achieved by using tyrosine kinase inhibitors (TKI). Most TKI compete for the ATP binding site on tyrosine kinase on the IGF-IR, preventing receptor autophosphorylation [86]. Due to the conservation of this binding site between the IGF-IR and IR, these TKI may also block signaling through IR. As demonstrated in mouse models, blocking IR and IGF-IR may be necessary to inhibit tumor growth in the setting of hyperinsulinemia [57]. Inhibiting IGF-IR phosphorylation may lead to increased phosphorylation of the epidermal growth factor receptor (EGFR) signaling pathway and resistance to monotherapy. Therefore, using IGF-IR TKI in conjunction with EGFR TKI could be a way to overcome this resistance [87]. Other agents targeting downstream signaling molecules, including dual inhibitors of PI3K and mTOR that overcome the issue of resistance to blocking either PI3K or mTOR individually, improves the antiproliferative activity of EGFR inhibitors in cell studies [88].
Figure 3.
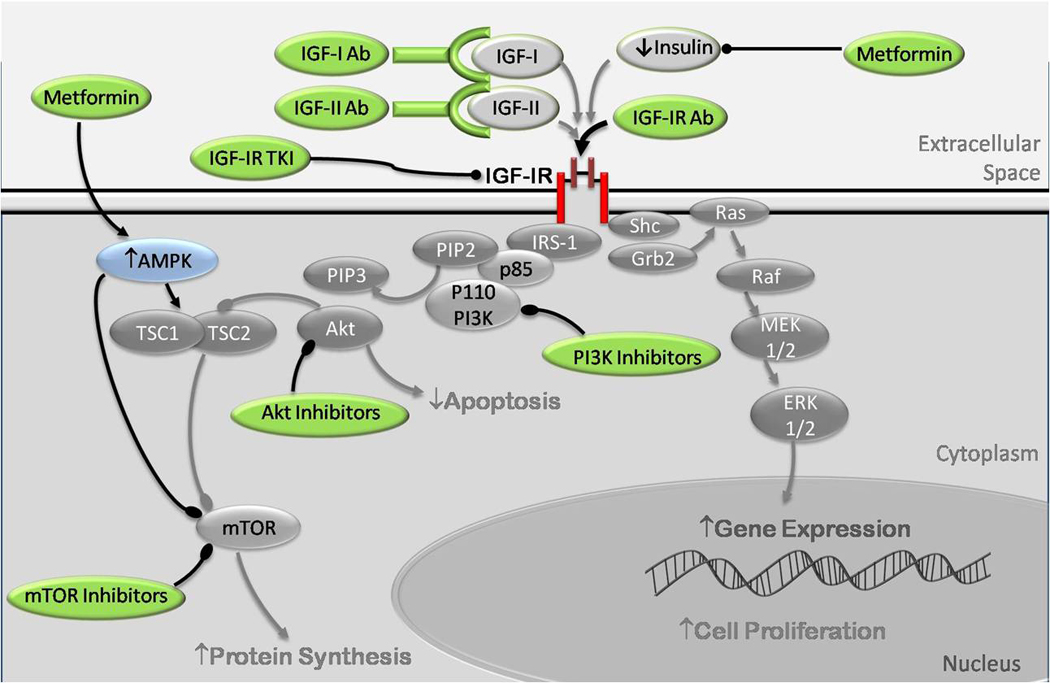
Anti-Cancer therapy targeting the insulin, IGF-I and IGF-II signaling. Monoclonal antibodies (Ab) have been developed to target IGF-I and IGF-II, preventing their interaction with the IR and IGF-IR. Antibodies have also been developed that bind to the IGF-IR, preventing binding of insulin, IGF-I or IGF-II. Tyrosine kinase inhibitors (TKI) bind to the IGF-IR and prevent phosphorylation and activation of the receptor. These TKI may also cross react with the IR. Inhibitors of PI3K, Akt and mTOR have also been developed that inhibit downstream signaling. Metformin acts in two ways. Firstly it decreases hepatic gluconeogenesis, thus decreasing circulating insulin levels. Secondly, it is an activator of AMP kinase (AMPK), resulting in inhibition of mTOR by direct phosphorylation and also by activation of the TSC1/TSC2 complex, leading to decreased protein synthesis.
