

Abstract
Among the identified microRNAs (miRs) thus far, ~50% of mammalian miRs are clustered in the genome and transcribed as polycistronic primary transcripts. However, whether clustered miRs mediate non-redundant and cooperative functions remains poorly understood. In this study, we first identified activation of the promoter of miR-144/451 by GATA-4, a critical transcription factor in the heart. Next, we observed that ectopic expression of miR-144 and -451 individually augmented cardiomyocyte survival, which was further improved by overexpression of miR-144/451, compared to control cells in response to simulated ischemia/reperfusion. In contrast, knockdown of endogenous miR-144 and -451 revealed opposite effects. Using luciferase reporter assay and western blot analysis, we also validated that both miR-144 and miR-451 target CUG triplet repeat-binding protein 2 (CUGBP2), a ubiquitously expressed RNA-binding protein, known to interact with COX-2 3′-UTR and inhibit its translation. Accordingly, protein levels of CUGBP2 were greatly reduced and COX-2 activity was markedly increased in miR-144-, miR-451- and miR-144/451-overexpressing cardiomyocytes, compared to GFP-cells. Furthermore, inhibition of COX-2 activity by either NS-398 or DUP-697 partially offset protective effects of the miR-144/451 cluster. Together, these data indicate that both partners of the miR-144/451 cluster confer protection against simulated I/R-induced cardiomyocyte death via targeting CUGBP2-COX-2 pathway, at least in part. Thus, both miR-144 and miR-451 may represent new therapeutic agents for the treatment of ischemic heart disease.
Keywords: microRNA, Cardiomyocyte, Apoptosis, Oxidative stress, COX-2
Introduction
Accumulating evidence has established cardiomyocyte death as a critical component in the pathogenesis of ischemia-reperfusion injury and heart failure [1, 2]. Thus, inhibition or prevention of cardiac cell death is a desirable endpoint. Given the intricacy of cell death signaling cascade, multiple levels of intervention will likely be required to modulate this process. Recent discovered microRNAs (miRs), targeting hundreds of cellular proteins, may widely influence the signaling networks associated with cell death/survival [3, 4]. For example, miR-21 has been confirmed to target programmed cell death 4 (PDCD4), phosphatase and tensin homology deleted from chromosome 10 (PTEN), sprouty1/2 (SPRY1/2), and superoxide dismutase 2 (SOD2), which activate Akt, ERK-MAPK, and other survival signaling pathways in cardiomyocytes [5–8]. Therefore, it should be an attractive therapeutic strategy to identify a new miR that suppresses aberrant cell loss and promotes cell survival following myocardial injury.
MiR-144 and miR-451 are closely clustered and evolutionally conserved [9]. They are processed from a single gene locus that is regulated by the essential hematopoietic transcription factor GATA-1 [9]. Furthermore, maturation of miR-144 and miR-451 seems to be posttranscriptionally regulated in a sequence-specific manner [10, 11]. As a result, expression levels of mature miR-144 and miR-451 are different in cells. Previously, miR-144 and miR-451 were identified to have erythroid-specific expression in zebra fish [12]. Knockdown of miR-451 in zebra fish embryos, but not miR-144, significantly impaired erythroid maturation [12]. Nonetheless, the miR-144/451 null mice were born without any structural defects [13]. Further study indicated that these miR-144/451 null erythrocytes were sensitive to oxidative stress [13]. In cancer cells, miR-144 was shown to reduce TRAIL-induced apoptosis by targeting caspase-3 [14]; and miR-451 was confirmed to target macrophage migration inhibitory factor (MIF) and the calcium-binding protein 39 (CAB39)/AMPK signaling pathway [15, 16]. Importantly, both miR-144 and miR-451 are predicted to target CUG triplet repeat-binding protein 2 (CUGBP2), a RNA binding protein which interacts with COX-2 mRNA 3′-UTR and inhibits its translation [17]. Collectively, these data suggest that the miR-144/451 cluster may play a critical role in modulation of cell death/survival. Recently, we and others observed that miR-451 (currently, no information available on alteration of miR-144 in the heart) was significantly downregulated in ex vivo ischemic/reperfused animal hearts and human failing hearts [18, 19]. However, miR-451 was upregulated in human infarcted hearts with less than 7 days old, but no changes with more than 4 weeks old [20]. Whether transiently increased miR-451 represents an adaptive response or reduced miR-451 contributes to the process of cardiac disease is not clear. Additionally, it is not known as to whether overexpression of the miR-144/451 cluster in cardiomyocytes augments each other’s phenotype.
In the present study, we employed gain-of-function and loss-of-function approaches to determine the functional role of the miR-144/451 cluster in cardiomyocyte death under simulated ischemic conditions. For the first time, we found that GATA-4 directly regulated the miR-144/451 expression in cardiomyocytes. Upregulation of the miR-144/451 cluster was associated with cardioprotection against hypoxic stress via targeting the CUGBP2-COX-2 signaling pathway. We also observed that there was a functional cooperation within the miR-144/451 cluster to protect against simulated ischemia/reperfusion-induced cardiomyocyte death. Thus, results from the present study will potentially advance our understanding of how miR orchestrates the complex genetic networks responsible for cardiovascular homeostasis and disease.
Methods and Materials
miR Mimics/Inhibitors and Plasmid Constructs
miR mimics/inhibitors to miR-144 or miR-451, and miR mimic/inhibitor controls were purchased from the Dharmacon (www.dharmacon.com). The plasmid harboring GATA-4 and the adenoviral vector encoding GATA-4 were provided by Dr. Molkentin (Cincinnati Children’s Hospital Medical Center). The miR-144/451 promoter containing one cis-GATA-4 motif, or two cis-GATA-4 motifs was amplified by PCR, and subcloned into the pGL3-basic vector (Promega, Madison, WI) at the KpnI and BglII sites. A CUGBP2 3′-UTR segment including one miR-144 binding site or two miR-451 binding sites was amplified by PCR from mouse genomic DNA and inserted into the pMIR-REPORT™ luciferase miRNA expression reporter vector (Ambion, Inc.) at the SpeI and HindIII sites. All plasmid constructs were verified by sequencing. The sequences of miR-144/451 promoter and CUGBP2 3′-UTR segment, as well as all primers are listed in supplemental data.
Cell Culture and Construction of Adenoviral Vectors
Cell lines used in this study were purchased from the American Type Culture Collection (ATCC). H9c2 (rat ventricular cell line) and HEK293 (human embryonic kidney cell line) were cultured in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% fetal bovine serum and 100 μg/ml penicillin/streptomycin. Primary miR-144, miR-451, miR-144/451 DNA was PCR-amplified from mouse genomic DNA using high fidelity AccuPrime Taq DNA polymerase (Invitrogen). After sequencing, the amplified fragment was first subcloned in pcDNA3 (Invitrogen), then inserted under the CMV promoter into the AdEasy-1/Shuttle backbone, similar to our previous construction of adenoviral vectors [18, 21]. Antisense miR-144, -451, and -144/451 adenoviruses (named AdmiR-144AS, AdmiR-451AS, and AdmiR-144/451AS, respectively) were generated by cloning the miR-144, -451, or -144/451 primary DNA in the reverse orientation relative to the CMV promoter. The adenovirus expressing RNAi targeted at mouse and rat GATA4 (AdG4i) was generated as previously described [22]. Recombinant viruses were amplified in HEK 293 cells, and further purified using the Adeno-X™ Maxi Purification Kit (Clontech) and titered using the Adeno-X Rapid Titer kit (Clontech).
