

Abstract
Werner syndrome (WS) is a rare, segmental progeroid syndrome caused by defects in the WRN gene, which encodes a RecQ helicase. WRN has roles in many aspects of DNA metabolism including DNA repair and recombination. In this study, we exploited two different recombination assays previously used to describe a role for the structure-specific endonuclease ERCC1-XPF in mitotic and targeted homologous recombination. We constructed CHO cell lines isogenic with the cell lines used in these previous studies by depleting WRN using shRNA vectors. When intrachromosomal, mitotic recombination was assayed in WRN depleted CHO cells, a hyperrecombination phenotype was observed, and a small number of aberrant recombinants was generated. Targeted homologous recombination was also examined in WRN depleted CHO cells using a plasmid-chromosome targeting assay. In these experiments, loss of WRN resulted in a significant decrease in nonhomologous integration events and ablation of recombinants requiring random integration of the corrected targeting vector. Aberrant recombinants were also recovered, but only from WRN depleted cells. The pleiotropic recombination phenotypes conferred by WRN depletion, reflected in distinct homologous and nonhomologous recombination pathways, suggest a role for WRN in processing specific types of homologous recombination intermediates as well as an important function in nonhomologous recombination.
Keywords: Werner syndrome, homologous and nonhomologous recombination, ERCC1
1. INTRODUCTION
Werner syndrome (WS) is a rare, autosomal progeroid syndrome caused by defects in Werner helicase (WRN), a member of the RecQ family of DNA helicases, five of which exist in humans (reviewed in Brosh, 2002, 2007; Bohr, 2008). WS patients display many phenotypes associated with aging sooner than the normal population, including wrinkled skin, graying of the hair, cataracts, stooped posture, diabetes and arthritis. WS patients typically begin to show these aging phenotypes after puberty and usually die in the fifth decade of life. In addition to accelerated aging, these patients have an increased risk of cancer, in particular for sarcomas (Martin, 1978; Salk, 1982; Martin et al., 1999). Cells from WS patients grow more slowly, senesce earlier, and are hypersensitive to a variety of DNA damaging agents (Martin et al., 1970; Ogburn et al., 1997; Poot et al., 1992, 1999). WRN has been shown to interact with numerous proteins and is thought to function in multiple pathways of DNA metabolism including replication, transcription, telomere maintenance, and DNA repair and recombination (Brosh, 2002, 2007). The exact mechanisms of action of WRN in each of these pathways are areas of active study.
With respect to its roles in recombination, WRN has been implicated in homologous recombination mainly through studies of ectopically integrated gain-of-function recombination reporter constructs in cells from WS patients. When such a substrate was used to measure spontaneous and cis-platinum (cisPt)-induced homologous recombination, the recovery of recombinants was suppressed in WRN deficient cells (Prince et al., 2001; Saintigny et al., 2002; Swanson et al., 2004; Dhillon et al., 2007). The study by Prince et al. (2001), which measured spontaneous mitotic recombination, concluded that while WS and control cells initiated mitotic recombination at similar rates, WRN deficient cells were defective in successfully resolving recombination intermediates into products. This study also analyzed some of the few recombinants obtained from spontaneous, intrachromosomal mitotic recombination and reported a significant increase in the proportion of crossover-type recombination events in WRN deficient cells relative to control cell lines. However, since there is substantial evidence that WRN suppresses homologous recombination at early stages (Baynton et al., 2003; Cheng et al., 2006; Bachrati et al., 2008; Franchitto et al., 2008) the absence of WRN would be predicted to lead to increased HR and a hyperrecombinogenic phenotype. Chen et al. (2003) were able to demonstrate modest, but statistically significant, increases in extrachromosomal homologous recombination in isogenic hTERT-immortalized WS cells complemented with either helicase- or exonuclease-deficient mutant WRN proteins relative to wild-type controls. Curiously, however, isogenic cells not expressing WRN protein at all reported lower HR than cells complemented with wild-type WRN or either mutant WRN protein, and cells expressing a mutant WRN protein lacking both helicase and exonuclease activities demonstrated levels of recombination not statistically different from isogenic wild-type controls. These results were interpreted as suggesting that balanced exonuclease and helicase activities of WRN were required for normal HR, and that WRN played a structural role in addition to its enzymatic activities in optimizing HR.
The role of WRN in nonhomologous end-joining (NHEJ) was also addressed in this study (Chen et al., 2003) using the same isogenic panel of hTERT-immortalized WS cell lines. A V(D)J recombination assay measuring coding joining and signal joining showed that WRN was required for efficient NHEJ, and that both enzymatic activities of WRN contribute to optimal NHEJ. A function for WRN in NHEJ had been proposed previously based on biochemical studies demonstrating physical and functional interaction between WRN and the Ku heterodimer, which binds to broken ends where it may act to recruit WRN (Cooper et al., 2000; Walker et al., 2001). Functionally, WRN exonuclease activity is stimulated by interaction with Ku both in terms of its processivity and its ability to digest past adducts (Cooper et al., 2000; Li and Comai, 2000; Orren et al., 2001; Karmakar et al., 2002a). WRN may be phosphorylated by and interact with DNA-PKcs through its interaction with Ku (Karmakar et al., 2002b). FEN-1 activity is stimulated by WRN (Brosh et al., 2001), and WRN interacts with XRCC4-ligase IV resulting in a stimulation of its exonuclease activity, which may act to prepare a suitable substrate for XRCC4-ligase IV from noncohesive DNA ends (Kusumoto et al., 2008). These results, taken together, suggest a role for WRN in pathway choice between HR and NHEJ, possibly via competition for WRN recruitment (Li et al., 2002; Baynton et al., 2003; Cheng et al., 2004). WRN may also play a structural role in NHEJ by regulating protein-protein interactions (Chen et al., 2003).
In addition to its roles in recombination, WRN has been implicated in the repair of DNA interstrand crosslinks (ICLs), consistent with the hypersensitivity of WS cells to agents that induce ICLs (Poot et al., 2001, 2002). Zhang et al. (2005) demonstrated a requirement for WRN in an in vitro crosslink repair assay by immunodepletion of cell extracts and complementation of repair by recombinant WRN protein, showing a specific protein-protein association of WRN with the Cdc5L component of the Pso4 complex required for psoralen ICL repair. From results generated using this system the authors concluded that WRN was necessary to stabilize the ICL repair intermediate through bubble formation, which facilitated recruitment of ERCC1-XPF to the site of the ICL. It is of interest that for the recently described progeroid syndrome XFE, clinically associated with some defective alleles of XPF and observed in nullizygous ERCC1 mice, it has been suggested that failure to efficiently repair endogenously generated ICLs and the accumulation of unrepaired lesions may be sufficient to explain the premature aging observed as well as other aspects of its complex phenotype (Neidernhofer et al., 2006). Since it is well established that homologous recombination is critically involved in ICL repair in mammalian cells (reviewed in Dronkert and Kanaar, 2001; Li and Heyer, 2008; Hinz, 2010), we wished to investigate the role of WRN in a system that would allow a direct comparison of WRN and ERCC1 function in recombination.
In the work reported here, stable “knock-down” Chinese hamster ovary (CHO) cell lines depleted for WRN by shRNA were constructed and used to examine the role of WRN in homologous recombination. Two different recombination assays were utilized: a direct repeat recombination reporter system for interrogating intrachromosomal homologous recombination (i.e., mitotic recombination), and a plasmid-chromosome gene targeting recombination system for analyzing a strand invasion recombination pathway. These recombination assays were previously used to demonstrate the function of ERCC1 in homologous recombination (Sargent et al., 1997; Adair et al., 2000). Results from these experiments, as well as others (Sargent et al., 2000; Neidernhofer et al., 2001), definitively implicated ERCC1 as required for both mitotic and targeted homologous recombination pathways. ERCC1 null cells were deficient in generating crossover-type and crossover with co-conversion recombination products; also, aberrant recombinants with deletions and rearrangements, not recovered at all in wild-type cells, represented a large proportion of the recombinants generated (Sargent et al., 1997; Adair et al., 2000). Using stable CHO WRN knock-down cell lines isogenic with those used in these previously reported studies of ERCC1, our results indicate that WRN depletion results in pleiotropic effects on recombination, and demonstrate that WRN deficient cells are hyperrecombinogenic for mitotic (i.e., intrachromosomal) homologous recombination, but not for targeted homologous recombination. Additionally, WRN deficient cells exhibited unique recombination phenotypes in both recombination assays, suggesting a role for WRN in resolving specific types of homologous recombination intermediates. Our results indicate that WRN may be involved in pathway choice for the resolution of mitotic recombination intermediates, and confirm that it is also required for efficient nonhomologous recombination.
2. MATERIALS AND METHODS
2.1. Cloning and sequencing of CHO WRN cDNA
Construction of stable WRN knock-down cell lines in CHO cells required hamster WRN cDNA sequence to design shRNAs. Therefore, we compared complete mRNA sequences for human (GenBank accession number AF091214) and mouse (GenBank accession number AF091215) WRN to design degenerate primers for amplifying overlapping sections of sequence covering the first 3 kb of coding sequence (total coding sequence 4373 bp). PCR products were amplified using cDNA from CHO cell line ATS-49tg, cloned (TA cloning system, Invitrogen) and sequenced. Target sequence for shRNA molecules was selected based on previously synthesized shRNA sequences specific for human WRN mRNA (from R. Legerski, U.T. M.D. Anderson Cancer Center, Houston TX). A second set of CHO-specific primers was designed to cover the areas selected for shRNA targeting using the obtained sequence and PCR products were again amplified from CHO cDNA, cloned and sequenced. This verified sequence was used to generate the shRNA sequences specific for targeting CHO WRN mRNA (see Figure 1A).
Fig. 1.