Ultimately, examining the expression of genes and proteins in tumors is envisaged to determine the susceptibility of individual tumors to therapy targeted at specific pathways and proteins. Further animal and human studies are necessary to determine the importance of the IR and IGF-IR signaling pathways in tumor development in the setting of hyperinsulinemia and the mitogenic and metabolic effects of blocking these pathways in individuals with insulin resistance and T2DM.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Emily Jane Gallagher, Mount Sinai School of Medicine, Division of Endocrinology, Diabetes and Bone Diseases, Box #1055, One Gustave L. Levy Place, New York, NY 10029, USA.
Derek LeRoith, Mount Sinai School of Medicine, Division of Endocrinology, Diabetes and Bone Diseases, Box #1055, One Gustave L. Levy Place, New York, NY 10029, USA.
References
- 1.Vigneri P, et al. Diabetes and cancer. Endocr Relat Cancer. 2009;16(4):1103–1123. doi: 10.1677/ERC-09-0087. [DOI] [PubMed] [Google Scholar]
- 2.Calle EE, et al. Overweight, Obesity, and Mortality from Cancer in a Prospectively Studied Cohort of U.S. Adults. N Engl J Med. 2003;348(17):1625–1638. doi: 10.1056/NEJMoa021423. [DOI] [PubMed] [Google Scholar]
- 3.Flegal KM, et al. Prevalence and Trends in Obesity Among US Adults, 1999–2008. JAMA. 2010;303(3):235–241. doi: 10.1001/jama.2009.2014. [DOI] [PubMed] [Google Scholar]
- 4.Hemkens L, et al. Risk of malignancies in patients with diabetes treated with human insulin or insulin analogues: a cohort study. Diabetologia. 2009;52(9):1732–1744. doi: 10.1007/s00125-009-1418-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Currie C, Poole C, Gale E. The influence of glucose-lowering therapies on cancer risk in type 2 diabetes. Diabetologia. 2009;52(9):1766–1777. doi: 10.1007/s00125-009-1440-6. [DOI] [PubMed] [Google Scholar]
- 6.Heuson J-C, Legros N, Heimann R. Influence of Insulin Administration on Growth of the 7,12-Dimethylbenz(a)anthracene-induced Mammary Carcinoma in Intact, Oophorectomized, and Hypophysectomized Rats. Cancer Res. 1972;32(2):233–238. [PubMed] [Google Scholar]
- 7.Corpet DE, et al. Insulin injections promote the growth of aberrant crypt foci in the colon of rats. Nutr Cancer. 1997;27(3):316–320. doi: 10.1080/01635589709514543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tran TT, et al. Direct Measure of Insulin Sensitivity with the Hyperinsulinemic-Euglycemic Clamp and Surrogate Measures of Insulin Sensitivity with the Oral Glucose Tolerance Test: Correlations with Aberrant Crypt Foci Promotion in Rats. Cancer Epidemiol Biomarkers Prev. 2003;12(1):47–56. [PubMed] [Google Scholar]
- 9.Yakar S, et al. Increased Tumor Growth in Mice with Diet-Induced Obesity: Impact of Ovarian Hormones. Endocrinology. 2006;147(12):5826–5834. doi: 10.1210/en.2006-0311. [DOI] [PubMed] [Google Scholar]
- 10.Gunter MJ, et al. Insulin, insulin-like growth factor-I, endogenous estradiol, and risk of colorectal cancer in postmenopausal women. Cancer Res. 2008;68(1):329–337. doi: 10.1158/0008-5472.CAN-07-2946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gunter MJ, et al. A Prospective Evaluation of Insulin and Insulin-like Growth Factor-I as Risk Factors for Endometrial Cancer. Cancer Epidemiology Biomarkers & Prevention. 2008;17(4):921–929. doi: 10.1158/1055-9965.EPI-07-2686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ma J, et al. Prediagnostic body-mass index, plasma C-peptide concentration, and prostate cancer-specific mortality in men with prostate cancer: a long-term survival analysis. The Lancet Oncology. 2008;9(11):1039–1047. doi: 10.1016/S1470-2045(08)70235-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ma J, et al. A Prospective Study of Plasma C-Peptide and Colorectal Cancer Risk in Men. J.Natl. Cancer Inst. 2004;96(7):546–553. doi: 10.1093/jnci/djh082. [DOI] [PubMed] [Google Scholar]