Adult Rat Ventricular Myocyte Isolation and Adenovirus-Mediated Gene Transfer
Rat ventricular myocytes were isolated from adult male Sprague-Dawley rats (6–8-wk old) and plated on laminin-coated glass coverslips or dishes, as described previously [21, 23]. After 1–2 hours, attached cardiomyocytes were infected with adenoviruses in diluted media, at a multiplicity of infection (MOI) of 500, for 2 hours before addition of suitable volume of culture media. Transfection efficiency, determined by GFP gene expression in cultured cardiomyocytes under fluorescence microscopy, was more than 95% after 48-h by this method. For detection of miR and protein expression, cultured cardiomyocytes were harvested after 60-h of adenoviral infection.
Total RNA Extraction and Quantitative RT-PCR
Total RNA was isolated from cultured cells using the miRNeasy Mini kit (Qiagen), according to the manufacturer’s protocol. The concentration of RNA was determined by a NanoDrop ND-1000 Spectrophotometer (NanoDrop Tech., Rockland, DE). All RT reactions, including no-template controls and RT minus controls, were run in triplicate in a GeneAmp PCR 9700 Thermocycler (Applied Biosystems). U6 was used as an internal control. Relative expression was calculated using the comparative threshold cycle (Ct) method, as previously described [18].
Simulated Ischemia/Reperfusion Treatment and Cell Survival Assay
For H9c2 cells, at 48 h post-transfection with miR mimics or inhibitors, the cells were subjected to simulated ischemia/reperfusion. Specifically, the medium was replaced with ischemic buffer containing 1.3 mMCaCl2, 5 mM KCl, 0.3 mMKH2PO4, 0.5 mM MgCl2, 0.4 mM MgSO4, 128 mM NaCl, 4 mM NaHCO3, and10 mM HEPES, pH 6.8, and placed in a chamber mimicking the hypoxic (1% O2) and hypercapnic conditions (20% CO2) [24]. Following 8-h hypoxia, the cells were re-oxygenated for 3 hours in DMEM with 1% serum at 37°C. For adult cardiomyocytes, at 60 h post-infection, cardiomyocytes were cultured in ischemia buffer and placed into a hypoxic chamber (37°C, 1% O2, 20% CO2 and 79% N2) for 1-h, followed by 3-h reperfusion under normal culture conditions. We chose the simulated ischemia/reperfusion protocol instead to use hydrogen peroxide treatment, because it may mimic the conditions occurred during ischemia/reperfusion-induced injury in vivo. In particular, the ischemic buffer was designed to reflect the ionic constituents and lack of glucose in cells during ischemia. Cardiomyocyte viability assessment was performed with addition of the soluble tetrazolium salt, MTS, to cells (CellTiter 96 AQueous One Solution Cell Proliferation Assay Kit, Promega), as per manufacturer’s instructions. For apoptosis analysis, DNA fragmentation was determined by an ELISA kit (Roche Applied Science, Indianapolis, IN). Cardiomyocytes were lysed by gently dispersing the pellet to prevent shearing of cells and release of nuclear DNA. The extract was then centrifuged at low speed and assayed according to the manufacturer’s instructions. Fold increase was obtained by dividing the measured absorbance of an experimental group by the absorbance of the positive control, provided in the kit. Caspase-3 activity was determined in cardiomyocyte lysates (100 μg) using Caspase-3/CPP32 Fluorometric Assay kit (BioVision, Inc, www.BioVision.com).
COX-2 Activity and PGE2 Immunoassay
Cardiomyocytes were harvested with lysis buffer. COX-2 activity was assayed colorimetrically by monitoring the appearance of oxidized N,N,N′,N′-tetramethyl-p-phenylenediamine (TMPD) at 590 nm, according to the manufacturer’s instructions (Cayman Chemical). For PGE2 immunoassay, cardiomyocyte lysates were diluted 10-fold with the enzyme immunoassay (EIA) buffer, and the amount of PGE2 in the diluted lysate was determined using a PGE2 EIA kit from Cayman Chemical. The values were expressed as pg/mg protein.
Western Blot Analysis
Protein samples were extracted from cultured cells, with procedures as described in detail elsewhere [18, 23]. Equal amounts of protein were subjected to SDS-PAGE. Binding of the primary antibody was detected by peroxidase-conjugated secondary antibodies and enhanced chemiluminescence (Amersham), and bands were quantified with densitometry. The source of antibodies and dilutions used were as follows: goat anti- GATA-4 (1:250 dilution), mouse anti-CUGBP2 (1:250 dilution) and mouse anti-COX-2 antibodies (1:250 dilution) (Santa Cruz Bitotech. Inc). α-actin (1:1000 dilution, Sigma) was probed in each membrane as a loading control.
Luciferase Reporter Assay
For miR-144/451 promoter assay, H9c2 cells were plated in 12-well plates and transiently transfected with 1 μg of miR-144/451 promoter-luciferase reporter plasmid and 0.5 or 1 μg of pGATA4 using the DharmaFECT Duo Transfection Reagent (Thermo Fisher Scientific Inc) according to the manufacturer’s protocol. The total amount of DNA for each transfection was kept at 2 μg/well by the addition of empty vector when needed. Cells were harvested 48 h later, and luciferase assays were performed on a Monolight 3010 luminometer (Pharmingen) with a luciferase assay kit (Promega, Madison, WI).
For CUGBP2-3′UTR assay, H9c2 cells were cotransfected in 12-well plates using the DharmaFECT Duo Transfection Reagent according to the protocol of the manufacturer, with 0.4 μg of the CUGBP2-3′UTR luciferase reporter vector and 0.08 μg of the control vector pMIR-REPORT (Ambion, Inc.). For each well, 100-nM (final) miR-144, -451 or scrambled miR control was used. Cell lysates were prepared 72-h later, and luciferase activity was measured, and expressed as relative light units using a luciferase assay kit (Promega). β-galactosidase activity was measured with a commercially available kit (Promega). 3′UTR activity of each construct was expressed as the ratio of luciferase/β-galactosidase activity. All transfections were performed in triplicate from three independent experiments.
Statistical Analysis
Data are presented as mean ± SD. Comparisons were made by Student’s t test as appropriate. A P value of <0.05 was considered statistically significant.
Results
GATA-4 Directly Regulates the MiR-144/451 Cluster Expression in Cardiomyocytes
The GATA family of zinc finger transcription factors has been implicated in regulating cell growth, differentiation and survival [25–28]. Of the six GATA family members, GATA-1/2/3 are predominant in blood and ectodermal derivatives, whereas GATA-4/5/6 are expressed in heart and endodermal derivatives [25]. Recently, an elegant study has shown that the miR-144/451 cluster bearing miR-144 and miR-451 is directly activated by GATA-1 in erythroid cells [9]. Additionally, analysis of multiple mouse tissues for microRNA expression indicated that miR-144 and miR-451 are mostly expressed in blood and heart [9]. We therefore hypothesize that the miR-144/451 cluster is regulated by GATA-4 in cardiomyocytes. To test this hypothesis, we first overexpressed GATA-4 in adult rat cardiomyocytes. Compared to AdGFP-infected group, infection with AdGATA-4 resulted in 2.3-fold increases in GATA-4 protein levels (Figs. 1A and B), in which a single transcript of the miR-144/451 cluster was upregulated by 2.0-fold (Fig. 1C). Stem-loop real-time PCR further demonstrated that mature miR-144 and miR-451 were increased by 1.9-fold and 2.5-fold, respectively (Fig. 1C). On the contrary, downregulation of endogenous GATA-4 by small interference RNA (AdG4i) caused 35% decrease in the levels of miR-144/451 transcript, 19% reduction in mature miR-144, and 25% decrease in mature miR-451 (Figs 1D–F). Together, these results suggest that the miR-144/451 cluster is potential downstream target of GATA-4.