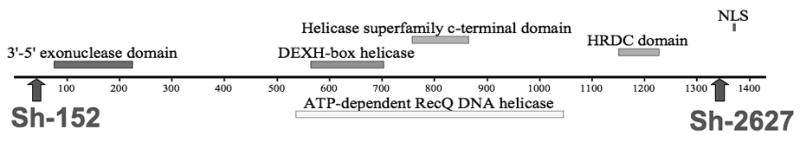
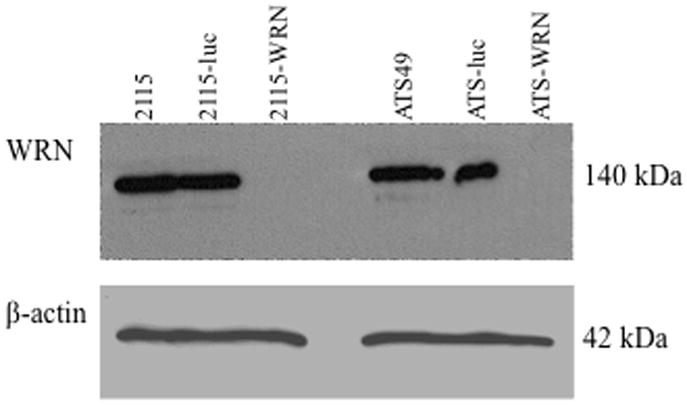
shRNA knock-down of CHO WRN at the protein level. (A) Diagram of WRN protein with conserved protein domains identified by boxes above and below the line. The locations of the two shRNA sequences are indicated by arrows. Sh-152 was used in the pSilencer-puro vector and was transfected into CHO cell lines first. Sh-2627 was cloned into the hygromycin version of the vector. Maximum knock-down of CHO WRN was achieved by sequential transfection of both shRNA vectors. (B) Western blot demonstrating knock-down in parental, control and WRN deficient cell lines. WRN was depleted in two CHO cell lines: GS21-15 and ATS-49tg to generate lines 2115-WRN and ATS-WRN respectively. A shRNA to luciferase was transfected into the same two CHO cell lines to generate non-specific shRNA control cell lines 2115-luc and ATS-luc.. βactin served as a loading control antibody.
2.2. Cell lines and culture conditions
Two CHO cell lines were used to derive control and WRN deficient cell lines: GS21-15 (Sargent et al., 1997) and ATS-49tg (Adair et al., 1989). GS21-15 was derived from ATS-65tg, a CHO cell line hemizygous for APRT and containing a spontaneous inactivating transversion mutation abolishing an EcoRV restriction site in exon 2 (Merrihew et al., 1995). Gene targeting followed by selection for “pop-out” recombinants was used to insert a yeast FLP recombination target (FRT) in intron 2 generating cell line RMP41 from ATS-65tg. The final APRT duplication in GS21-15 was created by utilizing the site-specific FLP recombination system to target the vector pGS73 as described (Merrihew et al., 1995). ATS-49tg is also hemizygous for APRT but contains an spontaneous, inactivating 3-bp deletion eliminating a MboII restriction site in exon 5 (Smith and Adair, 1996).
WRN was depleted in GS21-15 and ATS-49tg using shRNA. The same vector backbone with two different selectable markers (pSilencer-puromycin and -hygromycin, Ambion) was used with two different shRNA duplexes (IDT, Inc.) with homology to the 5′ region beginning at base 152 (AGCTTTTCCAAAAAATGGGTCCATCGTTTACAGTTATCTCTTGAATAACTGTA AACGATGGACCG) and the 3′ region beginning at base 2627 (AGCTTTTCCAAAAAACTAGGCATCACTTTCTTGATCTCTTGATCTCTTGAATC AAGAAAGTGATGCCTAGCG) of the CHO WRN cDNA sequence. The shRNA vectors were transfected sequentially (FuGENE, Roche) and independent puromycin- and then hygromycin-resistant clones were selected, expanded into larger cultures and ultimately screened for WRN depletion by western blot. The pSilencer-puromycin vector was used again with an shRNA duplex sequence to the firefly luciferase gene (AGCTTTTCCAAAAAAACGGATTACCAGGGATTTCTCTCTTGAAGAAATCCCT GGTAATCCGTG) to generate isogenic control cell lines (designated 2115-luc and ATS-luc) for off-target effects. Independent clones of 2115-luc and ATS49-luc were screened by transfection with pGL2-Luc (FuGENE, Roche) and measurement of luciferase expression using the luciferase assay (Promega) to verify shRNA function. shRNA duplexes were designed as required by the Ambion system to contain a HindIII overhang on the 5′ end and a BamHI overhang on the 3′ end to facilitate cloning into the pSilencer vector.
All cell lines were maintained as monolayers in alpha-modified minimal essential medium (α-MEM), containing 2mM L-glutamine, penicillin (50 U/ml), streptomycin (50 μg/ml) and 10% fetal bovine serum, in a 37°C incubator (5% CO2/95% air). 2115-luc and ATS-luc lines were maintained in the this media with the addition of 7 μg/ml puromycin (Sigma). 2115-WRN and ATS-WRN were maintained with the addition of 50 μg/ml hygromycin (EMD) and 7 μg/ml puromycin.
2.3. Growth curve and cisPt survival
Growth characteristics were analyzed by plating each cell line (2115-luc, 2115-WRN, ATS-luc, ATS-WRN) in 60-mm dishes in triplicate for each time point (0, 4, 5, 6, 7, 8 days) with 20,000 cells per dish. After 2 hours, the zero time point plates were harvested and the cells counted using a Coulter Counter (Beckman Coulter Z4). All other plates were harvested and counted on the specified day.
CisPt sensitivity was assayed by clonogenic survival. Each cell line (2115-luc, 2115-WRN, ATS-luc, ATS-WRN) was plated to 100-mm dishes, and after two days media was removed and the appropriate dose of cisPt (in serum-free media) was added for 1 hour. Each plate was then harvested and a specific number of cells plated in triplicate in standard media (supplemented with appropriate selection). After 7–9 days, plates were fixed with methanol, stained with crystal violet, and colonies counted to determine survival. Each cell line was assayed twice.
2.4. Western blotting
Transformed clones were screened by immunoblotting to choose the best candidate for further experiments. Protein extracts were prepared from cell pellets by lysis in RIPA buffer (150 mM NaCl, 50 mM Tris, 1 mM EDTA, 1% NP40) for 10 minutes at 4°C followed by centrifugation to pellet cellular debris. Concentrations were determined by BCA assay (Pierce). SDS-PAGE was performed on 30 μg protein extracts in 7.5% SDS-acrylamide gels. After electrophoresis and transfer to PVDF membrane, blots were blocked for 1 hour with 5% dry milk in TTBS and primary antibody was added and incubated for 16–18 hours at 4°C. WRN primary antibody (Santa Cruz Biotech) was used at 1:800 dilution and the loading control β-actin primary antibody (Abcam) was used at 1:5000 dilution. Appropriate HRP-conjugated secondary antibody (Sigma) was used at 1:2000–1:5000 dilution and detection was performed with Pierce Pico West detection reagents (Pierce).
2.5. Intrachromosomal recombination assay
2.5.1. Fluctuation experiments
Cell lines derived from GS21-15 (2115-luc and 2115-WRN) were used in the intrachromosomal mitotic recombination assay. Fluctuation assays were performed to determine recombination rates in these cell lines. For each experiment, 12 independent populations were seeded by plating ~ 50 cells into the wells of a 12-well plate in non-selective media. After 12–14 days or when the wells were nearly confluent, each population was transferred to a T-75 flask. The cells were allowed to grow for 5 days at which point each population was again transferred to a 150-mm dish and allowed to grow 2 more days to reach near confluency. Each population was then trypsinized into 20 ml non-selective media and passed through a 23-gauge needle before counting. APRT−/TK− cells were selected by plating 1×106 cells in ten 100-mm dishes containing media with 10% dialyzed fetal bovine serum supplemented with 0.4 mM 8-azaadenine (8AA) and 1μM ganciclovir (Ganc) and appropriate selective agents (puromycin for 2115-luc or puromycin+hygromycin for 2115-WRN). APRT− cells were selected by plating 1×106 cells in five 100-mm dishes containing 0.4 mM 8-azaadenine (8AA) and the appropriate selection (puro or puro+hygro). Plating efficiency was determined by plating 200 cells in four 60-mm plates containing media with 10% dialyzed fetal bovine serum and puro or puro+hygro. Plating efficiency plates were fixed and stained after 8 days. Selection plates were examined after 12–14 days and two independent colonies per population per selection were picked into T-25 flasks containing media with appropriate selection (puro or puro+hygro). After colonies were picked, all plates were fixed and stained.
2.5.2. Computational determination of recombination rate
Recombination rates were calculated using the Ma-Sandri-Sarkar Maximum Likelihood Estimator (MSS-MLE) originally conceived by Sarkar et al. (1992) and refined by Ma et al. (1992). This method is recognized as the most robust method for estimation of m (number of mutations per population) as it utilizes all the data from the fluctuation experiment, not just the median or mean, and is valid over all values for r and m (Foster, 2006). Additionally, this method allows for statistical comparisons of data (Stewart, 1994). Although fluctuation analysis was originally designed to determine mutation rates, the method can be used to calculate recombination rates after removing populations whose representative clone was determined to arise by random point mutation from the analysis. A web-based computer algorithm (FALCOR) was used to compute m with this method (Hall et al., 2009). Using FALCOR, the total colony count (r) per population per selection was used to generate an m value for each population. Plating efficiency for each population was used to compute the viable cells per population (PE * total number of cells harvested) and the viable cells per selection (PE * number of cells plated * number of plates) per population. These values were used to calculate the fraction of the culture plated (viable cells per selection/viable cells per population), which corrects for the sampling and plating efficiency variations between populations (Jones, 1993; Stewart et al., 1990; Foster, 2006). Then the corrected m (mact) was calculated for each population per selection using the equation mact = mobs * (z−1)/zln(z) where z=fraction of the culture plated.
Stewart (1994) reported that when m was obtained with the MSS-MLE method, ln(m) is normally distributed. Therefore, 95% confidence limits can be calculated using the t distribution: ln(m) ± tσ, where σ is the estimated standard deviation calculated by the equation: σ =1.225 * antilog of ln(m)−0.315/√C and t is obtained at α(2) = 0.05 and (C-1) degrees of freedom (Rosche and Foster, 2000) with C = number of populations. The value of ln(m) and antilog of ln(m) is now the average of all populations within the selection. Using these methods, an estimate of the recombination rate (μ) can be calculated using the equation (specific for asynchronous populations): μ = 0.6932 * m/N, where m is the antilog of ln(m) averaged for all populations within the selection and N is the average total number of viable cells in the populations. Confidence limits for μ are applied by substituting the values for ln(m) ± tσ into the above equation for μ.