- 14.Jenab M, et al. Serum C-peptide, IGFBP-1 and IGFBP-2 and risk of colon and rectal cancers in the European Prospective Investigation into Cancer and Nutrition. Int J Cancer. 2007;121(2):368–376. doi: 10.1002/ijc.22697. [DOI] [PubMed] [Google Scholar]
- 15.Cust AE, et al. Serum levels of C-peptide, IGFBP-1 and IGFBP-2 and endometrial cancer risk; results from the European prospective investigation into cancer and nutrition. Int J Cancer. 2007;120(12):2656–2664. doi: 10.1002/ijc.22578. [DOI] [PubMed] [Google Scholar]
- 16.Gunter MJ, et al. Insulin, insulin-like growth factor-I, and risk of breast cancer in postmenopausal women. J Natl Cancer Inst. 2009;101(1):48–60. doi: 10.1093/jnci/djn415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Verheus M, et al. Serum C-peptide levels and breast cancer risk: Results from the European prospective investigation into cancer and nutrition (EPIC) Int J Cancer. 2006;119(3):659–667. doi: 10.1002/ijc.21861. [DOI] [PubMed] [Google Scholar]
- 18.Eliassen AH, et al. Circulating insulin and c-peptide levels and risk of breast cancer among predominately premenopausal women. Cancer Epidemiol Biomarkers Prev. 2007;16(1):161–164. doi: 10.1158/1055-9965.EPI-06-0693. [DOI] [PubMed] [Google Scholar]
- 19.Calle E, Kaaks R. Overweight, obesity and cancer: epidemiological evidence and proposed mechanisms. Nat Rev Cancer. 2004;4(8):579–591. doi: 10.1038/nrc1408. [DOI] [PubMed] [Google Scholar]
- 20.Baillargeon J-P, Nestler JE. Polycystic Ovary Syndrome: A Syndrome of Ovarian Hypersensitivity to Insulin? J Clin Endocrinol Metab. 2006;91(1):22–24. doi: 10.1210/jc.2005-1804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Papa V, Pezzino V, Costantino A, et al. Elevated insulin-receptor content in human breast cancer. Journal of Clin Invest. 1990;86(5):1503–1510. doi: 10.1172/JCI114868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lee AV, Jackson JG, Gooch JL, et al. Enhancement of Insulin-Like Growth Factor Signaling in Human Breast Cancer: Estrogen Regulation of Insulin Receptor Substrate-1 Expression in Vitro and in Vivo. Mol Endocrinol. 1999;13(5):787–796. doi: 10.1210/mend.13.5.0274. [DOI] [PubMed] [Google Scholar]
- 23.Dunnwald LK, Rossing MA, Li CI. Hormone receptor status, tumor characteristics, and prognosis: as prospective cohort of breast cancer patients. Breast Cancer Res. 2007;9(1):R6. doi: 10.1186/bcr1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bowker SL, et al. Increased cancer-related mortality for patients with type 2 diabetes who use sulfonylureas or insulin. Diabetes Care. 2006;29(2):254–258. doi: 10.2337/diacare.29.02.06.dc05-1558. [DOI] [PubMed] [Google Scholar]
- 25.Alimova IN, Liu B, Fan Z, et al. Metformin inhibits breast cancer cell growth, colony formation and induces cell cycle arrest in vitro. Cell Cycle. 2009;8(6):909–915. doi: 10.4161/cc.8.6.7933. [DOI] [PubMed] [Google Scholar]
- 26.Zakikhani M, Dowling R, Fantus IG, Sonenberg N, Pollak M. Metformin is an AMP kinase-dependent growth inhibitor for breast cancer cells. Cancer research. 2006;66(21):10269–10273. doi: 10.1158/0008-5472.CAN-06-1500. [DOI] [PubMed] [Google Scholar]
- 27.Baxter RC, Bryson JM, Turtle JR. Somatogenic Receptors of Rat Liver: Regulation by Insulin. Endocrinology. 1980;107(4):1176–1181. doi: 10.1210/endo-107-4-1176. [DOI] [PubMed] [Google Scholar]