Figure 1.
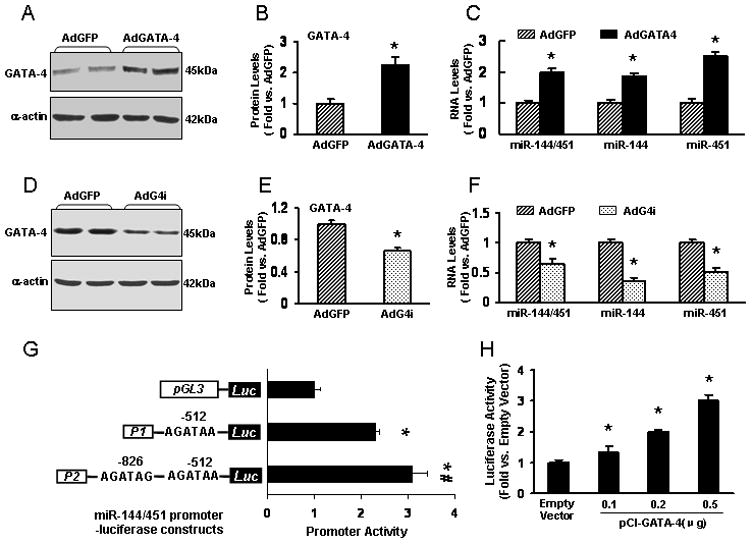
MiR-144/451 cluster is regulated by GATA-4 in cardiomyocytes. (A) Immunoblots for detection of GATA-4 expression in AdGATA-4-infected adult rat cardiomyocytes. α-actin was used as loading control. (B) Immunoblots were quantified by densitometry, and results are presented as means ± SD (n=4). (C) A single transcript of the miR-144/451 cluster, mature miR-144 and mature miR-451 were determined in AdGATA-4-infected cardiomyocytes by real-time PCR. *, p<0.05 vs. GFP controls, n=4. (D–F) GATA-4 interference RNA (AdG4i) infection resulted in decreased GATA-4 expression, which further reduced the levels of miR-144/451, mature miR-144 and miR-451 in adult cardiomyocytes. *, p<0.05 vs. GFP controls, n=3. (G) Both -512 and -826 GATA sites were responsible for GATA-4-induced activation of the miR-144/451 promoter in H9c2 cells. (G) GATA-4 dose-dependently activated the miR-144/451 promoter (P2) in H9c2 cells. *, p<0.05 vs. empty vector controls. Similar results were observed in three additional, independent experiments.
Next, we sought to investigate whether GATA-4 directly regulates the miR-144/451 promoter activity. Two miR-144/451 promoter-luciferase plasmids were generated which contain one (P1) or two (P2) putative GATA binding sites at positions -512 and -826 of the miR-144/451 promoter region (Fig. 1G and supplemental data). These luciferase reporter constructs were co-transfected into H9c2 cardiomyocytes with either pCI-GATA-4 or pCI control. β–gal vector was also co-transfected as an internal control. 48 hours after transfection, we conducted luciferase activity assays and observed that co-transfection of GATA-4 strongly augmented the luciferase activity from both P1 and P2 constructs (Fig. 1G). Interestingly, the promoter activity was significantly increased in cells co-transfected with P2 construct, compared with P1 plasmid (Fig. 1G). These data indicate that both of the putative GATA-4 binding sites in the miR-144/451 promoter region are responsive to GATA-4. We also observed that GATA-4 activation of miR-144/451 promoter is dose-dependent (Fig. 1H). Thus, several lines of evidence suggest that the miR-144/451 locus is a direct target of GATA-4 in cardiomyocytes.
Both MiR-144 and MiR-451 Protect against Simulated Ischemia/Reperfusion-Induced Cell Death
Since GATA-4 acts as a robust regulator of cardiomyocyte survival [27, 28], it is reasonable to speculate that the miR-144/451 cluster may confer cardio-protection against stress-induced cell death. To test this, we first transfected miR mimics/inhibitors to miR-144 or miR-451 in rat H9c2 cardiomyocytes (Figs. 2A and B). 48 h later, cells were subjected simulated ischemia for 8 h followed by 3-h reoxygenation. Cell viability was measured by MTS incorporation, and showed that overexpression of miR-144 and miR-451 augmented cell survival upon simulated ischemia/reperfusion (sI/R), compared to miR mimic controls (Figs. 2C and D). Conversely, knock-down of miR-144 and miR-451 by miR inhibitors showed detrimental effects (Figs. 2A and B). Furthermore, H9c2 cells co-transfected with miR-144 and miR-451 mimics in combination exhibited a strikingly increased survival rate compared to H9c2 cells transfected with a single miR mimic (Fig. 2E), suggesting additive effects of the miR-144/451 cluster. In addition, H9c2 cells transfected with GATA-4 showed a marked resistance to sI/R (Fig. 2F), which is consistent with previous findings [27]. However, protective effects of GATA-4 were partially offset by either miR-144 inhibitor or miR-451 inhibitor, and completely eliminated by co-transfection with combined miR inhibitors to miR-144 and miR-451(Fig. 2F). These data further suggest that functional cooperativity between miR-144 and miR-451 to protect cardiomyocytes from ischemia/reperfusion-induced cell death.
Figure 2.
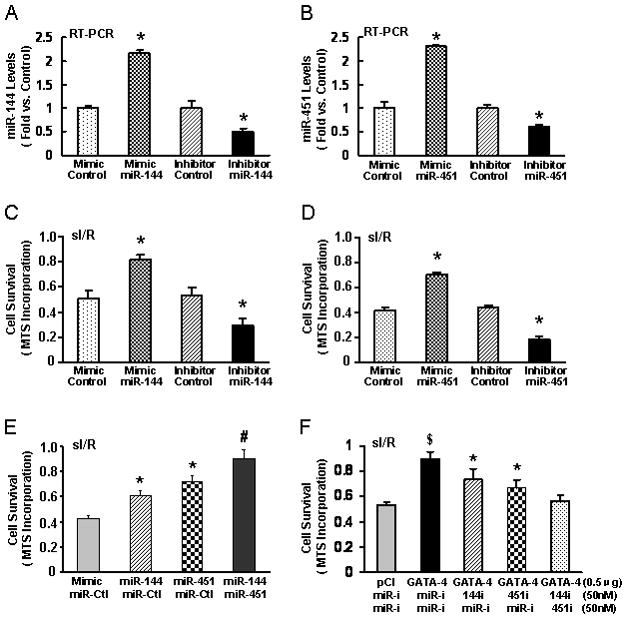
Effects of miR mimics/inhibitors to miR-144 or miR-451 on simulated ischemia/reperfusion (sI/R)-induced cell death. Stem-loop real-time PCR determined the levels of (A) miR-144 and (B) miR-451 in H9c2 cells transfected with miR-144 and miR-451 mimics. (C and D) Transfection of miR-144 and miR-451 increased cell survival upon sI/R treatment, whereas knockdown of these miRs by inhibitors showed detrimental effects (*p< 0.05 versus control). (E) Co-transfection of mimics to miR-144 and miR-451 in H9c2 cells demonstrated additive effects in protection against sI/R-triggered cell death (*p< 0.05 vs. control, # p< 0.05 vs. either miR-144 or miR-451 transfection). (F)Protective effects of GATA-4 on sI/R-induced cell death were partially abrogated by co-transfection with either miR-144 or miR-451 inhibitor, and completely blocked by co-transfection with both miR inhibitors. The total amount of DNA (0.5 μg) + miR(100nM) for each group was kept equal by the addition of empty vector and miR controls. ($, p< 0.05 vs. empty vector pCI; *p< 0.05 vs. GATA-4+ miR inhibitor control). Similar results were observed in two additional, independent experiments. miR-i stands for miR-inhibitor. Ctl stands for control.