2.5.3. Analysis of recombination products
Two independent colonies per population per selection were picked although only one was used for subsequent genetic analysis while the other was reserved in case of loss of the first clone. All clones were maintained in culture in T-75 flasks until each was nearly confluent. Genomic DNA was prepared from cell pellets (Promega Wizard SV Genomic DNA Purification kit). Southern analysis was performed to determine the structures of the APRT recombinants using a fragment containing the entire APRT gene as probe. Each clone was digested with BamHI, BamHI/HindIII and HindIII (New England Biolabs) before Southern blotting, and diagnostic restriction patterns were used to determine recombinant structures (see Sargent et al. 1997 for descriptions of diagnostic patterns used for determining recombinant types). Any clone determined to have the pattern of a gene conversion (i.e., retaining the original heteroallellic APRT duplication) was further tested by digestion with EcoRV and Southern blotting to verify conversion of the EcoRV site in the downstream APRT allele. If any clone retained the EcoRV restriction site, it was assumed to have arisen by a random mutation elsewhere in APRT and was discarded from the analysis of recombination rates.
2.6. Plasmid-chromosome targeted recombination assay
The gene correction targeting system for hamster APRT and targeting vector pAG6ins0.9 have been described previously (Adair et al., 2000). Cells (ATS-luc and ATS-WRN) were electroporated with SalI linearized pAG6ins0.9 as previously described (Adair et al., 2000; Nairn and Adair, 2006) and APRT+ recombinants were selected by plating into ALASA media prepared by supplementing α-MEM + 10% FBS with 25 μM alanosine, 50 μM azaserine, and 100 μM adenine. A small subset of cells was plated in HAT (α-MEM + 10% FBS containing 100 μM hypoxanthine, 2 μM amethopterin, 50 μM thymidine) to select for non-targeted integration events arising from nonhomologous recombination (i.e., GPT+ clones). Another small subset of cells was plated into non-selective media to determine plating efficiencies (PE). All media was supplemented with appropriate selection agents for maintaining shRNA vectors (either puro for ATS-luc or puro+hygro for ATS-WRN). After 10–12 days, PE and HAT plates were fixed and stained. ALASA plates were examined 14–16 days after plating and all colonies picked into individual flasks and maintained in selection media. All clones were maintained in culture until each was nearly confluent in a T-75 flask. Genomic DNA was prepared from cell pellets (Promega Wizard SV Genomic DNA Purification kit). Southern blotting using a fragment containing the entire APRT gene as probe was used to determine the structure of each recombinant at the APRT locus, based on diagnostic restriction digest patterns as previously described (Adair et al., 1989, 2000; Nairn and Adair, 2006).
3. RESULTS
3.1. WRN deficient cell lines exhibit slow growth and cisPt sensitivity
Stable WRN deficient cell lines and isogenic wild-type control cell lines were generated by shRNA vector knockdown in two CHO parental cell lines. Two shRNA vectors with shRNA sequences targeting different regions of the CHO WRN gene were used in combination to generate the WRN deficient lines (Figure 1A), while control cell lines were generated by transfection of a shRNA vector bearing the shRNA to luciferase (non-specific control). It was necessary to stably transform the WRN deficient cell lines with two shRNA vectors sequentially because initial transformation with the pSilencer-Puro WRNsh-152 vector resulted in only about 60% depletion of WRN protein as determined by immunoblotting (data not shown). The GS21-15-derived cell lines contain an intrachromosomal recombination reporter substrate at the endogenous APRT allele and were used for mitotic recombination (intrachromosomal homologous recombination) assays (2115-luc and 2115-WRN). The same shRNA vectors were used to generate stably transformed cell lines showing significant WRN depletion and a wild-type control by transformation of CHO cell line ATS-49tg, and these derived cell lines were used in plasmid-chromosome targeted gene correction recombination assays (ATS-luc and ATS-WRN).
Results of western blots (Fig. 1B) show nearly complete knockdown of WRN protein in both WRN deficient cell lines stably transformed with the two shRNA vectors. Both WRN deficient cell lines (2115-WRN and ATS-WRN) had slower growth characteristics compared to their respective isogenic controls. This difference was more pronounced in the ATS-49tg-derived cell lines where the WRN deficient cell line (ATS-WRN) had 30% fewer cells than the control cell line (ATS-luc) at days 5 and 6 before reaching maximal density. The 2115-derived WRN deficient cell line lagged behind its isogenic control at 5 days by 30% but only by 10% at 6 days before reaching maximal density (data not shown).
The WRN deficient cell lines were tested for sensitivity to the crosslinking compound cis-platinum (cisPt). As shown in Figure 2, both WRN deficient lines (light bars) were significantly more sensitive to cisPt than their isogenic control lines (dark bars) although the sensitivity was more pronounced in the ATS-49-derived line. These data are consistent with previously reported results documenting a slow growth phenotype and sensitivity to cisPt in WRN deficient cells, both for primary cells from WS patients and SV40 immortalized lines (Poot et al., 2001; Saintigny et al., 2002; Dhillon et al., 2007).
Fig. 2.
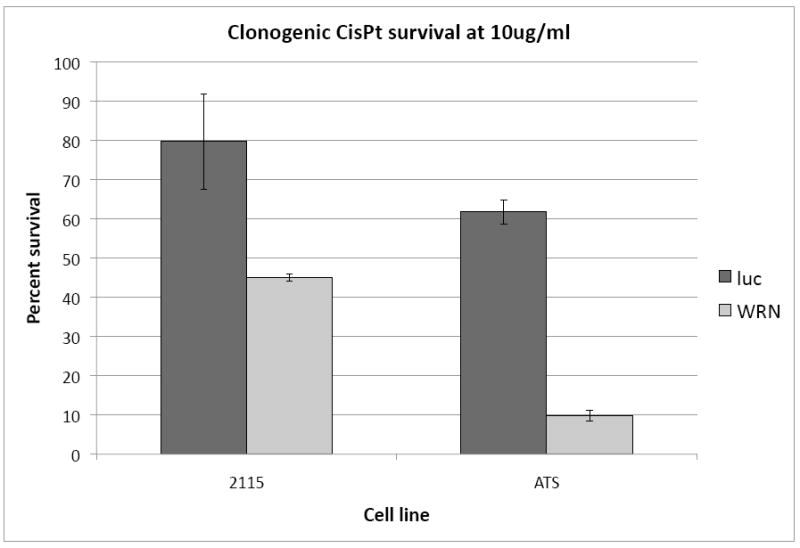
Sensitivities of control and WRN deficient cell lines to 10μg/ml cisplatin (CisPt). Clonogenic survival assays were performed on control (dark bars) and WRN deficient (light bars) cell lines from both CHO backgrounds: GS21-15 and ATS-49tg. Error bars indicate standard deviations.
3.2. WRN deficient cells are hyperrecombinogenic for intrachromosomal mitotic recombination and exhibit an abnormal distribution of recombination events
The intrachromosomal mitotic recombination assay was performed using a recombination substrate consisting of a partial tandem duplication fixed at the endogenous APRT locus in the CHO cell line GS21-15 (Figure 3A). The upstream copy of APRT is inactivated by a point mutation resulting in the loss of a unique EcoRV restriction site in exon 2, and a deletion of a portion of exon 5, whereas the downstream copy of APRT is complete and active. Both copies of APRT contain the FLP recombinase target (FRT) sequence in an intronic region of the gene that does not affect APRT function (Merrihew et al., 1995). Two selectable markers, TK and GPT, are located between the two APRT heteroallelic duplications.
Fig. 3.

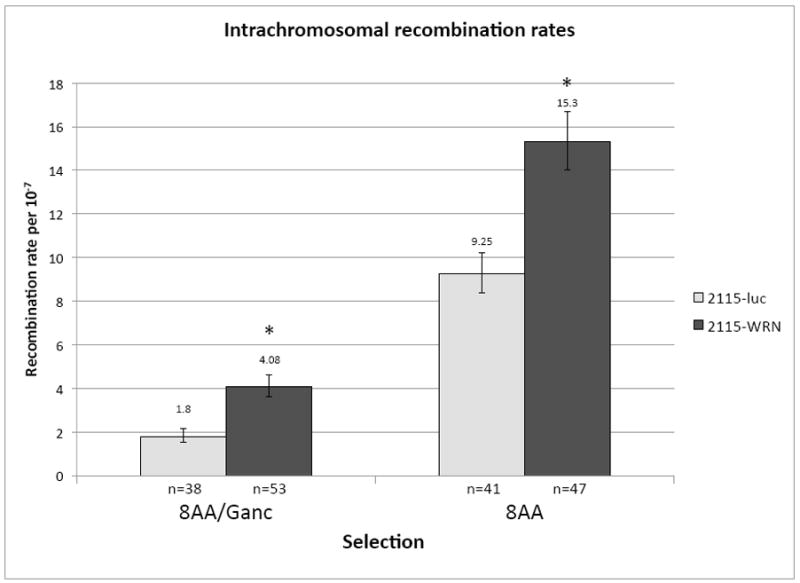
Intrachromosomal recombination assay. (A) Diagram of endogenous recombination substrate fixed at the endogenous APRT locus in GS21-15-derived CHO cell lines. The upstream copy of APRT is inactivated by a transversion mutation in exon 2 abolishing an EcoRV restriction site (thick black vertical line) and a deletion of the terminal portion of exon 5 (black diamond). The downstream copy of APRT is wild-type and retains the EcoRV restriction site (RV). Two selectable markers lie between the APRT copies: TK (checked box) and GPT (grey box). Sizes of regions are shown below. Chromosomal sequence is denoted by thick lines while the thin line between the upstream APRT and TK indicates sequence retained from the targeting plasmid used to generate this construct. Various restriction sites used to distinguish recombination products are also indicated (H: HindIII, B: BamHI, RV: EcoRV). (B) Recombination rates using intrachromosomal recombination assay for control (light bars) and WRN deficient (dark bars) GS21-15-derived cell lines. Recombination rates were calculated using fluctuation analysis and the MSS-MLE method. Error bars indicate 95% confidence limits. 8AA/Ganc was used to select for APRT−/TK− and 8AA to select for APRT− recombinants. The number of independent populations used is indicated below each bar. * indicates p<0.05.