- 28.Chen W, et al. Phenotypes and genotypes of insulin-like growth factor 1, IGF-binding protein-3 and cancer risk: evidence from 96 studies. Eur J Hum Genet. 2009;17(12):1668–1675. doi: 10.1038/ejhg.2009.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rinaldi S, et al. Serum levels of IGF-I, IGFBP-3 and colorectal cancer risk: results from the EPIC cohort, plus a meta-analysis of prospective studies. Int J Cancer. 2010;126(7):1702–1715. doi: 10.1002/ijc.24927. [DOI] [PubMed] [Google Scholar]
- 30.Major JM, et al. Insulin-Like Growth Factor-I and Cancer Mortality in Older Men. J Clin Endocrinol Metab. 2010;95(3):1054–1059. doi: 10.1210/jc.2009-1378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dunn SE, et al. Dietary Restriction Reduces Insulin-like Growth Factor I Levels, Which Modulates Apoptosis, Cell Proliferation, and Tumor Progression in p53-deficient Mice. Cancer Res. 1997;57(21):4667–4672. [PubMed] [Google Scholar]
- 32.Wu Y, et al. Reduced Circulating Insulin-like Growth Factor I Levels Delay the Onset of Chemically and Genetically Induced Mammary Tumors. Cancer Res. 2003;63(15):4384–4388. [PubMed] [Google Scholar]
- 33.Wu Y, et al. Circulating Insulin-like Growth Factor-I Levels Regulate Colon Cancer Growth and Metastasis. Cancer Res. 2002;62(4):1030–1035. [PubMed] [Google Scholar]
- 34.Fukuda R, et al. Insulin-like Growth Factor 1 Induces Hypoxia-inducible Factor 1-mediated Vascular Endothelial Growth Factor Expression, Which is Dependent on MAP Kinase and Phosphatidylinositol 3-Kinase Signaling in Colon Cancer Cells. J. Biol.Chem. 2002;277(41):38205–38211. doi: 10.1074/jbc.M203781200. [DOI] [PubMed] [Google Scholar]
- 35.Heron-Milhavet L, LeRoith D. Insulin-like Growth Factor I Induces MDM2-dependent Degradation of p53 via the p38 MAPK Pathway in Response to DNA Damage. J. Biol.chem. 2002;277(18):15600–15606. doi: 10.1074/jbc.M111142200. [DOI] [PubMed] [Google Scholar]
- 36.Meyer GE, et al. Insulin-like growth factor I stimulates motility in human neuroblastoma cells. Oncogene. 2001;20(51):7542–7550. doi: 10.1038/sj.onc.1204927. [DOI] [PubMed] [Google Scholar]
- 37.Canonici A, et al. Insulin-like growth factor-I receptor, E-cadherin and alphav integrin form a dynamic complex under the control of alpha-catenin. Int J Cancer. 2008;122(3):572–582. doi: 10.1002/ijc.23164. [DOI] [PubMed] [Google Scholar]
- 38.Wang YZ, Wong YC. Sex hormone-induced prostatic carcinogenesis in the Noble rat: The role of insulin-like growth factor-1 (IGF-1) and vascular endothelial growth factor (VEGF) in the development of prostate cancer. Prostate. 1998;35(3):165–177. doi: 10.1002/(sici)1097-0045(19980515)35:3<165::aid-pros2>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- 39.Nielsen FC. The molecular and cellular biology of insulin-like growth factor II. Progress in Growth Factor Research. 1992;4(3):257–290. doi: 10.1016/0955-2235(92)90023-b. [DOI] [PubMed] [Google Scholar]
- 40.Jelinic P, Shaw P. Loss of imprinting and cancer. The Journal of Pathology. 2007;211(3):261–268. doi: 10.1002/path.2116. [DOI] [PubMed] [Google Scholar]
- 41.Sakatani T, et al. Loss of Imprinting of Igf2 Alters Intestinal Maturation and Tumorigenesis in Mice. Science. 2005;307(5717):1976–1978. doi: 10.1126/science.1108080. [DOI] [PubMed] [Google Scholar]
- 42.Kaneda A, et al. Enhanced sensitivity to IGF-II signaling links loss of imprinting of IGF2 to increased cell proliferation and tumor risk. Proceedings of the National Academy of Sciences. 2007;104(52):20926–20931. doi: 10.1073/pnas.0710359105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jogie-Brahim S, Feldman D, Oh Y. Unraveling Insulin-Like Growth Factor Binding Protein-3 Actions in Human Disease. Endocr Rev. 2009;30(5):417–437. doi: 10.1210/er.2008-0028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Perks CM, et al. IGF-II and IGFBP-2 differentially regulate PTEN in human breast cancer cells. Oncogene. 2007;26(40):5966–5972. doi: 10.1038/sj.onc.1210397. [DOI] [PubMed] [Google Scholar]