Overexpression of the miR-144/451 Cluster Protects Adult Cardiomyocytes against Simulated Ischemia/Reperfusion-Induced Cellular Injury
To gain insights into the functional significance of miR-144/451 in the ischemic heart, gain-of-function and loss-of function approaches were employed using cultured adult rat cardiomyocytes. We generated six adenoviral vectors bearing primary miR-144, miR-451 and miR-144/451 in the sense or antisense direction, designated as AdmiR-144, AdmiR-451, AdmiR-144/451 and AdmiR-144AS, AdmiR-451AS, AdmiR-144/451AS, respectively. Following infection of cardiomyocytes with these recombinant adenoviral vectors for 48-h, we observed nearly 95% infection efficiency (Fig. 3A). Importantly, there were no apparent morphological alterations or differences in the number of adherent cells and rod-shaped cells among the groups (Fig. 3A). After 60-h of adenoviral infection, stem-loop real-time PCR clearly showed overexpression of the exogenous miR-144 in AdmiR-144 and AdmiR-144/451-infected cardiomyocytes, but not in AdmiR-451-infected cells (Fig. 3B). Similarly, miR-451 was overexpressed by ~3-fold in AdmiR-451- and AdmiR-144/451-infected cardiomyocytes, but not in AdmiR-144-cells (Fig. 3C). Conversely, the endogenous miR-144 was successfully knocked-down in AdmiR-144AS and AdmiR-144/451AS-cells by ~40% (Fig. 3B). In the same manner, miR-451 was significantly decreased in AdmiR-451AS- and AdmiR-144/451AS-infected cardiomyocytes (Fig. 3C). We next examined the effects of miR-144, -451, and -144/451 on cell survival upon 1-h simulated ischemia, followed by 3-h reperfusion (Fig. 3D). Cell viability analysis showed that ectopic expression of miR-144 and -451 augmented cell survival by 15%, which was further improved by overexpression of miR-144/451 (Fig. 3E). On the contrary, knock-down of endogenous miR-144 and -451 revealed opposite effects (i.e. reduced survival by ~20% in miR-144AS- and miR-451AS-cells; by ~35% in miR-144/451AS-cells, relative to GFP-cells, Fig. 3E). Similarly, release of lactate dehydrogenase (LDH) upon sI/R, a biochemical marker for necrotic cell death, was significantly suppressed in both miR-144- and miR-451-cardiomyocytes, and further dampened in miR-144/451-cells, compared to GFP-controls (Fig. 3F). Conversely, downregulation of miR-144, miR-451, and miR-144/451 revealed opposite effects (Fig. 3F). Furthermore, upon simulated I/R, AdmiR-144- and miR-451-infected myocytes exhibited a significant decrease in histone-associated DNA fragmentation and caspase-3 activity, compared to Ad.GFP-infected myocytes (Figs. 3G and H). In contrast, infection with AdmiR-144AS and miR-451AS significantly increased I/R-induced DNA fragmentations and caspase-3 activities (Figs. 3G and H). Synergistically, overexpression of the miR-144/451 cluster exhibited less cellular damage (i.e., DNA fragmentation and caspase-3 activity) than single miR-overexpressing cells (Figs. 3G and H). Taken together, these data indicate that both miR-144 and miR-451 have protective effects in cardiomyocytes against simulated I/R-triggered cellular injury and the miR-144/451 cluster mediates cooperative actions.
Figure 3.
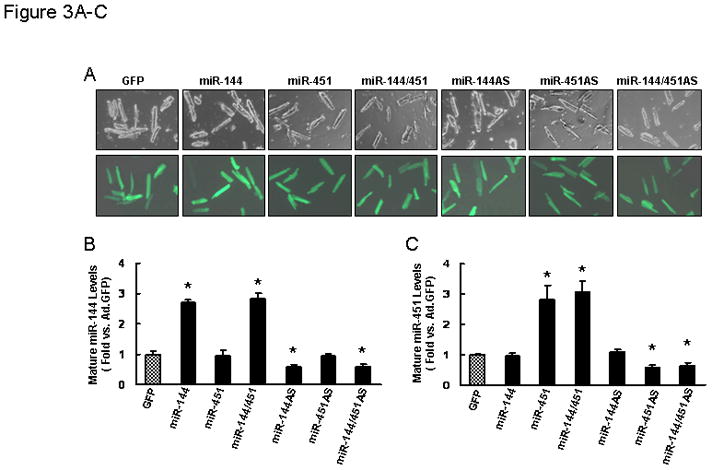
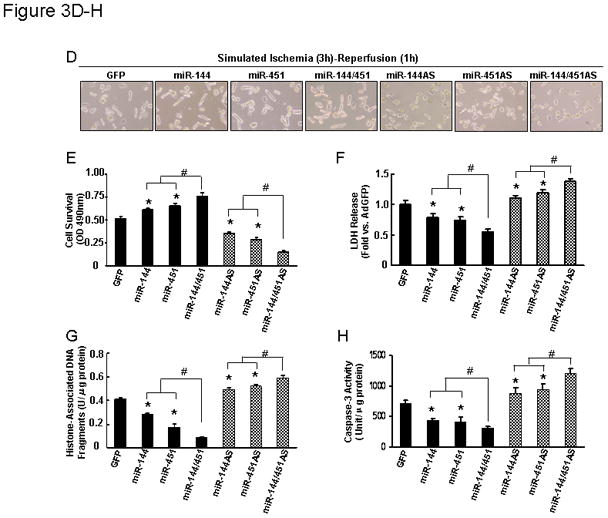
Effects of miR-144 and miR-451 on simulated ischemia/reperfusion-induced adult cardiomyocyte injury. (A) More than 95% of adult rat cardiomyocytes were infected by recombinant adenoviral vectors at a MOI of 500 for 48 h. (B) Mature miR-144 and (C) mature miR-451 levels were determined by stem-loop real-time PCR. Overexpression of miR-144 and miR-451 showed: (D and E) resistant to simulated I/R-induced cell death, (F) decreased LDH release, (G) reduced DNA fragmentation and (H) Caspase-3 activity. Conversely, downregulation of miR-144 and miR-451 exhibited opposite effects (D–H). As expected, increased or decreased both miRs within the miR-144/451 cluster revealed synergistic effects. *, p<0.05 vs. AdGFP control; #, p<0.05 vs. single miR-cells. Similar results were observed in three additional, independent experiments.