As a loss-of-function assay, this recombination reporter system allows facile detection of a wide range of outcomes and can easily distinguish conservative gene conversions from non-conservative recombination events such as crossovers, as well as revealing mutations and deletions. It is also fixed at the endogenous hemizygous APRT locus and thus is controlled for position effects. Plating cells into 8-azaadenine-containing media (8AA) selects for APRT− recombinants arising from either gene conversion or crossover-type events. Gene conversions arise from processing of recombination intermediates to result in the unidirectional transfer of the EcoRV mutation from the upstream copy of APRT to the downstream copy, retaining two APRT copies in recombinants. Crossover-type events (also called “pop-outs”) arising from single-reciprocal exchange (SRE) or from single-strand annealing (SSA) result in retention of only one copy of APRT in recombinants. Plating cells into 8-azaadenine + ganciclovir (8AA/Ganc) selects for only crossover-type events (APRT−/TK−), but these recombinants can present structures with or without co-conversion of the proximal GPT gene (Sargent et al., 1997).
Fluctuation analysis (as described in section 2.5.2) was used to determine recombination rates for the wild-type control and WRN deficient cell lines derived from isogenic GS21-15 CHO cells (i.e., 2115-luc and 2115-WRN) in both 8AA/Ganc and 8AA selections (Figure 3B). Populations whose representative clone was determined to have arisen by a random mutation were subtracted before rate calculations. The spontaneous recombination rates were significantly higher (p<0.05) in 2115-WRN relative to 2115-luc cells for both selections, indicating that in this intrachromosomal recombination assay depletion of WRN results in a hyperrecombination phenotype.
One independent clone isolated from each population in fluctuation experiments was expanded in culture and its isolated DNA was analyzed by Southern blotting to determine the structures of independent recombinants (Supplemental Figure 1). The distributions of recombinants generated in wild-type 2115-luc control cells were very similar to those from previously reported data for wild-type GS21-15 cells (Sargent et al., 1997). For APRT− clones selected in 8AA alone, gene conversions (GC) represented 77% (30/39) and crossover-type recombinants (XO) represented 23% (9/39) of all APRT− recombinants recovered from 2115-luc cells. These proportions compared to 83% (88/105) GCs and 16% XOs (17/105) in GS21-15 cells (Sargent et al., 1997). For APRT−/TK− clones selected in 8AA/Ganc, 100% of the 37 2115-luc and 72 GS21-15 recombinants recovered were XOs. APRT−/TK− recombinants were further classified into two groups: simple crossover-type events (XO) and crossovers accompanied by co-conversion of the adjacent GPT sequence (XO+GPT), thought to arise by processing of a heteroduplex intermediate formed when the upstream and downstream APRT alleles anneal during strand annealing (Sargent et al., 1997). In 2115-luc cells, 84% (31/37) of the APRT−/TK− recombinants were simple crossovers and 16% (6/37) were XO+GPT co-conversion with crossover recombinants, compared with 75% (51/68) simple crossovers and 25% (17/68) XO+GPT recombinants in GS21-15 cells.
The wild-type control cell line 2115-luc used in our study was derived by simple transfection of GS21-15 with a shRNA vector targeting firefly luciferase and is therefore isogenic to it. When we compared previously published results (Sargent et al., 1997) for recombinant distributions in GS21-15, obtained from analogous experiments of the same design, with our data for 2115-luc, the differences for each recombinant class were not statistically significant using a one-sided Fisher’s exact test. We therefore compared the recombinant distributions in WRN deficient 2115-WRN cells to the combined distributions of APRT− and APRT−/TK− recombinants generated in GS21-15 and 2115-luc, as presented in Table 1.
Table 1.
Distribution of recombination products obtained from spontaneous mitotic intrachromosomal recombination assay.
| 8AA selection (APRT−) | |||
|---|---|---|---|
| Events | Gene Structure | 2115-WRN | GS21-15a +2115-luc |
| Gene conversion (GC) | 27/47 (58%) | 118/144 (82%) | |
| Crossover (XO) | 18/47 (38%) | 26/144 (18%) | |
| Aberrant | 2/47 (4%) | 0/144 (0%) | |
| 8AA/Ganc selection (APRT−/TK−) | |||
| Events | Gene Structure | 2115-WRN | GS21-15a +2115-luc |
| Crossover (XO) | 49/54 (91%) | 82/105 (78%) | |
| Crossover with GPT co- conversion (XO+GPT) | 4/54 (7%) | 23/105 (22%) | |
| Aberrant | 1/54 (2%) | 0/105 (0%) | |
Recombination products for isogenic GS21-15 cell line as previously published (Sargent et al., 1997)
Results in Table 1 show that selection in 8AA alone generated both GC and XO APRT− recombinants in wild-type cells. In 2115-WRN cells, GCs were reduced from 82% to 58% of all recombinants generated, and XOs were increased from 18% to 38% of recombinants; in addition, 2 aberrant recombinants (4%) were recovered from 2115-WRN cells. These differences in recombinant distribution are statistically significant (by Fischer’s one-sided exact test) for the reduction in GC events (p < 0.001) and the increase in XO events (p = 0.005), and approach 95% confidence for statistical significance (p = 0.06) for generation of the aberrant recombinant class in the 2115-WRN cell line. For selection of APRT−/TK− recombinants using 8AA/Ganc, all recombinants in wild-type cells were XOs; however, XOs could be further classified as simple crossovers or crossovers accompanied by co-conversion of the GPT marker (XO+GPT). In wild-type cells, the proportion of XO+GPT recombinants represented 22% of all recombinants. By contrast, in 2115-WRN cells XO+GPT recombinants represented only 7% of recombinants (p = 0.02). In addition, one aberrant recombinant was obtained from 2115-WRN cell populations using this selection, representing 2% of recombinants; this was not statistically significant (p = 0.34).
3.3. WRN deficient cells exhibit an altered gene targeting phenotype and reduced nonhomologous recombination
In the experiments described above, interrogation of intrachromosomal homologous recombination between directly repeated APRT heteroalleles revealed a hyperrecombinogenic phenotype and some different recombination outcomes in WRN deficient cells, which were similar in some respects to those previously observed in ERCC1 null cells (Sargent et al., 1997). In particular, WRN deficiency resulted in aberrant recombinants not recovered from wild-type cells and also apparently affected the resolution of recombination intermediates containing a heteroduplex loop, leading to a significant change in the proportions of simple XO and XO+GPT recombinants for APRT−/TK− clones obtained from 2115-WRN populations. The heteroduplex loop intermediate proposed to lead to XO and XO+GPT recombinant classes (Sargent et al., 1997) is processed in a similar fashion to recombination intermediates requiring removal of terminal nonhomologous DNA sequences blocking the ends of homology in gene targeting vectors (Adair et al., 2000; Niedernhofer et al., 2001). Also, both types of intermediates require ERCC1-XPF for proper resolution (Sargent et al., 1997, 2000; Adair et al., 2000; Niedernhofer et al., 2001; Talbert et al., 2008). These considerations caused us to next examine the role of WRN in a plasmid-chromosome targeted gene correction assay at the CHO APRT locus using a targeting vector which presented to the cell such an end-blocked homologous recombination intermediate.
In this assay, the endogenous hemizygous APRT allele in the CHO cell line ATS-49tg is inactivated by a 3-bp mutation abolishing an MboII restriction site in exon 5. This allele is targeted by electroporation of the targeting vector (pAG6ins0.9) containing a complete copy of APRT interrupted by a ~ 900-bp insertion in exon 3 (Figure 4A). Upon linearization of pAG6ins0.9 in the inserted sequence in exon 3 and electroporation into ATS-49tg cells, successful plasmid-chromosome homologous recombination requires the precise removal of the lengthy end-blocking sequences from the homologous arms of the targeting vector. APRT+ clones, selected by plating cells into ALASA-containing media, can arise by gene conversion, targeted integration (single crossover after removal of terminal nonhomologies), or vector correction (removal of terminal nonhomologies followed by gap repair and random, nonhomologous integration of the repaired vector) as diagramed in Figure 4B. The targeting vector also contains the selectable GPT marker allowing for detection of non-targeted (i.e., nonhomologous) recombination events arising through NHEJ, by plating a small sample of electroporated cells into HAT containing media. Calculation of the ratio of frequencies of APRT:GPT recombinants gives a normalized gene targeting efficiency allowing comparison between cell lines and from experiment to experiment (Adair et al., 1989, 2000; Nairn and Adair, 2006).
Fig. 4.


Plasmid-chromosome targeted recombination assay. (A) Diagram of pAG6ins0.9 targeting vector. This vector contains a full-length copy of the CHO APRT gene (dark grey bar) interrupted by 945bp of heterologous sequence (black bar) at the XhoI site located in exon 3. The plasmid was linearized by digestion with SalI to generate an ends-in targeting configuration where 271nt and 670nt flank the double-strand break. The vector backbone contains a GPT allele used to select for non-targeted recombinatiton events. The target APRT locus (light grey) in ATS-49tg has an inactivating 3bp deletion mutation in exon 5, which destroys a MboII restriction site. Relevant restriction sites are shown: B-BamHI; Bg-BglII; RV-EcoRV; H-HindIII; Mb-MboII; S-SalI; X-XhoI. (B) Plating cells into ALASA media selects for APRT+ rcombinants. Three possible classes of recombinants are diagramed in addition to an aberrant class.
shRNA control and WRN deficient cell lines derived from ATS-49tg (i.e., ATS-luc and ATS-WRN respectively) were electroporated with SalI-linearized pAG6ins0.9 and frequencies of APRT+ recombinants and GPT+ transformants were calculated (Table 2). No significant difference in the APRT+ recombination frequency was observed between wild-type control ATS-luc and WRN deficient ATS-WRN cell lines (0.83 × 10−6 vs. 0.78 × 10−6); however there was a significant decrease (p = 0.014) in the frequency of GPT+ transformants generated in WRN deficient cells compared to wild-type control cells, indicating decreased efficiency of random integration by nonhomologous recombination in ATS-WRN cells. This decrease in GPT transformation frequency artificially drove the normalized APRT targeting efficiency higher in WRN deficient cells compared to the control (1.57 per 103 vs. 0.73 per 103 GPT integrations).
Table 2.