- 45.Stacey SN, et al. Common variants on chromosomes 2q35 and 16q12 confer susceptibility to estrogen receptor-positive breast cancer. Nat Genet. 2007;39(7):865–869. doi: 10.1038/ng2064. [DOI] [PubMed] [Google Scholar]
- 46.Neuhausen SL, et al. Genetic variation in insulin-like growth factor signaling genes and breast cancer risk among BRCA1 and BRCA2 carriers. Breast Cancer Res. 2009;11(5):R76. doi: 10.1186/bcr2414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Garner CP, Ding YC, John EM, et al. Genetic variation in IGFBP2 and IGFBP5 is associated with breast cancer in populations of African descent. Hum Genet. 2008;123(37):247–255. doi: 10.1007/s00439-008-0468-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Levine AJ, et al. Coordination and communication between the p53 and IGF-1-AKT-TOR signal transduction pathways. Genes Dev. 2006;20(3):267–275. doi: 10.1101/gad.1363206. [DOI] [PubMed] [Google Scholar]
- 49.Moorehead RA, et al. Insulin-like Growth Factor-II Regulates PTEN Expression in the Mammary Gland. Journal of Biological Chemistry. 2003;278(50):50422–50427. doi: 10.1074/jbc.M306894200. [DOI] [PubMed] [Google Scholar]
- 50.Silha JV, et al. Insulin-Like Growth Factor (IGF) Binding Protein-3 Attenuates Prostate Tumor Growth by IGF-Dependent and IGF-Independent Mechanisms. Endocrinology. 2006;147(5):2112–2121. doi: 10.1210/en.2005-1270. [DOI] [PubMed] [Google Scholar]
- 51.Butt A, et al. Insulin-like Growth Factor-binding Protein-3 Modulates Expression of Bax and Bcl-2 and Potentiates p53-independent Radiation-induced Apoptosis in Human Breast Cancer Cells. J. Biol.Chem. 2000;275(50):39174–39181. doi: 10.1074/jbc.M908888199. [DOI] [PubMed] [Google Scholar]
- 52.Buckbinder L, et al. Induction of the growth inhibitor IGF-binding protein 3 by p53. Nature. 1995;377(6550):646–649. doi: 10.1038/377646a0. [DOI] [PubMed] [Google Scholar]
- 53.Ingermann AR, Yang YF, Paisley TE, et al. Identification of a novel cell death receptor mediating IGFBP-3-induced antitumor effects in breast and prostate cancer. J Biol Chem. 2010 doi: 10.1074/jbc.M110.122226. DOI: 10.1074/jbc.M110.122226 ( http://www.jbc.org) [DOI] [PMC free article] [PubMed]
- 54.Rinaldi S, Peeters PHM, Berrino F, et al. IGF-I, IGFBP-3 and breast cancer risk in women: The European Prospective Investigation into Cancer and Nutrition (EPIC) Endocr Relat Cancer. 2006;13(2):593–605. doi: 10.1677/erc.1.01150. [DOI] [PubMed] [Google Scholar]
- 55.Rinaldi S, Kaaks R, Zeleniuch-Jacuotte A, et al. Insulin-Like Growth Factor-I, IGF Binding Protein-3, and Breast Cancer in Young Women: A Comparison of Risk Estimates Using Different Peptide Assays. Cancer Epidemiology Biomarkers & Prevention. 2005;14(1):48–52. [PubMed] [Google Scholar]
- 56.Fukudu I, Hizuka N, Itoh E, et al. Acid-labile subunit in growth hormone excess and deficiency in adults: evaluation of its diagnostic value in comparison with insulin-like growth factor (IGF)-1 and IGF-binding protein-3. Andocr J. 2002;49(3):379–386. doi: 10.1507/endocrj.49.379. [DOI] [PubMed] [Google Scholar]
- 57.Novosyadlyy R, et al. Insulin-mediated acceleration of breast cancer development and progression in a nonobese model of type 2 diabetes. Cancer Res. 2010;70(2):741–751. doi: 10.1158/0008-5472.CAN-09-2141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhang X, Yee D. Insulin-like Growth Factor Binding Protein-1 (IGFBP-1) Inhibits Breast Cancer Cell Motility. Cancer Res. 2002;62(15):4369–4375. [PubMed] [Google Scholar]