Both miR-144 and miR-451 Directly Target CUGBP2 Gene
To elucidate potential mechanisms of the miR-144/451 cluster in protection of cardiomyocytes against simulated I/R injury, it will be essential to identify putative targets of miR-144 and miR-451. Recent studies have documented that CUG triplet repeat-binding protein 2 (CUGBP2) is repressed by miR-451 (but not miR-144) during erythroid maturation of MEL cells [17], although CUGBP2-3′ UTR contains predicted binding sites for both miR-144 and miR-451 (Fig. 4A and supplemental data). However, we observed that CUGBP2 was downregulated by ~20% in AdmiR-144-infected cardiomyocytes and by ~30% in Ad.miR-451-cardiomyocytes (Fig. 4B). More strikingly, protein levels of CUGBP2 were decreased by ~80% in AdmiR-144/451-cells (Fig. 4B). Conversely, knockdown of the endogenous miR-144 by AdmiR-144AS infection yielded 1.3-fold increase in protein levels of CUGBP2, while AdmiR-451AS infection resulted in 1.5-fold increase of CUGBP2 protein (Fig. 4B). As expected, downregulation of both miRs by AdmiR-144/451AS infection caused 2.3-fold upregulation of CUGBP2 (Fig. 4B). Notably, real-time PCR demonstrated that CUGBP2 messenger RNA copy numbers were similar among these groups (data not shown). These results suggest that both miR-144 and miR-451 dampen CUGBP2 levels by impeding its translation, rather than by breaking down its messenger RNAs.
Figure 4.
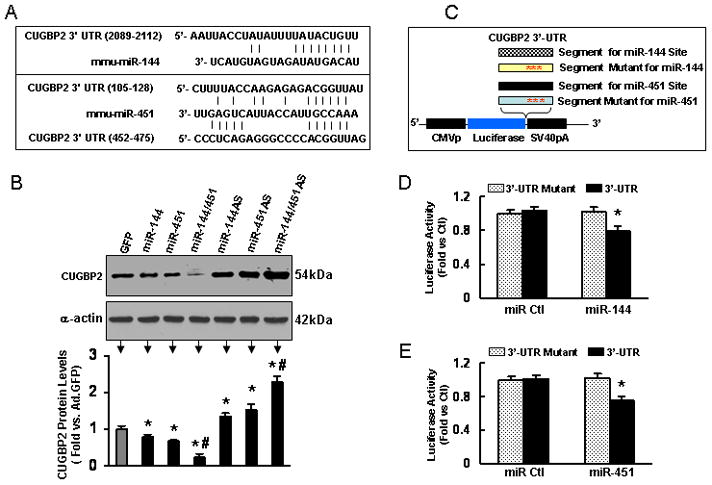
Both miR-144 and miR-451 directly target CUGBP2 gene. (A) CUGBP2 3 ′-UTR contains miR-144 and miR-451 interacting sites. (B) Protein levels of CUGBP2 were reduced in miR-144-, miR-451-, and miR-144/451-cardiomyocytes; whereas upregulated in miR-144AS-, miR-451AS-, and miR-144/451AS-cardiomyocytes. (C) Diagram of plasmid construction. A segment of CUGBP2 3 ′ UTR or a mutated segment was cloned downstream of the luciferase-encoding region. (D) Dual luciferase activity assay of H9c2 cells co-transfected with the plasmid containing the segment of CUGBP2 3 ′ UTR for miR-144 and either miR-144 or a control oligoribonucleotide showed that miR-144 inhibited luciferase activity, compared with controls. (E) Luciferase activity was suppressed by miR-451, but not miR control. *, p<0.05 vs. controls. Similar results were observed in two additional, independent experiments.
To further validate whether both miR-144 and miR-451 directly recognize the 3′-UTR of CUGBP2, we generated serial constructs harboring the 3′UTR segment of CUGBP2 and its mutant fused downstream to the luciferase coding sequence(Fig. 4C; see supplemental data for a complete list of primers used). Co-transfection of miR-144 strongly inhibited the luciferase activity; whereas no effect was observed in cells co-transfected with a construct containing a mutated segment of CUGBP2 3′UTR (miR-144 seed sequence AUACUGU was mutated to GACACAA, Fig. 4D). This effect was specific because there was no change in luciferase reporter activity when a negative control miR was co-transfected with either reporter construct (Fig. 4D). Similarly, co-transfection of miR-451 resulted in greater reduction of the luciferase activity, compared to miR controls (Fig. 4E). Collectively, these data indicate that CUGBP2 transcript may represent a genuine target of both miR-144 and miR-451.
Overexpression of the miR-144/451 Cluster Augments COX-2 Activity
As a ubiquitously expressed RNA-binding protein, CUGBP2 has been implicated in the regulation of mRNA splicing, editing, stability and translation [29, 30]. More interestingly, CUGBP2 is known to interact with COX-2 3′-UTR and inhibit its mRNA translation [29, 30], similar to the function of microRNA. Therefore, we examined whether the miR-144/451 cluster regulates protein levels of COX-2 in cultured cardiomyocytes. Western-blotting results showed that COX-2 protein was significantly increased in AdmiR-144- and AdmiR-451-infected cardiomyocytes by 1.5-fold and 1.7-fold, respectively, compared to AdGFP controls (Fig. 5A). On the other hand, infection with AdmiR-144/451 robustly induced COX-2 expression (2.5-fold vs. AdGFP, Fig. 5A). In contrast, down-regulation of either or both of the miRs by AdmiR-144AS, AdmiR-451AS, and AdmiR-144/451AS markedly decreased COX-2 protein levels (by 50%, 75%, and 92%, vs. AdGFP, respectively, Fig. 5A). Accordingly, COX-2 activity measured by monitoring the appearance of oxidized N, N, N′, N′-tetramethyl-p-phenylenediamine (TMPD) at 590 nm was significantly increased in miR-144-, miR-451- and miR-144/451-cardiomyocytes; whereas remarkably decreased in miR-144AS-, miR-451AS- and miR-144/451AS-cells (Fig. 5B). We also measured levels of PGE2, a downstream target of COX-2, in these adenoviral vector-infected cardiomyocytes. Consistently, COX-2-dependent PGE2 synthesis was correlated well with COX protein level/ activity in these cells, as indicated in Figs. 5B and 5C. Together, these results suggest that the miR-144/451 cluster regulates the CUGBP2-COX-2 signaling pathway in cardiomyocytes.
Figure 5.
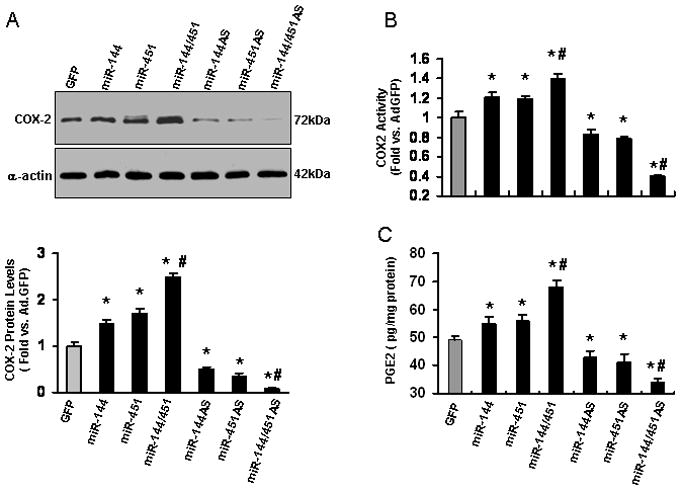
Both partners of the miR-144/451 cluster upregulated (A) the COX-2 protein levels and (B) activity as well as (C) PGE2 levels in cardiomyocytes infected with various adenovirual vectors as indicated. n=4, *, p<0.05 vs. AdGFP control; #, p<0.05 vs. single miR-cells.