Frequencies of targeted recombination in control and WRN deficient cell lines after electroporation with the targeting vector pAG6ins0.9.
| Cell line | Total No. cells electroporated | Frequency of APRT+ recombinants (per 106) ± SEM | Frequency of GPT+ transfectants (per 104) ± SEM | Ratio of APRT+:GPT+ (per 103) | Clones obtained |
|---|---|---|---|---|---|
| ATS-WRN | 47.6 × 107 | 0.78 ± 0.36 | 4.95 ± 0.82 | 1.57 | 44 |
| ATS-luc | 40.8 × 107 | 0.83 ± 0.14 | 11.4 ± 2.0 | 0.73 | 45 |
The structures of the APRT alleles in each recombinant clone obtained from these experiments were analyzed by Southern blotting (45 clones from ATS-luc and 44 clones from ATS-WRN). The four potential recombination outcomes can be distinguished by specific restriction enzyme digestion patterns (Supplemental Fig. 2 and Adair et al., 2000; Nairn and Adair, 2006). Table 3 tabulates the proportions of each class of recombinant for control ATS-luc and WRN deficient ATS-WRN cell lines. Targeting of ATS-WRN and ATS-luc cells with pAG6ins0.9 yielded similar proportions of gene conversion and targeted integration events. However, no vector correction events were obtained from ATS-WRN cells in contrast to a small proportion (9%) in ATS-luc cells. Additionally, 9% of recombinants in ATS-WRN cells were determined to be aberrant integration/deletion events. These last two results were similar to those obtained from isogenic ERCC1 null cells where in the absence of ERCC1, no vector correction events were obtained and aberrant recombinants were recovered (Adair et al., 2000). However, unlike in ERCC1 null cells, aberrant targeted recombinants represented only a small proportion of the total.
Table 3.
Proportions of targeted recombination events with pAG6ins0.9
| Cell line |
APRT+ recombinants |
||||
|---|---|---|---|---|---|
| Total analyzed | Target gene conversions | Targeted insertions | Targeting vector corrections | Aberrant | |
| ATS-WRN | 44 | 25/44 (57%) | 15/44 (34%) | 0/44 (0%) | 4/44 (9%) |
| ATS-luc | 45 | 25/45 (56%) | 16/45 (39%) | 4/45 (9%) | 0/45 (0%) |
3.4. Characterization of aberrant APRT recombinant structures
Both in the experiments reported here, and in analogous experiments for ERCC1 null cell lines previously reported (Sargent et al., 1997; Adair et al., 2000), aberrant APRT recombinants were recovered from WRN or ERCC1 deficient but not wild-type cell lines in two different recombination assays. Although the proportions of aberrant recombinants recovered relative to total recombinants were substantially higher in ERCC1 deficient cells compared to WRN deficient cells, we analyzed the structures of recombinants from the WRN deficient cell lines to determine similarities and differences, which might reveal insights into the mechanisms by which they arose.
In 2115-WRN cells from 8AA/Ganc and 8AA selections, a small number (total of 3) of aberrant recombinants with small deletions or insertions were recovered (Table 1). The structures of these three aberrant recombinants obtained from 2115-WRN cells were deduced by Southern blotting (Supplemental Fig 3) and are shown in Figure 5A. The one aberrant recombinant obtained from the 8AA/Ganc selection was determined to have arisen as a crossover-type event with an insertion of 0.7kb near the 5′ end of the APRT gene. The two remaining aberrant recombinants were obtained after 8AA selection alone. One was determined to be a crossover-type event with a small deletion (0.2kb) outside the APRT gene, while the other was a gene conversion but with the majority of the GPT gene reversed in position with the 5′ end of the downstream APRT allele accompanied by small deletions in the area of the GPT insertion. This reversed position inactivated the downstream APRT heteroallele as well as GPT. In the analogous experiments with ERCC1 null cells, 15 of 33 aberrant recombinants obtained from both selections were analyzed in detail to determine their molecular structures. Of those analyzed, 14 were found to be deletions and one a more complex rearrangement event (Sargent et al., 1997).
Fig. 5.


Aberrant events recovered from WRN deficient cells. (A) Aberrant events recovered from mitotic recombination assay. Three aberrant events (one from 8AA/Ganc selection and two from 8AA selection) were recovered and the deduced structure of each is shown. (B) Structures of aberrant recombinants obtained in plasmid-chromosome targeted recombination. Four aberrant events were recovered from targeting WRN deficient ATS-WRN cells with pAG6ins0.9. The deduced structures of each are shown.
The deduced structures of the four aberrant recombinant clones recovered from the plasmid-chromosome targeted recombination assay in the ATS-WRN cells were determined by examination of Southern blots (Supplemental Fig 4) and are shown in Figure 5B. Two of the aberrant clones obtained arose from triplication events presumably resulting from dimerization of the targeting vector. In one clone, the upstream APRT was wild-type, the middle APRT gene retained the full nonhomologous insert sequence present in the targeting vector, and the downstream APRT retained only a portion of the nonhomologous insert and additionally had a small deletion encompassing the XhoI site. In the second event of this type, the downstream APRT was wild-type while the upstream APRT retained the full nonhomologous insert sequence and the middle copy had a portion of the GPT gene rearranged into the 5′ region of the APRT, similar to what was observed in one aberrant recombinant recovered from the mitotic recombination assay of 2115-WRN cells. Additionally, a large insertion was present between the first two APRT copies. The remaining two aberrant recombinants were targeted insertions with deletions: one with a small deletion (0.8kb) in the downstream APRT gene, and the other with a large deletion (~3.9kb) encompassing GPT and half of the upstream APRT gene. These aberrant events were similar to those obtained in the ERCC1 experiments, but unlike ERCC1 null derived aberrant recombinants where all deletions occurred in the downstream APRT allele (Adair et al., 2000), those obtained from WRN deficient cells had deletions in either upstream or downstream copies. One triplication event was recovered from ERCC1 null cells, but again the triplications obtained from WRN deficient cells differed slightly in that they contained deletions and insertions in addition to the triplication event.
4. DISCUSSION
In this study, we investigated the role of the WRN helicase in spontaneous intrachromosomal mitotic and targeted homologous recombination using assays previously developed in CHO cells to examine the role of ERCC1-XPF in recombination. Using shRNA vectors, we generated WRN deficient cell lines in the same parental CHO cell lines used in these previous studies (Sargent et al., 1997; Adair et al., 2000). Thus, the results reported here for WRN deficient cells are directly comparable to results previously obtained in the isogenic ERCC1 null cell lines and their common repair proficient parental lines. The two WRN deficient cell lines we constructed, 2115-WRN and ATS-WRN, each exhibited slight growth defects compared to their isogenic parental cell lines, and were also each more sensitive to the ICL-forming agent cisPt than isogenic controls, in agreement with previous experiments using WRN depleted human fibroblasts (Swanson et al., 2004; Dhillon et al., 2007) and lymphoblastoid cells from WS patients (Poot et al., 2001), consistent with a postulated role for WRN in ICL repair.
We first examined the role of WRN in spontaneous intrachromosomal recombination (i.e., mitotic recombination) using a recombination reporter previously used to investigate intrachromosomal recombination in isogenic wild-type and ERCC1 deficient CHO cell lines (Sargent et al., 1997). This reporter construct is fixed at the endogenous hamster APRT locus and thus controls for position effects; it also allows for loss-of-function selection, facilitating the detection of a wider range of endpoints than those detected in gain-of-function assays, including point mutations, deletions, and rearrangements. In this study, we report a hyperrecombination phenotype in the absence of WRN when spontaneous intrachromosomal mitotic recombination was assayed using this recombination reporter system. This hyperrecombinogenic intrachromosomal recombination phenotype is consistent with results from similar experiments in yeast Sgs1 and Srs2 mutants (Aguilera and Klein, 1988; Aboussekhra et al., 1992; Gangloff et al., 1994; Ira et al., 2003). Sgs1 is the sole RecQ helicase in budding yeast, and strains without this helicase display a hyperrecombination phenotype for both spontaneous and induced homologous recombination, indicating that this protein plays a role in suppressing homologous recombination (Watt et al., 1996; Friedl et al., 2001). Although not a true RecQ helicase, Srs2 has 3′–> 5′ helicase activity and also acts to suppress recombination and maintain genomic stability (Aguilera and Klein, 1988; Watt et al., 1996). Srs2 has been implicated in regulation of HR through removal of Rad51 from ssDNA (Krejci et al., 2003; Veaute et al., 2003), and Srs2 mutants exhibit increased frequencies of intrachromosomal recombination (Doe et al., 2000; Dupaigne et al., 2008). It is notable that Srs2 has been shown to have a strong antirecombinogenic function when nonhomologous ends must be removed by the endonuclease complex Rad1-Rad10 (Paques and Haber, 1997), analogous to the situation for the mammalian recombination pathways investigated in our study. To date no human homologue of Srs2 has been identified, although it has been suggested that one of the RecQ family helicases may perform similar functions in humans (Marini and Krejci, 2010). The mitotic hyperrecombination phenotype we observed in WRN deficient 2115-WRN cells is also consistent with the notion that WRN plays a role in suppressing homologous recombination in response to DNA damage and replication fork perturbation (Fukuchi et al., 1989; Cheng et al., 1990; Yamagata et al., 1998; Pichierri et al., 2001; Bachrati et al., 2008).
Our finding that the absence of WRN confers a hyperrecombinogenic intrachromosomal recombination phenotype contrasts with the suppression of intrachromosomal recombination reported by Prince et al. (2001) using a different mitotic recombination assay in SV-40 immortalized WS patient fibroblasts. In their study, the authors concluded that WRN deficient cells exhibited a recombination-dependent reduction in colony formation due to the non-viability of gene conversion recombinants; this reduction in viable recombinants was suggested to be responsible for > 20-fold reduction in the rate of mitotic recombination in WS-derived cell lines relative to controls. The ectopically integrated intrachromosomal recombination substrate used in their study, pNeoA, was designed as a gain-of-function recombination reporter and is restricted to generating recombinants dependent on a limited region (592 bp) of perfect homology shared by the two NEO heteroalleles. Our recombination reporter, on the other hand, is a loss-of-function substrate reporting a wider range of events, and offers 3 regions of perfect homology ranging from 0.6 – 4.5 kb (Sargent et al., 1997). This intrachromosomal recombination substrate is capable of generating viable conversion and crossover-type recombinants in WRN deficient cells as evidenced by our results. Furthermore, the characteristics of (i) the substrate residing at a fixed location in the genome, as opposed to the ectopic pNeoA substrate, and (ii) the ability to use isogenic cell lines as controls, greatly strengthens comparisons of WRN deficient cells to wild-type controls in our experiments.