- 59.Lu S, Archer MC. Insulin-like growth factor binding protein-1 over-expression in transgenic mice inhibits hepatic preneoplasia. Molecular Carcinogenesis. 2003;36(3):142–146. doi: 10.1002/mc.10105. [DOI] [PubMed] [Google Scholar]
- 60.Russel-Jones DL, Umpleby AM, Shojaee-Moradie F, et al. The effect of an intravenous infusion of IGF-I and insulin on IGFBP-1, IGFBP-3, acid labile subunit, free and bound IGF-I, catecholamines and potassium in normal volunteers during an amino acid and glucose clamp. Clin Endocrinol. 1997;47(6):685–691. doi: 10.1046/j.1365-2265.1997.3161133.x. [DOI] [PubMed] [Google Scholar]
- 61.Pandini G, et al. IGF-II Binding to Insulin Receptor Isoform A Induces a Partially Different Gene Expression Profile from Insulin Binding. Annals of the New York Academy of Sciences. 2004;1028:450–456. doi: 10.1196/annals.1322.053. (Signal Transduction and Communication in Cancer Cells) [DOI] [PubMed] [Google Scholar]
- 62.Belfiore A, et al. Insulin receptor isoforms and insulin receptor/insulin-like growth factor receptor hybrids in physiology and disease. Endocr Rev. 2009;30(6):586–623. doi: 10.1210/er.2008-0047. [DOI] [PubMed] [Google Scholar]
- 63.Sciacca L, Costantino A, Pandini G, et al. Insulin receptor activation by IGF-II in breast cancers: evidence for a new autocrine/paracrine mechanism. Oncogene. 1999;18(15):2471–2479. doi: 10.1038/sj.onc.1202600. [DOI] [PubMed] [Google Scholar]
- 64.Samani AA, et al. The Role of the IGF System in Cancer Growth and Metastasis: Overview and Recent Insights. Endocr Rev. 2007;28(1):20–47. doi: 10.1210/er.2006-0001. [DOI] [PubMed] [Google Scholar]
- 65.Blanco-Aparicio C, et al. PTEN, more than the AKT pathway. Carcinogenesis. 2007;28(7):1379–1386. doi: 10.1093/carcin/bgm052. [DOI] [PubMed] [Google Scholar]
- 66.Di Cristofano A, Pandolfi PP. The Multiple Roles of PTEN in Tumor Suppression. Cell. 2000;100(4):387–390. doi: 10.1016/s0092-8674(00)80674-1. [DOI] [PubMed] [Google Scholar]
- 67.Reiling JH, Sabatini DM. Stress and mTORture signaling. Oncogene. 2006;25(48):6373–6383. doi: 10.1038/sj.onc.1209889. [DOI] [PubMed] [Google Scholar]
- 68.Jiang B-H, Liu L-Z. Role of mTOR in anticancer drug resistance: Perspectives for improved drug treatment. Drug Resistance Updates. 2008;11(3):63–76. doi: 10.1016/j.drup.2008.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Algire C, et al. Metformin blocks the stimulative effect of a high-energy diet on colon carcinoma growth in vivo and is associated with reduced expression of fatty acid synthase. Endocr Relat Cancer. 2010;17(2):351–360. doi: 10.1677/ERC-09-0252. [DOI] [PubMed] [Google Scholar]
- 70.Landman GW, et al. Metformin associated with lower cancer mortality in type 2 diabetes (ZODIAC-16) Diabetes Care. 2010;33(2):322–326. doi: 10.2337/dc09-1380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Jiralerspong S, et al. Metformin and Pathologic Complete Responses to Neoadjuvant Chemotherapy in Diabetic Patients With Breast Cancer. J Clin Oncol. 2009;27(20):3297–3302. doi: 10.1200/JCO.2009.19.6410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.DiGiovanni J, et al. Deregulated expression of insulin-like growth factor 1 in prostate epithelium leads to neoplasia in transgenic mice. Proceedings of the National Academy of Sciences of the United States of America. 2000;97(7):3455–3460. doi: 10.1073/pnas.97.7.3455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Carboni JM, et al. Tumor Development by Transgenic Expression of a Constitutively Active Insulin-Like Growth Factor I Receptor. Cancer Res. 2005;65(9):3781–3787. doi: 10.1158/0008-5472.CAN-04-4602. [DOI] [PubMed] [Google Scholar]
- 74.Scotlandi K, et al. Expression of an IGF-I receptor dominant negative mutant induces apoptosis, inhibits tumorigenesis and enhances chemosensitivity in Ewing's sarcoma cells. Int J Cancer. 2002;101(1):11–16. doi: 10.1002/ijc.10537. [DOI] [PubMed] [Google Scholar]