Inhibition of COX-2 Activity Partly Offsets Protective Effects of the miR-144/451 Cluster
To further elucidate whether cardioprotective actions of the miR-144/451 cluster are mediated by COX-2, we pre-treated miR-overexpressing cardiomyocytes with two different COX-2 inhibitors NS-398 and DUP-697, followed by simulated ischemia (1h)-reperfusion (3h). In AdGFP-infected cardiomyocytes, pre-treatment with either NS-398 or DUP-697 significantly reduced cell survival (Fig. 6A) and increased both DNA fragmentation and Caspase-3 activity (Figs. 6B and C) upon simulated I/R. Interestingly, upon addition of COX-2 inhibitors NS-398/DUP-697, the protective action of miR-144 was completely eliminated in simulated I/R-treated cardiomyocytes (Figs. 6A-C). However, protective effects of miR-451 against simulated I/R stress were partly offset by either NS-398- or DUP-697-pretreatment. Although the miR-144/451 cluster strongly protected cardiomyocytes against simulated I/R-induced cell death/apoptosis, pre-inhibition of COX-2 by either NS-398- or DUP-697 also partially made this cluster ineffective (Figs. 6A–C). Thus, these results suggest that miR-144 protects against simulated I/R-induced cardiomyocyte death/apoptosis largely via the COX-2-dependent signaling pathway, whereas protective effects of miR-451 are partly associated with the COX-2 signaling.
Figure 6.
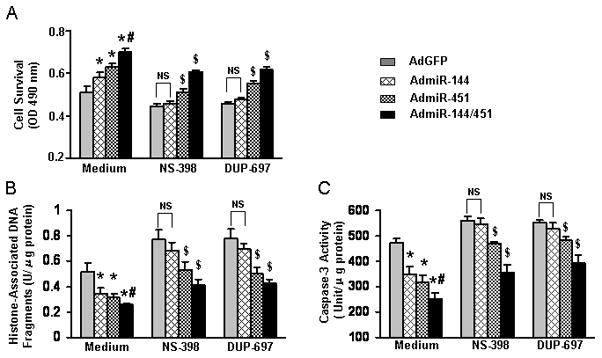
Effects of COX-2 inhibitors on miR-144/451-meadiated protection against simulated ischemia/reperfusion-induced (A) cardiomyocyte death, (B) DNA fragmentation and (C) Caspase-3 activity. n=4, *, p<0.05 vs. AdGFP control; #, p<0.05 vs. single miR-cells; $, p<0.05 vs. respective medium-controls.
GATA-4 Upregulates Protein Levels of COX-2 in Cardiomyocytes
Since GATA-4 regulates miR-144/451 expression, which dampens CUGBP2 protein levels and increases COX-2 levels in cardiomyocytes, we next determined whether CUGBP2-COX-2 signaling is affected in GATA-4-overexpressing cardiomyocytes. As shown in Figs. 7A and B, overexpression of GATA-4 significantly reduced CUGBP2 protein levels by ~40%, which consequently increased COX-2 protein levels by ~2-fold. In contrast, knockdown of endogenous GATA-4 demonstrated opposite effects (Figs. 7A and B). Our results further suggest that CUGBP2-COX-2 signaling is controlled by GATA-4 via its downstream target miR-144/451 (Fig. 7C). Importantly, these data also corroborate that GATA-mediated cardioprotection is associated with the miR-144/451 cluster, at least in part (Fig. 2F).
Figure 7.
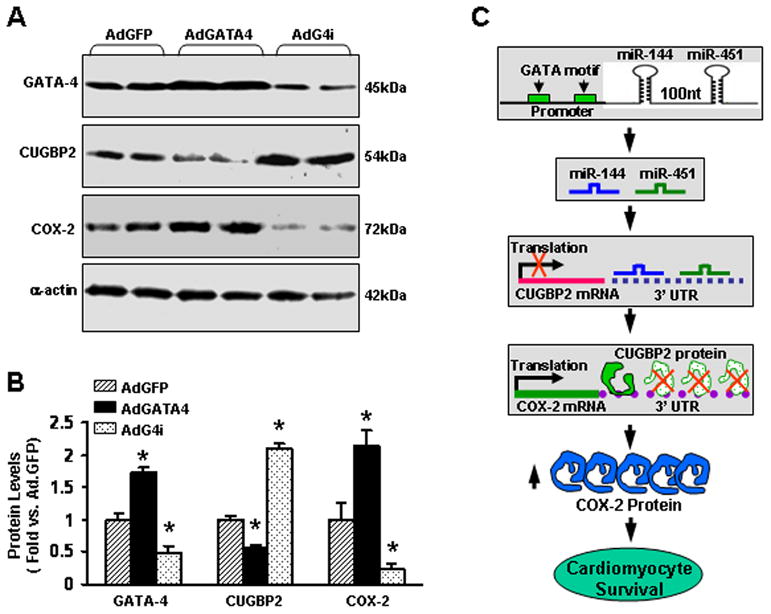
Effects of GATA-4 overexpression and knockdown on (A and B) CUGBP2 and COX-2 protein levels in adult rat cardiomyocytes. n=4, *, P<0.05 vs. AdGFP control. (C) Mechanisms of the miR-144/451 cluster-mediated protection against simulated ischema/reperfusion-induced cardiomyocyte injury.
Discussion
Among the identified miRs thus far, ~50% of mammalian miRs are clustered in the genome and transcribed as polycistronic primary transcripts, whereas ~40% are expressed as individual transcripts from intronic locations and ~10% from intergenic regions [31]. It is believed that distinct miRs within the same cluster may work in concert to regulate a cellular process [31]. However, two miRs co-expressed from miR-1/133 loci showed the intriguingly opposite effects on apoptosis [32]. Overexpression of miR-1 in H9c2 rat cardiomyocytes provokes apoptotic cell death by targeting Hsp60 and Hsp70. Conversely, miR-133 is protective against H2O2-induced apoptosis through targeting Caspase-9[32]. In the present study, we applied overexpression and knock-down approaches to explore the function of both miRs within the miR-144/451 cluster individually or in combination under simulated ischemic conditions. Our results for the first time demonstrate that both partners of the miR-144/451 cluster mediate protective role in cardiomyocytes. Importantly, overexpression of the entire miR-144/451 cluster resulted in cardioprotection to a synergistic extent.