When we examined the structures of the recombinants obtained from our intrachromosomal recombination experiments, we observed a significant shift toward recombination outcomes that resulted in crossover-type events as opposed to conversions in the absence of WRN (Table 1). This result is broadly consistent with conclusions of Prince et al. (2001) using pLrec, a different ecoptic reporter for intrachromosomal recombination than the pNeoA recombination substrate discussed above, although the actual proportions of recombinants we observed were different from their results. Their analysis of independent recombinants showed that 22% (2/9) WS cell-derived recombinants were conversions compared to 69% (11/16) from control cells; crossover-type “pop-out” recombinants arising from SSA or SRE, and unequal SCE recombinants accounted for 78% (7/9) of recombinants from WRN deficient cells and 31% (5/16) from control cells. By comparison, our results shown in Table 1 indicate that crossover-type recombinants increased significantly (p=0.005) from 18% (26/144) in isogenic wild-type cell lines to 38% (18/47) in WRN deficient 2115-WRN cells, while conversions decreased significantly (p<0.001) from 82% (118/144) in control cell lines to 58% (27/47) in 2115-WRN cells. We conclude from our data that WRN acts to suppress formation of crossover-type recombinants in favor of gene conversion recombinants either by inhibiting formation of SSA intermediates in a similar fashion to Srs2 activity in yeast (Paques and Haber, 1997), or by promoting the stability of strand invasion intermediates and facilitating branch migration analogous to Sgs1 activity (Bennett et al., 1999; Ashton and Hickson, 2010). Since WS cells exhibit translocations, deletions, and rearrangements (Fukuchi et al., 1985; 1989; Gebhart et al., 1988), such an activity for WRN in suppressing crossover-type recombinant formation may contribute to maintaining genomic stability.
Results in Table 1 also show that when selection against both APRT and TK was applied (8AA/Ganc selection), restricting selection to crossover-type recombinants, we were able to classify these recombinants into simple crossovers and crossovers with accompanying co-conversion of the GPT cassette 5′ to the downstream APRT heteroallele (Fig. 3A). Our results (Table 1) indicate a significant (p = 0.02) decrease in the generation of XO+GPT recombinants, from 22% (23/105) in wild-type control cells to 7% (4/54) in 2115-WRN cells. Although this phenotype is less pronounced in WRN deficient compared to ERCC1 deficient cells, which showed a decrease from 25% (17/68) in wild-type cells to 4% (1/24) in ERCC1 null cells (Sargent et al., 1997), this shared recombination deficiency phenotype suggests that WRN and ERCC1-XPF may participate in a common pathway for resolving these types of recombination intermediates. WRN may have an earlier role in heteroduplex processing compared to the demonstrated role for ERCC1-XPF in resolving nonhomologous sequences, perhaps acting to stabilize the heteroduplex loop. Sgs1 has been shown to unwind heteroduplex recombination intermediates that contain mismatches (Spell et al., 2004) and WRN acting in a similar manner may lead to stabilization of the relatively large heteroduplex loop long enough for ERCC1-XPF to process such an intermediate in favor of retaining heterologous sequence in recombinants and generating crossovers with co-conversions, explaining the decrease in XO+GPT events we observed in WRN knock-down cells.
To further investigate a putative function for WRN in processing recombination intermediates that require resolution of nonhomologous sequences, we used a plasmid-chromosome gene targeting assay previously developed to investigate the role of ERCC1-XPF in removing end-blocking nonhomologies from recombination intermediates (Adair et al., 2000). In this assay, the single APRT copy in the CHO chromosome is inactive due to a small deletion, and a wild-type copy of APRT, interrupted in an exon by a large insertion of nonhomologous sequence, is transfected into cells as a linear molecule with long terminal nonhomologies flanking a DSB and blocking the homologous arms of an “ends-in” targeting vector (Fig. 4A). Experiments using isogenic repair proficient and ERCC1 null cells in this recombination assay demonstrated a specific requirement for ERCC1 for normal processing of recombination intermediates requiring removal of long terminal nonhomologies (Adair et al., 2000).
We used CHO cell line ATS-49tg, the same parental cell line used to generate the ERCC1 null mutant used by Adair et al. (2000), to construct isogenic WRN deficient cells (ATS-WRN) by sequential transformation of sh152 and sh2627, and a control cell line (ATS-luc), as described for 2115-WRN and 2115-luc. Using this plasmid-chromosome recombination assay, WRN deficient cells showed neither impaired nor enhanced targeted recombination at the APRT locus, as reflected in the APRT homologous targeting frequency data (Table 2). This result is in contrast to results from our intrachromosomal mitotic recombination assay, in which WRN deficient cells exhibited a hyperrecombination phenotype. Although the two recombination assays have a number of similarities in the types of recombination intermediates generated, they may differ in mechanisms of pathway choice, and the channeling of intermediates into different pathways for resolution. The intrachromosomal mitotic recombination assay probably provides a more physiologically relevant system to assess recombination occurring spontaneously in mammalian cells, while the plasmid-chromosome gene targeting assay assesses homologous recombination in which a DSB in one of the recombination partners (the plasmid) initiates the formation of intermediates, and is designed to test the specific effects of strand invasion-blocking terminal nonhomologies on their resolution. Differences in results from different recombination assays have been noted in yeast systems as well; for example, while Sgs1 mutants are hyperrecombinogenic in spontaneous mitotic recombination assays, Lo et al. (2006) demonstrated that such cells did not exhibit any changes in DSB-induced allelic HR frequency.
The plasmid-chromosome assay, while not indicating a hyperrecombinogenic phenotype in the absence of WRN, did reveal a novel phenotype of WRN deficient cells in the frequency of untargeted, nonhomologous integration of the targeting vector into the CHO genome. Results in Table 2 show that in WRN deficient cells, such random integrations were significantly reduced compared to the isogenic control (p=0.014). This decrease in nonhomologous recombination was not observed in isogenic ERCC1 null cells in analogous experiments with the same targeting vector (Adair et al., 2000), and indicates that WRN is required for efficient nonhomologous recombination. Consistent with this observation, no recombinants obtained from WRN deficient cells were determined to have arisen by vector correction (0/44 clones), requiring precise removal of nonhomologous tails, gap repair, and integration of the now corrected vector elsewhere in the genome by nonhomologous recombination. While any of these steps may ultimately be the reason that no vector correction events were obtained, no effect was observed on targeted integration events in the absence of WRN, suggesting that removal of end-blocking nonhomologous sequences was normal in these cells. In isogenic ERCC1 null cells, no vector correction events were recovered either (0/67 clones), but there also was nearly complete ablation of targeted integration events, consistent with a role for ERCC1-XPF in removing nonhomologous sequences both from strand invasion and gap repair intermediates (Adair et al., 2000). We therefore conclude that the deficiency in generating this recombinant class in ATS-WRN cells is due to a defect in nonhomologous recombination, and our results provide additional, cell-based evidence for a functional role of WRN in promoting or facilitating NHEJ.
A small number of aberrant recombinants was recovered from WRN deficient cells in both recombination assays, suggesting that WRN is required for normal resolution of recombination intermediates formed during mitotic and targeted recombination. However, fewer aberrant events were recovered in the absence of WRN when compared to analogous experiments performed with isogenic ERCC1 null cells. WRN may act to stabilize or unwind recombination intermediates in order to facilitate the activities of other recombination components. In vitro experiments have demonstrated WRN unwinding activity on a variety of structures including bubbles, forks and Holliday junctions as well as G4 tetraplex, triple helix, and D-loop structures (Cheng et al., 2003). In addition, WRN may have activity similar to the proposed activity of Srs2 whereby long terminal nonhomologies are stabilized by the helicase prior to their removal by Rad1-Rad10 (Paques and Haber, 1997; Sugawara et al., 2000). In the absence of such stabilization, inappropriate removal of the nonhomologous tails may occur, possibly resulting in deletions or other types of aberrant structures. This function may be redundant with other helicases as evidenced by the relatively small number of aberrant recombinants recovered in the absence of WRN in our studies.
Our results using WRN deficient CHO cells implicate this helicase in several aspects of recombination in mammalian cells. Results from the experiments described here indicate that WRN is clearly required to suppress excessive intrachromosomal mitotic recombination in response to spontaneous DNA damage, and that WRN facilitates efficient NHEJ. In the absence of WRN, the balance may shift to favor recombination over NHEJ, resulting in inappropriate HR that potentially contributes to increased genomic instability observed in cells from WS patients. Our data also indicate that once mitotic recombination has been initiated, WRN likely acts to influence homologous recombination to favor gene conversions, an action that promotes genomic stability by suppressing crossover-type events that could result in deletions and possible loss of heterozygosity. Finally, our results demonstrate that WRN has a role in the correct resolution of recombination intermediates formed during mitotic recombination and strand invasion, as evidenced by the recovery of aberrant events in WRN deficient cells using both recombination assays. Our results thus provide evidence supporting a complex role for WRN in mammalian recombination, not only in regulating entry into recombination and NHEJ pathways, but also acting within specific HR pathways to promote faithful recombination in conjunction with other protein partners, consistent with WRN acting as a multifunctional protein that promotes genomic stability.