- 75.Lee C-T, et al. Recombinant adenoviruses expressing dominant negative insulin-like growth factor-I receptor demonstrate antitumor effects on lung cancer. Cancer Gene Ther. 2003;10(1):57–63. doi: 10.1038/sj.cgt.7700524. [DOI] [PubMed] [Google Scholar]
- 76.Bonnette SG, Hadsell DL. Targeted Disruption of the IGF-I Receptor Gene Decreases Cellular Proliferation in Mammary Terminal End Buds. Endocrinology. 2001;142(11):4937–4945. doi: 10.1210/endo.142.11.8500. [DOI] [PubMed] [Google Scholar]
- 77.Cox ME, et al. Insulin receptor expression by human prostate cancers. Prostate. 2009;69(1):33–40. doi: 10.1002/pros.20852. [DOI] [PubMed] [Google Scholar]
- 78.Novosyadlyy R, et al. Physical and functional interaction between polyoma virus middle T antigen and insulin and IGF-I receptors is required for oncogene activation and tumour initiation. Oncogene. 2009;28(39):3477–3486. doi: 10.1038/onc.2009.209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Zhang H, et al. Inhibition of cancer cell proliferation and metastasis by insulin receptor downregulation. Oncogene. 2010;29:2517–2527. doi: 10.1038/onc.2010.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Rensing KL, et al. Could recombinant insulin compounds contribute to adenocarcinoma progression by stimulating local angiogenesis? Diabetologia. 2010;53(5):966–970. doi: 10.1007/s00125-010-1687-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Fernandez AM, Kim JK, Yakar S, et al. Functional inactivation of the IGF-I and insulin receptors in skeletal muscle causes type 2 diabetes. Genes Dev. 2001;15(15):1926–1934. doi: 10.1101/gad.908001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Fierz Y, et al. Insulin sensitizing therapy attenuates type 2 diabetes-mediated mammary tumor progression. Diabetes. 2010;59(3):686–693. doi: 10.2337/db09-1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Carboni JM, et al. BMS-754807, a small molecule inhibitor of insulin-like growth factor-1R/IR. Molecular Cancer Therapeutics. 2009;8(12):3341–3349. doi: 10.1158/1535-7163.MCT-09-0499. [DOI] [PubMed] [Google Scholar]
- 84.Wang Y, Lipari P, Wang X, et al. A Fully Human Insulin-Like Growth Factor-I Receptor Antibody SCH 717454 (Robatumumab) Has Antitumor Activity as a Single Agent and in Combination with Cytotoxics in Pediatric Tumor Xenografts. Molecular Cancer Therapeutics. 2010;9(2):410–418. doi: 10.1158/1535-7163.MCT-09-0555. [DOI] [PubMed] [Google Scholar]
- 85.Karp DD, Paz-Ares LG, Novello S, et al. Phase II Study of the Anti-Insulin-Like Growth Factor Type 1 Receptor Antibody CP-751,871 in Combination With Paclitaxel and Carboplatin in Previously Untreated, Locally Advanced, or Metastatic Non-Small-Cell Lung Cancer. J Clin Oncol. 2009;27(15):2516–2522. doi: 10.1200/JCO.2008.19.9331. [DOI] [PubMed] [Google Scholar]
- 86.Clemmons DR. Modifying IGF1 activity: an approach to treat endocrine disorders, atherosclerosis and cancer. Nat Rev Drug Discov. 2007;6(10):821–833. doi: 10.1038/nrd2359. [DOI] [PubMed] [Google Scholar]
- 87.Haluska P, Carboni JM, TenEyck C, et al. HER receptor signaling confers resistance to the insulin-like growth factor-I receptor inhibitor, BMS-536924. Mol Cancer Ther. 2008;7:2589–2598. doi: 10.1158/1535-7163.MCT-08-0493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Fan Q-W, Cheng CK, Nicholaides TP, et al. A dual phosphoinositide-3-kinase α/mTOR inhibitor cooperates with blockade of epidermal growth factor receptor in PTEN-mutant glioma. Cancer Res. 2007;67(17):7960–7965. doi: 10.1158/0008-5472.CAN-07-2154. [DOI] [PMC free article] [PubMed] [Google Scholar]