While we confirmed that miR-144 and miR-451 shared GATA-4 target promoter and are processed from a single polycistronic precursor transcript, the endogenous mature levels of miR-451 and miR-144 were not equal with robust expression of miR-451 (~70-fold higher than miR-144) in cardiomyocytes. This pattern may be explained in part by the hairpin structure of miR-451 compared to miR-144. As described in two recent seminal papers [10, 11], mature miR-451 bears 17- nucleotide stem region and 6 terminal nucleotides spanning the loop region, whereas 20 nucleotides of mature miR-144 are limited to the stem only. This unusual feature makes miR-451 adopt a dicer-independent miR biogenesis and possibly make it more stable than miR-144. Nonetheless, miR-144 and miR-451 might be functionally related targeting same genes, or different genes within the same pathway. Although miR-144 has hundreds of potential targets and miR-451 has much fewer predicted targets (http://mirdb.org/; http://www.mirbase.org), we confirmed, by Western-blot and dual-luciferase reporter assays, that both miR-144 and miR-451 targeted CUGBP2 in cardiomyocytes. Our study further demonstrated that both the miRs of the miR-144/451 cluster upregulated COX-2-PGE2, a downstream singling effect of CUGBP2, which may contribute to their protective actions. While the role of COX-2 in ischemic heart disease and tissue response to hypoxia is still debated [33–35], a series of in vivo and in vitro studies have well established that COX-2 activity is necessary for the protective effects of late preconditioning, postconditioning, and bradykinin [33, 36, 37]. Additionally, constitutive expression of COX-2 in cardiomyocytes is known to confer protection against reperfusion injury [38], whereas COX-2 knockout mice demonstrate aggravated cardiac ischemia/reperfusion injury [39]. In support of these findings, the positive action of miR-144 and miR-451 was significantly abrogated by pre-treatment of cardiomyocytes with COX inhibitors NS-398 and DUP-697. Collectively, these results suggest that GATA-4-mediated miR-144/451 cluster work together to downregulate CUGBP2, thereby activating COX-2-PGE2 survival pathway in cardiomyocytes (Figure 7C).
In conclusion, we show here that GATA-4-regulated miR-144/451 cluster confers cardiomyocyte-protective effect against simulated ischemia/reperfusion-induced cellular injury. The underlying mechanisms are partially associated with activation of COX-2 signaling pathways. However, further studies are needed to fully elucidate the functional consequences of this miR cluster in vivo using transgenic and knockout approaches. Also, it will be of great interest to assess its potential therapeutic application for the treatment of ischemic heart disease.
Supplementary Material
Acknowledgments
Funding Sources: This study was supported by NIH grants HL-087861 and HL087861-03S1 (G.-C. Fan), HL089824 and HL081859 (Y. Wang), University of Cincinnati CEG grant #1007636 (G.-C. Fan).
Footnotes
Conflict of Interest Disclosures: None
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Elsässer A, Suzuki K, Schaper J. Unresolved issues regarding the role of apoptosis in the pathogenesis of ischemic injury and heart failure. J Mol Cell Cardiol. 2000;32:711–24. doi: 10.1006/jmcc.2000.1125. [DOI] [PubMed] [Google Scholar]
- 2.Regula KM, Kirshenbaum LA. Apoptosis of ventricular myocytes: a means to an end. J Mol Cell Cardiol. 2005;38:3–13. doi: 10.1016/j.yjmcc.2004.11.003. [DOI] [PubMed] [Google Scholar]
- 3.Baek D, Villén J, Shin C, Camargo FD, Gygi SP, Bartel DP. The impact of microRNAs on protein output. Nature. 2008;455:64–71. doi: 10.1038/nature07242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Selbach M, Schwanhäusser B, Thierfelder N, Fang Z, Khanin R, Rajewsky N. Widespread changes in protein synthesis induced by microRNAs. Nature. 2008;455:58–63. doi: 10.1038/nature07228. [DOI] [PubMed] [Google Scholar]
- 5.Cheng Y, Liu X, Zhang S, Lin Y, Yang J, Zhang C. MicroRNA-21 protects against the H(2)O(2)-induced injury on cardiac myocytes via its target gene PDCD4. J Mol Cell Cardiol. 2009;47:5–14. doi: 10.1016/j.yjmcc.2009.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sayed D, He M, Hong C, Gao S, Rane S, Yang Z, et al. MicroRNA-21 is a downstream effector of AKT that mediates its antiapoptotic effects via suppression of Fas ligand. J Biol Chem. 2010;285:20281–90. doi: 10.1074/jbc.M110.109207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jazbutyte V, Thum T. MicroRNA-21: from cancer to cardiovascular disease. Curr Drug Targets. 2010;11:926–35. doi: 10.2174/138945010791591403. [DOI] [PubMed] [Google Scholar]
- 8.Fleissner F, Jazbutyte V, Fiedler J, Gupta SK, Yin X, Xu Q, et al. Asymmetric Dimethylarginine Impairs Angiogenic Progenitor Cell Function in Patients With Coronary Artery Disease Through a MicroRNA-21-Dependent Mechanism. Circ Res. 2010;107:138–43. doi: 10.1161/CIRCRESAHA.110.216770. [DOI] [PubMed] [Google Scholar]
- 9.Dore LC, Amigo JD, Dos Santos CO, Zhang Z, Gai X, Tobias JW, et al. A GATA-1-regulated microRNA locus essential for erythropoiesis. Proc Natl Acad Sci U S A. 2008;105:3333–8. doi: 10.1073/pnas.0712312105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cheloufi S, Dos Santos CO, Chong MM, Hannon GJ. A dicer-independent miRNA biogenesis pathway that requires Ago catalysis. Nature. 2010;465:584–9. doi: 10.1038/nature09092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cifuentes D, Xue H, Taylor DW, Patnode H, Mishima Y, Cheloufi S, et al. A Novel miRNA Processing Pathway Independent of Dicer Requires Argonaute2 Catalytic Activity. Science. 2010;328:1694–8. doi: 10.1126/science.1190809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pase L, Layton JE, Kloosterman WP, Carradice D, Waterhouse PM, Lieschke GJ. MiR-451 regulates zebrafish erythroid maturation in vivo via its target gata2. Blood. 2009;113:1794–804. doi: 10.1182/blood-2008-05-155812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dos Santos CO, Yu D, Amigo J, Kandros E, Valentine ER, Shelat S, et al. The microRNA144/451 locus enhances nuclear FOXO3a activity to protect erythroid cells against oxidant stress. Blood. 2008:112. (Abstract) [Google Scholar]
- 14.Ovcharenko D, Kelnar K, Johnson C, Leng N, Brown D. Genome-scale microRNA and small interfering RNA screens identify small RNA modulators of TRAIL-induced apoptosis pathway. Cancer Res. 2007;67:10782–8. doi: 10.1158/0008-5472.CAN-07-1484. [DOI] [PubMed] [Google Scholar]