Supplementary Material
Acknowledgments
This work was supported by USPHS grants from the NCI (P01 CA097175) and NIEHS (P30 ES007784). J.J.R was supported by a training grant (T32 CA009480).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aboussekhra A, Chanet R, Adjiri A, Fabre F. Semidominant suppressors of Srs2 helicase mutations of Saccharomyces cerevisiae map in the RAD51 gene, whose sequence predicts a protein with similarities to procaryotic RecA proteins. Mol Cell Biol. 1992;12:3224–3234. doi: 10.1128/mcb.12.7.3224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adair GM, Nairn RS, Wilson JH, Seidman MM, Brotherman KA, MacKinnon C, Scheerer JB. Targeted homologous recombination at the endogenous adenine phosphoribosyltransferase locus in Chinese hamster cells. Proc Natl Acad Sci U S A. 1989;86:4574–4578. doi: 10.1073/pnas.86.12.4574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adair GM, Rolig RL, Moore-Faver D, Zabelshansky M, Wilson JH, Nairn RS. Role of ERCC1 in removal of long non-homologous tails during targeted homologous recombination. EMBO J. 2000;19:5552–5561. doi: 10.1093/emboj/19.20.5552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguilera A, Klein HL. Genetic control of intrachromosomal recombination in Saccharomyces cerevisiae. I. Isolation and genetic characterization of hyper-recombination mutations. Genetics. 1988;119:779–790. doi: 10.1093/genetics/119.4.779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashton TM, Hickson ID. Yeast as a model system to study RecQ helicase function. DNA Repair (Amst) 9:303–314. doi: 10.1016/j.dnarep.2009.12.007. [DOI] [PubMed] [Google Scholar]
- Bachrati CZ, Hickson ID. RecQ helicases: guardian angels of the DNA replication fork. Chromosoma. 2008;117:219–233. doi: 10.1007/s00412-007-0142-4. [DOI] [PubMed] [Google Scholar]
- Baynton K, Otterlei M, Bjoras M, von Kobbe C, Bohr VA, Seeberg E. WRN interacts physically and functionally with the recombination mediator protein RAD52. J Biol Chem. 2003;278:36476–36486. doi: 10.1074/jbc.M303885200. [DOI] [PubMed] [Google Scholar]
- Bennett RJ, Keck JL, Wang JC. Binding specificity determines polarity of DNA unwinding by the Sgs1 protein of S. cerevisiae. J Mol Biol. 1999;289:235–248. doi: 10.1006/jmbi.1999.2739. [DOI] [PubMed] [Google Scholar]
- Bohr VA. Rising from the RecQ-age: the role of human RecQ helicases in genome maintenance. Trends Biochem Sci. 2008;33:609–620. doi: 10.1016/j.tibs.2008.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brosh RM, Jr, von Kobbe C, Sommers JA, Karmakar P, Opresko PL, Piotrowski J, Dianova I, Dianov GL, Bohr VA. Werner syndrome protein interacts with human flap endonuclease 1 and stimulates its cleavage activity. EMBO J. 2001;20:5791–5801. doi: 10.1093/emboj/20.20.5791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brosh RM, Jr, Bohr VA. Roles of the Werner syndrome protein in pathways required for maintenance of genome stability. Exp Gerontol. 2002;37:491–506. doi: 10.1016/s0531-5565(01)00227-3. [DOI] [PubMed] [Google Scholar]
- Brosh RM, Jr, Bohr VA. Human premature aging, DNA repair and RecQ helicases. Nucleic Acids Res. 2007;35:7527–7544. doi: 10.1093/nar/gkm1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Huang S, Lee L, Davalos A, Schiestl RH, Campisi J, Oshima J. WRN, the protein deficient in Werner syndrome, plays a critical structural role in optimizing DNA repair. Aging Cell. 2003;2:191–199. doi: 10.1046/j.1474-9728.2003.00052.x. [DOI] [PubMed] [Google Scholar]
- Cheng RZ, Murano S, Kurz B, Shmookler Reis RJ. Homologous recombination is elevated in some Werner-like syndromes but not during normal in vitro or in vivo senescence of mammalian cells. Mutat Res. 1990;237:259–269. doi: 10.1016/0921-8734(90)90008-f. [DOI] [PubMed] [Google Scholar]
- Cheng WH, Opresko PL, von Kobbe C, Harrigan JA, Bohr VA. The human Werner syndrome as a model system for aging. In: Nystrom T, Osiewacz HD, editors. Topics in Current Genetics. Springer-Verlag; Berlin: 2003. pp. 239–268. [Google Scholar]
- Cheng WH, von Kobbe C, Opresko PL, Arthur LM, Komatsu K, Seidman MM, Carney JP, Bohr VA. Linkage between Werner syndrome protein and the Mre11 complex via Nbs1. J Biol Chem. 2004;279:21169–21176. doi: 10.1074/jbc.M312770200. [DOI] [PubMed] [Google Scholar]
- Cheng WH, Kusumoto R, Opresko PL, Sui X, Huang S, Nicolette ML, Paull TT, Campisi J, Seidman M, Bohr VA. Collaboration of Werner syndrome protein and BRCA1 in cellular responses to DNA interstrand cross-links. Nucleic Acids Res. 2006;34:2751–2760. doi: 10.1093/nar/gkl362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper MP, Machwe A, Orren DK, Brosh RM, Ramsden D, Bohr VA. Ku complex interacts with and stimulates the Werner protein. Genes Dev. 2000;14:907–912. [PMC free article] [PubMed] [Google Scholar]
- Dhillon KK, Sidorova J, Saintigny Y, Poot M, Gollahon K, Rabinovitch PS, Monnat RJ., Jr Functional role of the Werner syndrome RecQ helicase in human fibroblasts. Aging Cell. 2007;6:53–61. doi: 10.1111/j.1474-9726.2006.00260.x. [DOI] [PubMed] [Google Scholar]
- Doe CL, Dixon J, Osman F, Whitby MC. Partial suppression of the fission yeast rqh1(-) phenotype by expression of a bacterial Holliday junction resolvase. EMBO J. 2000;19:2751–2762. doi: 10.1093/emboj/19.11.2751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dronkert ML, Kanaar R. Repair of DNA interstrand cross-links. Mutat Res. 2001;486:217–247. doi: 10.1016/s0921-8777(01)00092-1. [DOI] [PubMed] [Google Scholar]
- Dupaigne P, Le Breton C, Fabre F, Gangloff S, Le Cam E, Veaute X. The Srs2 helicase activity is stimulated by Rad51 filaments on dsDNA: implications for crossover incidence during mitotic recombination. Mol Cell. 2008;29:243–254. doi: 10.1016/j.molcel.2007.11.033. [DOI] [PubMed] [Google Scholar]
- Foster PL. Methods for determining spontaneous mutation rates. Methods Enzymol. 2006;409:195–213. doi: 10.1016/S0076-6879(05)09012-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franchitto A, Pirzio LM, Prosperi E, Sapora O, Bignami M, Pichierri P. Replication fork stalling in WRN-deficient cells is overcome by prompt activation of a MUS81-dependent pathway. J Cell Biol. 2008;183:241–252. doi: 10.1083/jcb.200803173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedl AA, Liefshitz B, Steinlauf R, Kupiec M. Deletion of the SRS2 gene suppresses elevated recombination and DNA damage sensitivity in rad5 and rad18 mutants of Saccharomyces cerevisiae. Mutat Res. 2001;486:137–146. doi: 10.1016/s0921-8777(01)00086-6. [DOI] [PubMed] [Google Scholar]
- Fukuchi K, Tanaka K, Nakura J, Kumahara Y, Uchida T, Okada Y. Elevated spontaneous mutation rate in SV40-transformed Werner syndrome fibroblast cell lines. Somat Cell Mol Genet. 1985;11:303–308. doi: 10.1007/BF01534688. [DOI] [PubMed] [Google Scholar]
- Fukuchi K, Martin GM, Monnat RJ., Jr Mutator phenotype of Werner syndrome is characterized by extensive deletions. Proc Natl Acad Sci U S A. 1989;86:5893–5897. doi: 10.1073/pnas.86.15.5893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gangloff S, McDonald JP, Bendixen C, Arthur L, Rothstein R. The yeast type I topoisomerase Top3 interacts with Sgs1, a DNA helicase homolog: a potential eukaryotic reverse gyrase. Mol Cell Biol. 1994;14:8391–8398. doi: 10.1128/mcb.14.12.8391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gangloff S, Soustelle C, Fabre F. Homologous recombination is responsible for cell death in the absence of the Sgs1 and Srs2 helicases. Nat Genet. 2000;25:192–194. doi: 10.1038/76055. [DOI] [PubMed] [Google Scholar]
- Gebhart E, Bauer R, Raub U, Schinzel M, Ruprecht KW, Jonas JB. Spontaneous and induced chromosomal instability in Werner syndrome. Hum Genet. 1988;80:135–139. doi: 10.1007/BF00702855. [DOI] [PubMed] [Google Scholar]
- Hall BM, Ma CX, Liang P, Singh KK. Fluctuation analysis CalculatOR: a web tool for the determination of mutation rate using Luria-Delbruck fluctuation analysis. Bioinformatics. 2009;25:1564–1565. doi: 10.1093/bioinformatics/btp253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinz JM. Role of homologous recombination in DNA interstrand crosslink repair. Environ Mol Mutagen. 2010;51:582–603. doi: 10.1002/em.20577. [DOI] [PubMed] [Google Scholar]
- Ira G, Malkova A, Liberi G, Foiani M, Haber JE. Srs2 and Sgs1-Top3 suppress crossovers during double-strand break repair in yeast. Cell. 2003;115:401–411. doi: 10.1016/s0092-8674(03)00886-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones ME. An improved estimator of spontaneous mutation rates in Luria-Delbruck fluctuation experiments. Mutat Res. 1993;292:191–198. doi: 10.1016/0165-1161(93)90147-r. [DOI] [PubMed] [Google Scholar]
- Karmakar P, Piotrowski J, Brosh RM, Jr, Sommers JA, Miller SP, Cheng WH, Snowden CM, Ramsden DA, Bohr VA. Werner protein is a target of DNA-dependent protein kinase in vivo and in vitro, and its catalytic activities are regulated by phosphorylation. J Biol Chem. 2002;277:18291–18302. doi: 10.1074/jbc.M111523200. [DOI] [PubMed] [Google Scholar]
- Karmakar P, Snowden CM, Ramsden DA, Bohr VA. Ku heterodimer binds to both ends of the Werner protein and functional interaction occurs at the Werner N-terminus. Nucleic Acids Res. 2002;30:3583–3591. doi: 10.1093/nar/gkf482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krejci L, Van Komen S, Li Y, Villemain J, Reddy MS, Klein H, Ellenberger T, Sung P. DNA helicase Srs2 disrupts the Rad51 presynaptic filament. Nature. 2003;423:305–309. doi: 10.1038/nature01577. [DOI] [PubMed] [Google Scholar]