- 15.Bandres E, Bitarte N, Arias F, Agorreta J, Fortes P, Agirre X, et al. microRNA-451 regulates macrophage migration inhibitory factor production and proliferation of gastrointestinal cancer cells. Clin Cancer Res. 2009;15:2281–90. doi: 10.1158/1078-0432.CCR-08-1818. [DOI] [PubMed] [Google Scholar]
- 16.Godlewski J, Nowicki MO, Bronisz A, Nuovo G, Palatini J, De Lay M, et al. MicroRNA-451 regulates LKB1/AMPK signaling and allows adaptation to metabolic stress in glioma cells. Mol Cell. 2010;37:620–32. doi: 10.1016/j.molcel.2010.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Papapetrou EP, Korkola JE, Sadelain M. A genetic strategy for single and combinatorial analysis of miRNA function in mammalian hematopoietic stem cells. Stem Cells. 2010;28:287–96. doi: 10.1002/stem.257. [DOI] [PubMed] [Google Scholar]
- 18.Ren XP, Wu J, Wang X, Sartor MA, Qian J, Jones K, et al. MicroRNA-320 is involved in the regulation of cardiac ischemia/reperfusion injury by targeting heat-shock protein 20. Circulation. 2009;119:2357–66. doi: 10.1161/CIRCULATIONAHA.108.814145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Thum T, Galuppo P, Wolf C, Fiedler J, Kneitz S, van Laake LW, et al. MicroRNAs in the human heart: a clue to fetal gene reprogramming in heart failure. Circulation. 2007;116:258–67. doi: 10.1161/CIRCULATIONAHA.107.687947. [DOI] [PubMed] [Google Scholar]
- 20.Bostjancic E, Zidar N, Glavac D. MicroRNA microarray expression profiling in human myocardial infarction. Dis Markers. 2009;27:255–68. doi: 10.3233/DMA-2009-0671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fan GC, Chu G, Mitton B, Song Q, Yuan Q, Kranias EG. Small heat-shock protein Hsp20 phosphorylation inhibits beta-agonist-induced cardiac apoptosis. Circ Res. 2004;94:1474–1482. doi: 10.1161/01.RES.0000129179.66631.00. [DOI] [PubMed] [Google Scholar]
- 22.Kobayashi S, Lackey T, Huang Y, Bisping E, Pu WT, Boxer LM, et al. Transcription factor gata4 regulates cardiac BCL2 gene expression in vitro and in vivo. FASEB J. 2006;20:800–2. doi: 10.1096/fj.05-5426fje. [DOI] [PubMed] [Google Scholar]
- 23.Wang X, Zingarelli B, O’Connor M, Zhang P, Adeyemo A, Kranias EG, et al. Overexpression of Hsp20 prevents endotoxin-induced myocardial dysfunction and apoptosis via inhibition of NF-kappaB activation. J Mol Cell Cardiol. 2009;47:382–90. doi: 10.1016/j.yjmcc.2009.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Islamovic E, Duncan A, Bers DM, Gerthoffer WT, Mestril R. Importance of small heat shock protein 20 (hsp20) C-terminal extension in cardioprotection. J Mol Cell Cardiol. 2007;42:862–9. doi: 10.1016/j.yjmcc.2007.01.002. [DOI] [PubMed] [Google Scholar]
- 25.Charron F, Nemer M. GATA transcription factors and cardiac development. Semin Cell Dev Biol. 1999;10:85–91. doi: 10.1006/scdb.1998.0281. [DOI] [PubMed] [Google Scholar]
- 26.Liang Q, Molkentin JD. Divergent signaling pathways converge on GATA4 to regulate cardiac hypertrophic gene expression. J Mol Cell Cardiol. 2002;34:611–6. doi: 10.1006/jmcc.2002.2011. [DOI] [PubMed] [Google Scholar]
- 27.Suzuki YJ, Evans T. Regulation of cardiac myocyte apoptosis by the GATA-4 transcription factor. Life Sci. 2004;74:1829–38. doi: 10.1016/j.lfs.2003.10.002. [DOI] [PubMed] [Google Scholar]
- 28.Suzuki YJ, Nagase H, Day RM, Das DK. GATA-4 regulation of myocardial survival in the preconditioned heart. J Mol Cell Cardiol. 2004;37:1195–203. doi: 10.1016/j.yjmcc.2004.09.009. [DOI] [PubMed] [Google Scholar]
- 29.Mukhopadhyay D, Houchen CW, Kennedy S, Dieckgraefe BK, Anant S. Coupled mRNA stabilization and translational silencing of cyclooxygenase-2 by a novel RNA binding protein, CUGBP2. Mol Cell. 2003;11:113–26. doi: 10.1016/s1097-2765(03)00012-1. [DOI] [PubMed] [Google Scholar]
- 30.Sureban SM, Murmu N, Rodriguez P, May R, Maheshwari R, Dieckgraefe BK, et al. Functional antagonism between RNA binding proteins HuR and CUGBP2 determines the fate of COX-2 mRNA translation. Gastroenterology. 2007;132:1055–65. doi: 10.1053/j.gastro.2006.12.031. [DOI] [PubMed] [Google Scholar]
- 31.Kim VN, Han J, Siomi MC. Biogenesis of small RNAs in animals. Nat Rev Mol Cell Biol. 2009;10:126–39. doi: 10.1038/nrm2632. [DOI] [PubMed] [Google Scholar]
- 32.Xu C, Lu Y, Pan Z, Chu W, Luo X, Lin H, et al. The muscle-specific microRNAs miR-1 and miR-133 produce opposing effects on apoptosis by targeting HSP60, HSP70 and caspase-9 in cardiomyocytes. J Cell Sci. 2007;120:3045–52. doi: 10.1242/jcs.010728. [DOI] [PubMed] [Google Scholar]
- 33.Bolli R, Shinmura K, Tang XL, Kodani E, Xuan YT, Guo Y, et al. Discovery of a new function of cyclooxygenase (COX)-2: COX-2 is a cardioprotective protein that alleviates ischemia/reperfusion injury and mediates the late phase of preconditioning. Cardiovasc Res. 2002;55:506–19. doi: 10.1016/s0008-6363(02)00414-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Papanicolaou KN, Streicher JM, Ishikawa TO, Herschman H, Wang Y, Walsh K. Preserved heart function and maintained response to cardiac stresses in a genetic model of cardiomyocyte-targeted deficiency of cyclooxygenase-2. J Mol Cell Cardiol. 2010;49:196–209. doi: 10.1016/j.yjmcc.2010.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Streicher JM, Kamei K, Ishikawa TO, Herschman H, Wang Y. Compensatory hypertrophy induced by ventricular cardiomyocyte-specific COX-2 expression in mice. J Mol Cell Cardiol. 2010;49:88–94. doi: 10.1016/j.yjmcc.2010.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Penna C, Mancardi D, Tullio F, Pagliaro P. Postconditioning and intermittent bradykinin induced cardioprotection require cyclooxygenase activation and prostacyclin release during reperfusion. Basic Res Cardiol. 2008;103:368–77. doi: 10.1007/s00395-007-0695-7. [DOI] [PubMed] [Google Scholar]
- 37.Gres P, Schulz R, Jansen J, Umschlag C, Heusch G. Involvement of endogenous prostaglandins in ischemic preconditioning in pigs. Cardiovasc Res. 2002;55:626–32. doi: 10.1016/s0008-6363(01)00505-3. [DOI] [PubMed] [Google Scholar]
- 38.Inserte J, Molla B, Aguilar R, Través PG, Barba I, Martín-Sanz P, et al. Constitutive COX-2 activity in cardiomyocytes confers permanent cardioprotection Constitutive COX-2 expression and cardioprotection. J Mol Cell Cardiol. 2009;46:160–8. doi: 10.1016/j.yjmcc.2008.11.011. [DOI] [PubMed] [Google Scholar]
- 39.Camitta MG, Gabel SA, Chulada P, Bradbury JA, Langenbach R, Zeldin DC, et al. Cyclooxygenase-1 and -2 knockout mice demonstrate increased cardiac ischemia/reperfusion injury but are protected by acute preconditioning. Circulation. 2001;104:2453–8. doi: 10.1161/hc4401.098429. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.