- Kusumoto R, Dawut L, Marchetti C, Wan Lee J, Vindigni A, Ramsden D, Bohr VA. Werner protein cooperates with the XRCC4-DNA ligase IV complex in end-processing. Biochemistry. 2008;47:7548–7556. doi: 10.1021/bi702325t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, Comai L. Functional interaction between Ku and the werner syndrome protein in DNA end processing. J Biol Chem. 2000;275:39800. doi: 10.1074/jbc.C000289200. [DOI] [PubMed] [Google Scholar]
- Li B, Comai L. Displacement of DNA-PKcs from DNA ends by the Werner syndrome protein. Nucleic Acids Res. 2002;30:3653–3661. doi: 10.1093/nar/gkf488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Heyer WD. Homologous recombination in DNA repair and DNA damage tolerance. Cell Res. 2008;18:99–113. doi: 10.1038/cr.2008.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo YC, Paffett KS, Amit O, Clikeman JA, Sterk R, Brenneman MA, Nickoloff JA. Sgs1 regulates gene conversion tract lengths and crossovers independently of its helicase activity. Mol Cell Biol. 2006;26:4086–4094. doi: 10.1128/MCB.00136-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma WT. Analysis of the Luria-Delbruck distribution using discrete convolution powers. Journal of Applied Probability. 1992:29. [Google Scholar]
- Marini V, Krejci L. Srs2: the “Odd-Job Man” in DNA repair. DNA Repair (Amst) 2010;9:268–275. doi: 10.1016/j.dnarep.2010.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin GM, Sprague CA, Epstein CJ. Replicative life-span of cultivated human cells. Effects of donor’s age, tissue, and genotype. Lab Invest. 1970;23:86–92. [PubMed] [Google Scholar]
- Martin GM. Genetic syndromes in man with potential relevance to the pathobiology of aging. Birth Defects Original Article Series. 1978;14:5–39. [PubMed] [Google Scholar]
- Martin GM, Oshima J, Gray MD, Poot M. What geriatricians should know about the Werner syndrome. J Am Geriatr Soc. 1999;47:1136–1144. doi: 10.1111/j.1532-5415.1999.tb05240.x. [DOI] [PubMed] [Google Scholar]
- Merrihew RV, Sargent RG, Wilson JH. Efficient modification of the APRT gene by FLP/FRT site-specific targeting. Somat Cell Mol Genet. 1995;21:299–307. doi: 10.1007/BF02257465. [DOI] [PubMed] [Google Scholar]
- Nairn RS, Adair GM. Use of gene targeting to study recombination in mammalian cell DNA repair mutants. Methods Mol Biol. 2006;314:133–154. doi: 10.1385/1-59259-973-7:133. [DOI] [PubMed] [Google Scholar]
- Niedernhofer LJ, Essers J, Weeda G, Beverloo B, de Wit J, Muijtjens M, Odijk H, Hoeijmakers JH, Kanaar R. The structure-specific endonuclease Ercc1-Xpf is required for targeted gene replacement in embryonic stem cells. EMBO J. 2001;20:6540–6549. doi: 10.1093/emboj/20.22.6540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niedernhofer LJ, Garinis GA, Raams A, Lalai AS, Robinson AR, Appeldoorn E, Odijk H, Oostendorp R, Ahmad A, van Leeuwen W, Theil AF, Vermeulen W, van der Horst GT, Meinecke P, Kleijer WJ, Vijg J, Jaspers NG, Hoeijmakers JH. A new progeroid syndrome reveals that genotoxic stress suppresses the somatotroph axis. Nature. 2006;444:1038–1043. doi: 10.1038/nature05456. [DOI] [PubMed] [Google Scholar]
- Ogburn CE, Oshima J, Poot M, Chen R, Hunt KE, Gollahon KA, Rabinovitch PS, Martin GM. An apoptosis-inducing genotoxin differentiates heterozygotic carriers for Werner helicase mutations from wild-type and homozygous mutants. Hum Genet. 1997;101:121–125. doi: 10.1007/s004390050599. [DOI] [PubMed] [Google Scholar]
- Orren DK, Machwe A, Karmakar P, Piotrowski J, Cooper MP, Bohr VA. A functional interaction of Ku with Werner exonuclease facilitates digestion of damaged DNA. Nucleic Acids Res. 2001;29:1926–1934. doi: 10.1093/nar/29.9.1926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paques F, Haber JE. Two pathways for removal of nonhomologous DNA ends during double-strand break repair in Saccharomyces cerevisiae. Mol Cell Biol. 1997;17:6765–6771. doi: 10.1128/mcb.17.11.6765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pichierri P, Franchitto A, Mosesso P, Palitti F. Werner’s syndrome protein is required for correct recovery after replication arrest and DNA damage induced in S-phase of cell cycle. Mol Biol Cell. 2001;12:2412–2421. doi: 10.1091/mbc.12.8.2412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poot M, Hoehn H, Runger TM, Martin GM. Impaired S-phase transit of Werner syndrome cells expressed in lymphoblastoid cell lines. Exp Cell Res. 1992;202:267–273. doi: 10.1016/0014-4827(92)90074-i. [DOI] [PubMed] [Google Scholar]
- Poot M, Gollahon KA, Rabinovitch PS. Werner syndrome lymphoblastoid cells are sensitive to camptothecin-induced apoptosis in S-phase. Hum Genet. 1999;104:10–14. doi: 10.1007/s004390050903. [DOI] [PubMed] [Google Scholar]
- Poot M, Yom JS, Whang SH, Kato JT, Gollahon KA, Rabinovitch PS. Werner syndrome cells are sensitive to DNA cross-linking drugs. FASEB J. 2001;15:1224–1226. doi: 10.1096/fj.00-0611fje. [DOI] [PubMed] [Google Scholar]
- Poot M, Gollahon KA, Emond MJ, Silber JR, Rabinovitch PS. Werner syndrome diploid fibroblasts are sensitive to 4-nitroquinoline-N-oxide and 8-methoxypsoralen: implications for the disease phenotype. FASEB J. 2002;16:757–758. doi: 10.1096/fj.01-0906fje. [DOI] [PubMed] [Google Scholar]
- Prince PR, Emond MJ, Monnat RJ., Jr Loss of Werner syndrome protein function promotes aberrant mitotic recombination. Genes Dev. 2001;15:933–938. doi: 10.1101/gad.877001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosche WA, Foster PL. Determining mutation rates in bacterial populations. Methods. 2000;20:4–17. doi: 10.1006/meth.1999.0901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saintigny Y, Makienko K, Swanson C, Emond MJ, Monnat RJ., Jr Homologous recombination resolution defect in Werner syndrome. Mol Cell Biol. 2002;22:6971–6978. doi: 10.1128/MCB.22.20.6971-6978.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salk D. Werner’s syndrome: a review of recent research with an analysis of connective tissue metabolism, growth control of cultured cells, and chromosomal aberrations. Human Genetics. 1982;62:1–5. doi: 10.1007/BF00295598. [DOI] [PubMed] [Google Scholar]
- Sargent RG, Rolig RL, Kilburn AE, Adair GM, Wilson JH, Nairn RS. Recombination-dependent deletion formation in mammalian cells deficient in the nucleotide excision repair gene ERCC1. Proc Natl Acad Sci U S A. 1997;94:13122–13127. doi: 10.1073/pnas.94.24.13122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sargent RG, Meservy JL, Perkins BD, Kilburn AE, Intody Z, Adair GM, Nairn RS, Wilson JH. Role of the nucleotide excision repair gene ERCC1 in formation of recombination-dependent rearrangements in mammalian cells. Nucleic Acids Res. 2000;28:3771–3778. doi: 10.1093/nar/28.19.3771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkar S, Ma WT, Sandri GH. On fluctuation analysis: a new, simple and efficient method for computing the expected number of mutants. Genetica. 1992;85:173–179. doi: 10.1007/BF00120324. [DOI] [PubMed] [Google Scholar]
- Smith DG, Adair GM. Characterization of an apparent hotspot for spontaneous mutation in exon 5 of the Chinese hamster APRT gene. Mutat Res. 1996;352:87–96. doi: 10.1016/0027-5107(96)00007-3. [DOI] [PubMed] [Google Scholar]
- Spell RM, Jinks-Robertson S. Examination of the roles of Sgs1 and Srs2 helicases in the enforcement of recombination fidelity in Saccharomyces cerevisiae. Genetics. 2004;168:1855–1865. doi: 10.1534/genetics.104.032771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart FM, Gordon DM, Levin BR. Fluctuation analysis: the probability distribution of the number of mutants under different conditions. Genetics. 1990;124:175–185. doi: 10.1093/genetics/124.1.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart FM. Fluctuation tests: how reliable are the estimates of mutation rates? Genetics. 1994;137:1139–1146. doi: 10.1093/genetics/137.4.1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanson C, Saintigny Y, Emond MJ, Monnat RJ., Jr The Werner syndrome protein has separable recombination and survival functions. DNA Repair (Amst) 2004;3:475–482. doi: 10.1016/j.dnarep.2004.01.002. [DOI] [PubMed] [Google Scholar]
- Talbert LL, Coletta LD, Lowery MG, Bolt A, Trono D, Adair GM, Nairn RS. Characterization of CHO XPF mutant UV41: influence of XPF heterozygosity on double-strand break-induced intrachromosomal recombination. DNA Repair (Amst) 2008;7:1319–1329. doi: 10.1016/j.dnarep.2008.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veaute X, Jeusset J, Soustelle C, Kowalczykowski SC, Le Cam E, Fabre F. The Srs2 helicase prevents recombination by disrupting Rad51 nucleoprotein filaments. Nature. 2003;423:309–312. doi: 10.1038/nature01585. [DOI] [PubMed] [Google Scholar]
- Walker JR, Corpina RA, Goldberg J. Structure of the Ku heterodimer bound to DNA and its implications for double-strand break repair. Nature. 2001;412:607–614. doi: 10.1038/35088000. [DOI] [PubMed] [Google Scholar]
- Watt PM, Hickson ID, Borts RH, Louis EJ. SGS1, a homologue of the Bloom’s and Werner’s syndrome genes, is required for maintenance of genome stability in Saccharomyces cerevisiae. Genetics. 1996;144:935–945. doi: 10.1093/genetics/144.3.935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamagata K, Kato J, Shimamoto A, Goto M, Furuichi Y, Ikeda H. Bloom’s and Werner’s syndrome genes suppress hyperrecombination in yeast sgs1 mutant: implication for genomic instability in human diseases. Proc Natl Acad Sci U S A. 1998;95:8733–8738. doi: 10.1073/pnas.95.15.8733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang N, Kaur R, Lu X, Shen X, Li L, Legerski RJ. The Pso4 mRNA splicing and DNA repair complex interacts with WRN for processing of DNA interstrand cross-links. J Biol Chem. 2005;280:40559–40567. doi: 10.1074/jbc.M508453200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.